


Abstract
The fatal drug-drug interaction between sorivudine, an antiviral drug, and 5-fluorouracil (5-FU) has been shown to be caused by a mechanism-based inhibition. In this interaction, sorivudine is converted by gut flora to (E)-5-(2-bromovinyl)uracil (BVU), which is metabolically activated by dihydropyrimidine dehydrogenase (DPD), and the activated BVU irreversibly binds to DPD itself, thereby inactivating it. In an attempt to predict this interaction in vivo from in vitro data, inhibition of 5-FU metabolism by BVU was investigated by using rat and human hepatic cytosol and human recombinant DPD. Whichever enzyme was used, increased inhibition was observed that depended on the preincubation time of BVU and enzyme in the presence of NADPH and BVU concentration. The kinetic parameters obtained for inactivation represented by kinact andK′app were 2.05 ± 1.52 min−1, 69.2 ± 60.8 μM (rat hepatic cytosol), 2.39 ± 0.13 min−1, 48.6 ± 11.8 μM (human hepatic cytosol), and 0.574 ± 0.121 min−1, 2.20 ± 0.57 μM (human recombinant DPD). The drug-drug interaction in vivo was predicted quantitatively based on a physiologically based pharmacokinetic model, using pharmacokinetic parameters obtained from the literature and kinetic parameters for the enzyme inactivation obtained in the in vitro studies. In rats, DPD was predicted to be completely inactivated by administration of BVU and the area under the curve of 5-FU was predicted to increase 11-fold, which agreed well with the reported data. In humans, a 5-fold increase in the area under the curve of 5-FU was predicted after administration of sorivudine, 150 mg/day for 5 days. Mechanism-based inhibition of drug metabolism is supposed to be very dangerous. We propose that such in vitro studies should be carried out during the drug-developing phase so that in vivo drug-drug interactions can be predicted.
A drug-drug interaction between sorivudine, an antiviral drug, and 5-fluorouracil (5-FU)1, an anticancer drug, caused one of the most serious cases of toxicity ever seen in Japan. In 1993, 15 Japanese patients with cancer and herpes zoster died from 5-FU toxicity, which was strongly suggested to be caused by high blood concentrations attributable to the interaction between 5-FU and sorivudine (Pharmaceutical Affairs Bureau, 1994; Okuda et al., 1997).
The interaction between sorivudine and 5-FU is based on a “mechanism-based inhibition”, which differs from competitive or noncompetitive inhibition (Desgranges et al., 1986; Okuda et al., 1997). A mechanism-based inhibitor is metabolized by an enzyme to form a metabolite that covalently binds to the same enzyme, leading to irreversible inactivation of the enzyme. Sorivudine is converted by gut flora to (E)-5-(2-bromovinyl)uracil (BVU), which is metabolically activated to dihydro-BVU [5-(2-bromoethylidenyl)uracil], an allyl bromide type of reactive intermediate, in the presence of NADPH, by dihydropyrimidine dehydrogenase (DPD), a rate-limiting enzyme in the metabolism of 5-FU (Okuda et al., 1998). Then, the dihydro-BVU irreversibly binds to DPD itself. A similar inhibition mechanism is reported for macrolide antibiotics such as erythromycin, in which case P450 demethylates the macrolide antibiotic to a nitrosoalkane, which forms a stable, inactive complex with P450 (Periti et al., 1992).
This type of interaction deserves more attention than the more common type of inhibition because the inhibitory effect remains after elimination of the inhibitor from blood and tissue, and this can lead to serious side effects. Furthermore, in the prediction of such drug-drug interactions in vivo from in vitro studies, it is necessary to consider the exposure time of the enzyme to the inhibitor and the enzyme turnover (Ito et al., 1998). In the present study, as a case of drug-drug interaction involving mechanism-based inhibition, interaction between 5-FU and sorivudine in rat and human was investigated with our method for the quantitative prediction of in vivo interactions from in vitro data.
Materials and Methods
Chemicals.
[6-14C]5-FU (56 mCi/mmol) was purchased from Moravek Biochemicals Inc. (Brea, CA). BVU was purchased from Sigma Chemical Co. (St. Louis, MO). [2′-14C]BVU was prepared as reported previously, and the specific activity was 57.4 mCi/mmol (Okuda et al., 1997).
Enzyme Sources.
Rat hepatic cytosol was prepared from 7-week-old Sprague-Dawley rats (Lu et al., 1992; Okuda et al., 1997).
Twenty-one cancer patients (13 males and 8 females; mean age, 59.7 years; range, 29–76 years), hospitalized in the National Cancer Center Hospital, Tokyo, Japan, were entered into the present study. Informed consent was obtained from each patient before study entry. All patients underwent partial hepatectomy to remove liver metastases of colon cancer. Pathologically and histologically normal liver samples used in the study were obtained from normal portions of the removed tissue. All of the samples were rapidly frozen in liquid nitrogen and stored at −80°C before use. An equivalent mixture of the cytosol preparations from livers of five patients (three males and two females; 49–71 years) was used in the metabolic inhibition assay (Lu et al., 1992; Okuda et al., 1997). Human recombinant DPD (rhDPD) was expressed in Escherichia coli and purified (Ogura et al., 1998), which was stored at −80°C before use.
Quantification of the DPD Content in Human Liver Cytosol by Enhanced Chemiluminescence (ECL) Western Blot.
Frozen liver samples were homogenized in 4 volumes of 35 mM K-phosphate buffer, pH 7.4, containing 2.5 mM MgCl2 and 10 mM 2-mercaptoethanol (buffer A). The homogenate was centrifuged at 105,000g for 60 min to obtain a cytosolic fraction. SDS-polyacrylamide gel electrophoresis (Laemmli, 1970) and Western blots (Towbin et al., 1979) of human hepatic cytosols (50 μg of protein each) were performed by previously described methods. Detection of immunoreactive bands was performed with an ECL Western blotting analysis system (Amersham International Plc., Buckinghamshire, UK) with rabbit polyclonal antibody raised against purified rhDPD (Ogura et al., 1998) as the primary antibody. The DPD protein content was quantified from the Western blots with a Shimadzu model CS9000 scanning densitometer (Shimadzu, Kyoto, Japan) by referring to a calibration curve constructed by using different amounts of purified DPD. The calibration curve showed linearity from 6.25 to 100 ng of the purified protein. Data were obtained from at least three experiments.
Determination of DPD Activity.
The DPD activity of human liver cytosols was determined as reported previously with [14C]5-FU as a substrate (Okuda et al., 1997). The reaction mixture containing buffer A, 200 μM NADPH, 20 μM [14C]5-FU, and hepatic cytosol (30 μg of protein) in a final volume of 50 μl was incubated at 37°C for 5 min. The reaction was stopped by adding 5 μl of 0.3 N KOH and boiling at 100°C for 5 min. 5-FU and its metabolite, α-fluoro-β-alanine (FBAL), were separated by thin-layer chromotography (plate: Polygram CEL 300 PEI/UV254; Machery-Nagel, Germany; solvent: t-butanol/ethyl acetate/water, 4:3:2) and quantified by BAS-2000II (Fujifilm, Tokyo, Japan) to calculate the FBAL formation rate.
Inhibition Study.
The 5-FU metabolizing activity of the cytosol and rhDPD was measured as reported previously (Okuda et al., 1997). Preincubation mixture (final volume: 50 μl) consisted of rat hepatic cytosol (10%) or human hepatic cytosol (10%) or rhDPD (0.6 μg), BVU (0–20 μM for rat hepatic cytosol and rhDPD; 0–200 μM for human hepatic cytosol), 200 μM NADPH, 2.5 mM MgCl2, and 30% (v/v) glycerol-35 mM K-phosphate buffer (pH 7.4). The preincubation was performed at 37°C for 0, 0.5, 1, 2.5, 5, 10, or 20 min (rat hepatic cytosol and rhDPD) or 0, 0.5, 1, or 2.5 min (human hepatic cytosol). An aliquot (5 μl for rat hepatic cytosol and rhDPD; 10 μl for human hepatic cytosol) was added to the incubation mixture (buffer A containing 20 μM [14C]5-FU and 200 μM NADPH). The final volume of the incubation mixture was 50 μl, and it was incubated at 37°C for 2 min.
Analysis of Enzyme Inactivation Kinetics.
Kinetic parameters for enzyme inactivation were obtained as reported elsewhere (Ito et al., 1998). The logarithm of the remaining enzymatic activity (formation rate of FBAL) was plotted against the preincubation time. The apparent inactivation rate constant (kobs) was determined from the slope of the initial linear phase. The value of kobs was plotted against the BVU concentration and the parameters (kinact andK′app) were obtained by the nonlinear least-squares method (MULTI program; Yamaoka et al., 1981) (Waley, 1985; Silverman, 1988). The following equation was used:
Rat Plasma and Human Serum Protein Binding of BVU.
A total of 600 μl of plasma prepared from 7-week-old male Sprague-Dawley rats or human serum (Sigma Chemical Co., St. Louis, MO) was added with [2′-14C]BVU to obtain final concentrations of 1 and 3.5 μM. The mixture was immediately transferred into the sample reservoir of a Centrifree Micropartition System (molecular mass cut-off of 30,000 Da; Amicon, Inc., Beverly, MA) after incubation at 37°C for 3 min and ultracentrifuged. The protein binding ratio was calculated from the radioactivity of the filtrate and remaining solution. The BVU concentration was set at 1 and 3.5 μM in the protein binding assay because the maximum plasma concentration of BVU was approximately 2 μM in humans after repeated oral administration of sorivudine at a dose of 150 mg/day (Ogiwara et al., 1990). The protein binding assay with rat plasma was also performed at the same concentration of BVU as in the assay that used human serum.
All assays were performed in triplicate. Data were expressed as mean ± S.D.
Quantitative Prediction of 5-FU/Sorivudine Interaction.
The differential equations for active and inactive DPD in the liver (Eact and Einact, respectively) can be described as follows:
Equations for rats (5-FU administration).
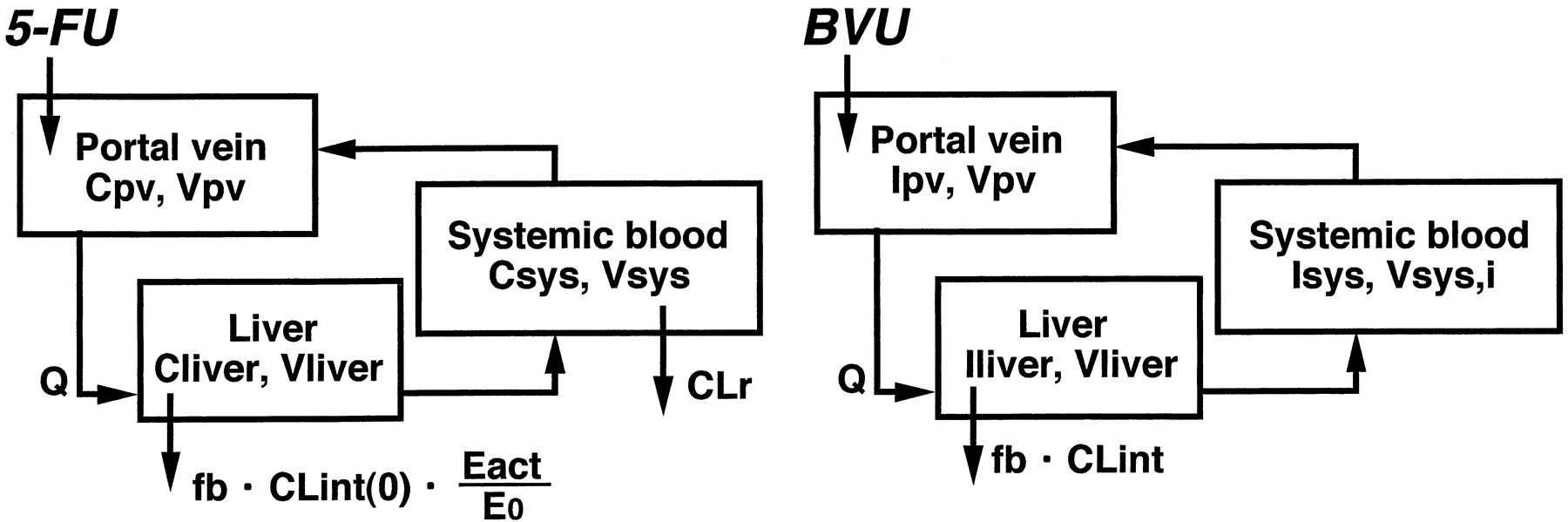
The differential equations for 5-FU (C) and BVU (I) can be expressed as follows according to the perfusion model (Fig.1):

Physiological model for the description of the time profiles of 5-FU and BVU concentrations in rats.
See Materials and Methods for the abbreviations used.
For 5-FU:
The following assumptions have been made in the above mass-balance equations:
- 1.
- 5-FU and BVU are administered orally.
- 2.
- BVU is eliminated only in the liver.
- 3.
- Distribution of 5-FU and BVU in the liver rapidly reaches equilibrium and the unbound concentrations in the hepatic vein are equal to those in the liver at equilibrium (well-stirred model).
- 4.
- The unbound molecule in the liver is subject to elimination.
- 5.
- CLint, the intrinsic clearance for hepatic elimination of BVU, is constant, independent of time.
- 6.
- Gastrointestinal absorption can be described by a first-order rate constant.
The pharmacokinetic parameters of 5-FU and BVU were determined from information in the literature (Tables1 and 2). By using the program STELLA II (High Performance Systems, Inc., Hanover, NH) and kinetic parameters for DPD inactivation determined in in vitro studies, the above differential equations were numerically solved to simulate the effects of BVU coadministration with oral 5-FU (200 μmol/kg). BVU (200 μmol/kg) was assumed to be orally administered 1 h before administration of 5-FU, and the time-courses of BVU blood concentration, active DPD content in the liver (Eact), and 5-FU blood concentration were simulated.
Pharmacokinetic parameters of 5-FU (200 μmol/kg dose) in rats
Pharmacokinetic parameters of BVU (200 μmol/kg dose) in rats
Equations for humans (tegafur administration).
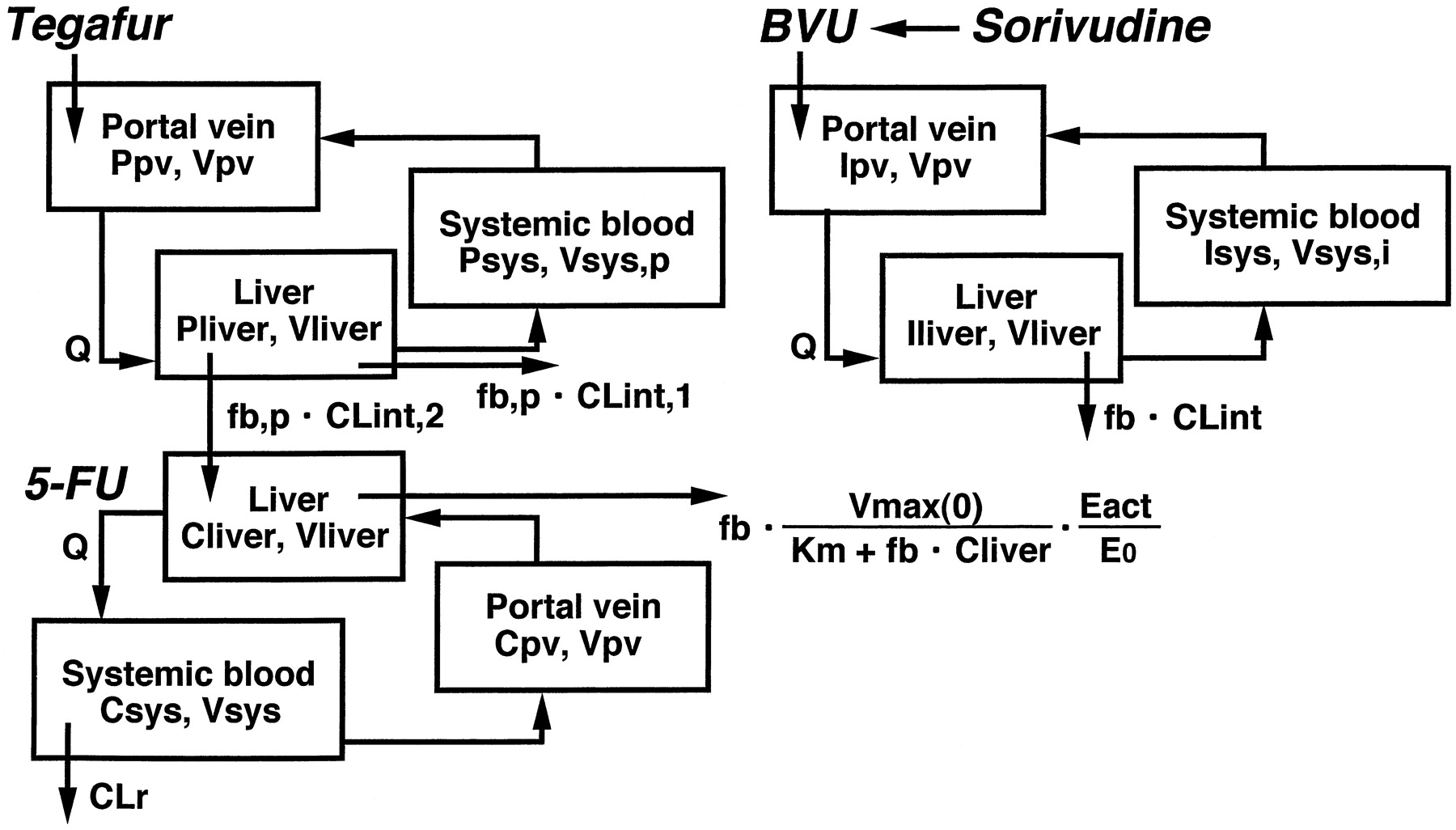
The differential equations for tegafur (P; a prodrug of 5-FU), 5-FU (C), and BVU (I) can be expressed as follows according to the perfusion model (Fig. 2):

Physiological model for the description of the time-profiles of tegafur, 5-FU, and BVU concentrations in humans.
Oral administration of tegafur (a prodrug of 5-FU) was assumed. See Materials and Methods for the abbreviations used.
For tegafur:
the same as equations 9 through 12,
where Pliver, Ppv, and Psys represent the concentration of tegafur in the liver, portal vein, and central compartment, respectively; CLint,1 represents the CLint for non-5-FU production from tegafur, and CLint,2 represents the CLint for 5-FU production from tegafur.
The following assumptions have been made in the above mass-balance equations for tegafur, 5-FU and BVU:
- 1.
- Some fraction of the orally administered tegafur is metabolized to 5-FU in the liver. Oral administration of BVU was assumed because sorivudine is metabolized to BVU by gut flora.
- 2.
- Tegafur and BVU are eliminated only in the liver.
- 3.
- Distribution of tegafur, 5-FU, and BVU in the liver rapidly reaches equilibrium, and the unbound concentrations in the hepatic vein are equal to those in the liver at equilibrium (well-stirred model).
- 4.
- The unbound molecule in the liver is subject to elimination.
- 5.
- CLint, the intrinsic clearance for hepatic elimination of BVU, is constant, independent of the time.
- 6.
- Gastrointestinal absorption can be described by a first-order rate constant.
The pharmacokinetic parameters of tegafur, 5-FU, and BVU were determined from information in the literature (Tables3 and 4). By using the program STELLA II and kinetic parameters for DPD inactivation determined in in vitro studies, the above differential equations were numerically solved to simulate the effects of sorivudine coadministration with oral tegafur (2500 μmol b.i.d. for 7 days). Sorivudine was assumed to be orally administered for 5 days, starting from the 3rd day of tegafur administration at a dose of 50 mg t.i.d. or 37 μmol t.i.d. as BVU; urinary excretion of unchanged sorivudine was 74.1% of the dose (Ogiwara et al., 1990) and BVU was assumed to correspond to the remaining 25.9%. The time-courses of BVU blood concentration, active DPD content in the liver (Eact), and 5-FU blood concentration were simulated.
Pharmacokinetic parameters of tegafur and 5-FU in humans
Pharmacokinetic parameters of BVU (37 μmol dose t.i.d.) in humans
Results
Correlation between 5-FU-Reducing Activity and DPD Content of Human Liver Cytosol.
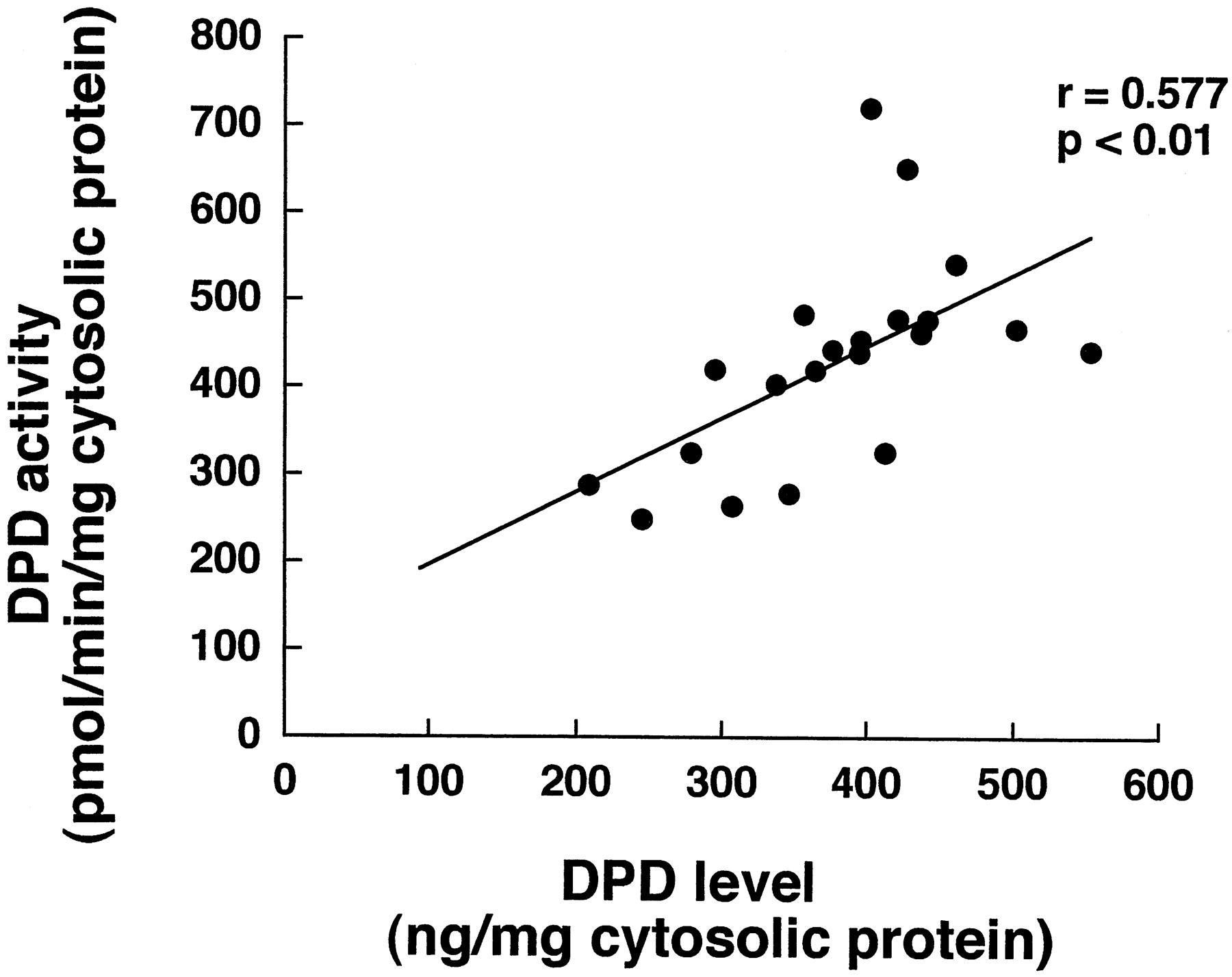
Figure 3 shows the correlation between DPD content and 5-FU-metabolizing activity, estimated from the FBAL formation rate, of 21 samples of human hepatic cytosol. 5-FU-metabolizing activity depended on the DPD content (r = 0.577; P < .01), confirming the involvement of DPD in 5-FU metabolism (Diasio and Harris, 1989). Furthermore, approximately a 3-fold interindividual difference, at maximum, was observed in the DPD activity.
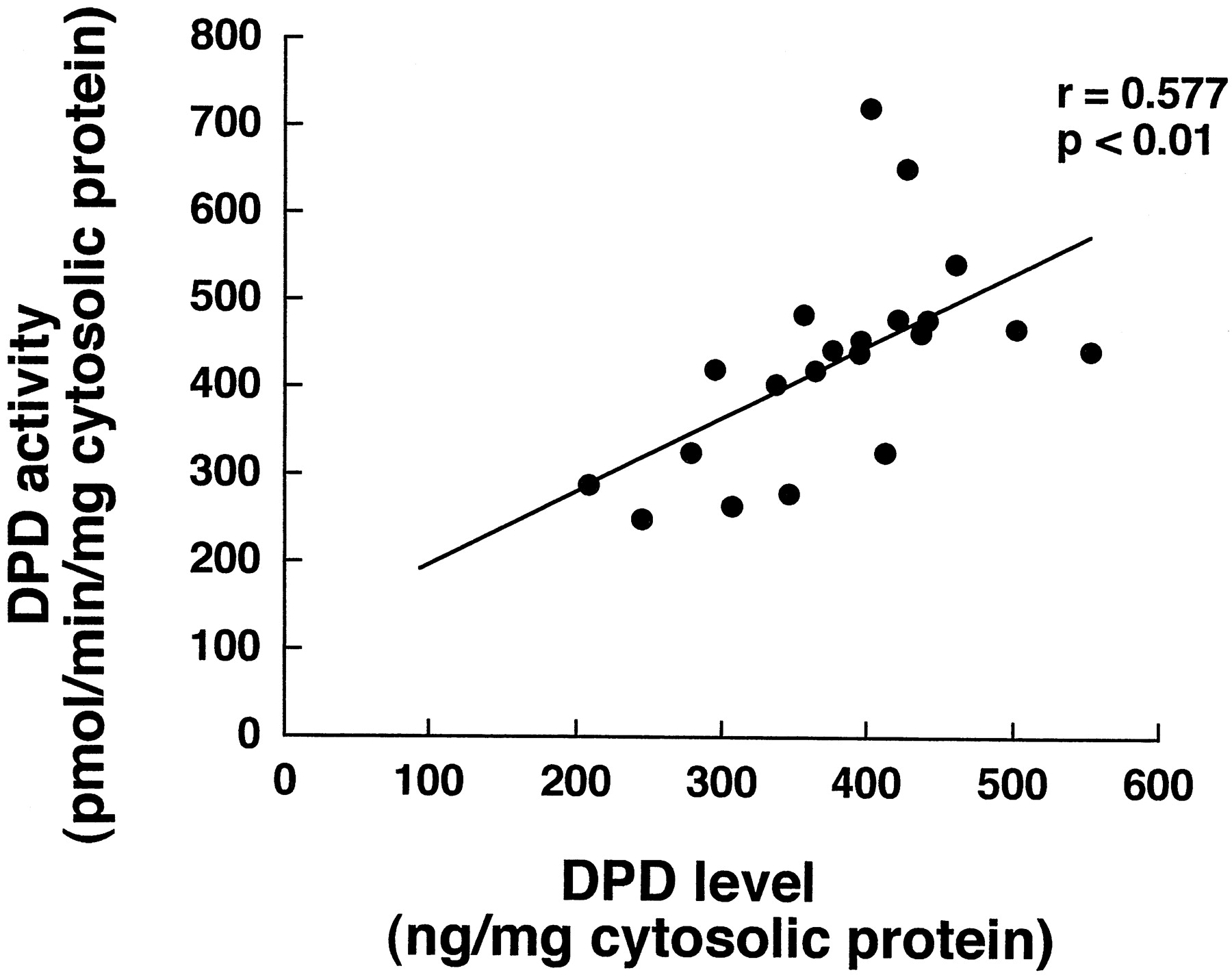
Correlation between 5-FU-reducing activity and human DPD level in 21 human liver samples.
The DPD activity of human liver cytosols was determined by using [14C]5-FU as a substrate. The DPD content of each sample was quantified by ECL Western blotting analysis with rabbit polyclonal antibody raised against purified rhDPD as the primary antibody.
Inhibition of 5-FU Metabolism In Vitro by BVU.
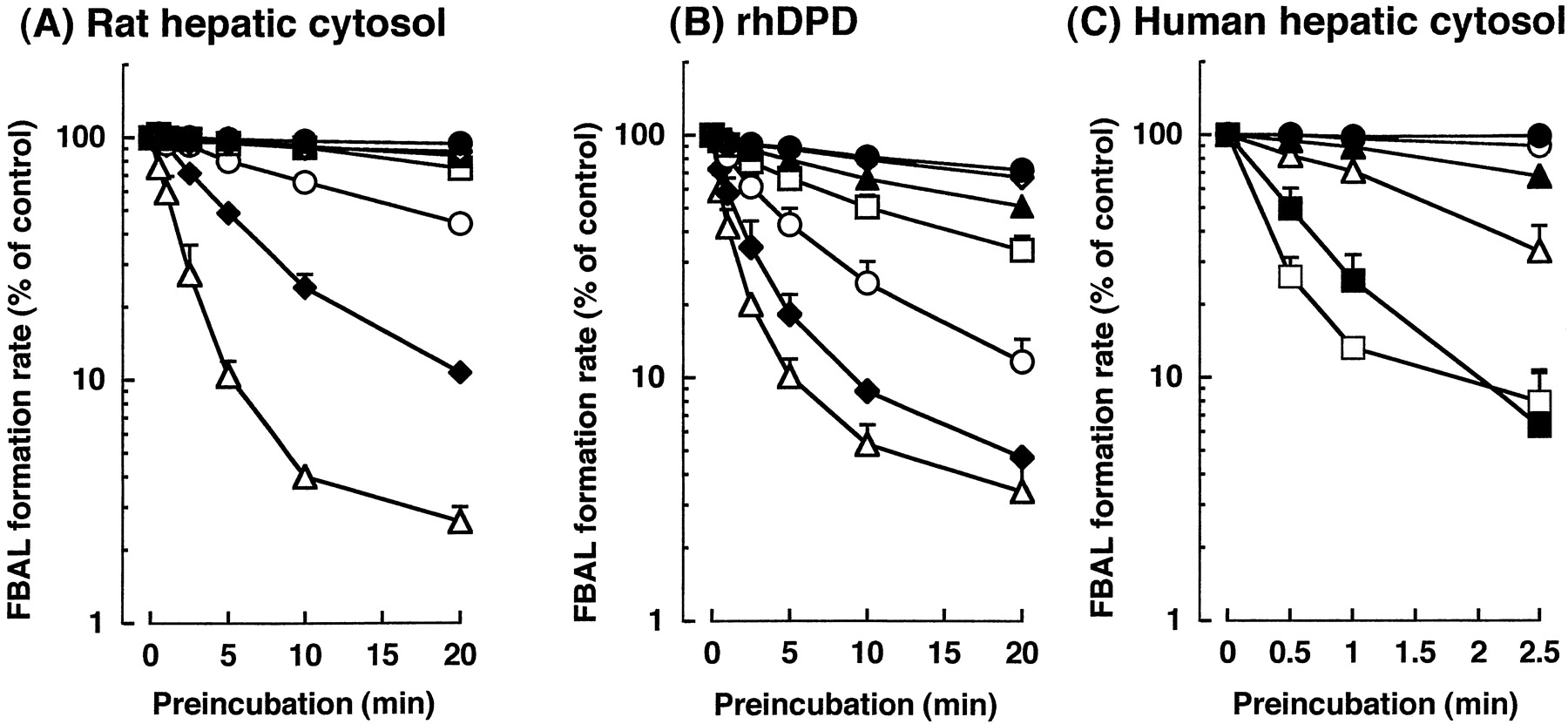
Figure 4 shows the effect of BVU concentration and preincubation time on 5-FU metabolism by rat hepatic cytosol, rhDPD, and human hepatic cytosol. Whichever enzyme was used, 5-FU metabolism was not inhibited without preincubation, even if the BVU concentration was increased. The degree of inhibition depended on the preincubation time and BVU concentration.

Inhibitory effects of BVU on 5-FU metabolism in rat hepatic cytosol (A), rhDPD (B), or human hepatic cytosol (C).
The 5-FU-metabolizing activity was measured by using [14C]5-FU as a substrate after preincubation of the cytosol or rhDPD with varying concentrations of BVU. BVU concentrations were: ●, 0; ⋄, 0.03; ▴, 0.1; ■, 0.3; ○, 1; ♦, 5; and ▵, 20 μM for (A) and (B); ●, 0; ○, 1; ▴, 3; ▵, 10; ▪, 50; and ■, 200 μM for (C).
Kinetic parameters for DPD inactivation, calculated from the data showing initial velocity, are summarized in Table5. Bothkinact andK′app obtained by using rhDPD were significantly lower than those obtained by using human hepatic cytosol (P < .001 by nonpaired t test). The parameters obtained by using rat and human hepatic cytosol were almost the same, showing no species difference.
Summary of kinetic parameters for BVU
Protein Binding of BVU in Rat Plasma and Human Serum.
The unbound fraction (fu) of BVU in rat plasma was 0.178 ± 0.006 and 0.152 ± 0.007 at 1 and 3.5 μM, respectively. The corresponding value in human serum was 0.236 ± 0.002 and 0.247 ± 0.007 at 1 and 3.5 μM, respectively.
Quantitative Prediction of 5-FU/BVU Interaction in Rats.
It was predicted that active DPD in the liver was immediately decreased after administration of BVU and that most of the DPD was inactivated 10 min after administration. The blood concentration of 5-FU was predicted to increase markedly compared with that in the control group, and the predicted area under the curve (AUC) increase was 11-fold, from 78 to 874 μM · h (Fig. 5).

Simulation of BVU effects on the hepatic DPD content and blood concentration of 5-FU in rats.
Time-courses of BVU blood concentration (A), active DPD content in the liver (B), and 5-FU blood concentration (C) were simulated according to eqs. 2 through 12. Dashed lines, control; solid lines, coadministration of BVU.
Quantitative Prediction of Tegafur/Sorivudine Interaction in Humans.
Blood concentration profiles of tegafur, 5-FU, and sorivudine in humans simulated by using the kinetic parameters used in the prediction were compared with the reported profiles (Nakajima et al., 1980; Ogiwara et al., 1990). Figure 6 shows the blood concentration profiles of tegafur and 5-FU after single oral administration of tegafur (300 mg) and that of BVU after oral administration of sorivudine (150 mg/day, t.i.d. for 5 days). The simulated and the reported profiles were comparable, indicating the validity of the kinetic parameters used in this simulation.

Comparison between observed (●) and simulated (dashed line) concentration profiles in humans.
Blood concentration profiles of tegafur (A) and 5-FU (B) after single oral administration of tegafur (300 mg) and that of BVU (C) after oral administration of sorivudine (150 mg/day for 5 days) were simulated by eqs. 9 through 21 by using the parameters in Tables 3 and 4 and compared with the reported profiles (Nakajima et al., 1980; Ogiwara et al., 1990).
Figure 7 shows the simulation with the obtained kinetic parameters for DPD inactivation by human hepatic cytosol or rhDPD. It was predicted that active DPD in the liver was immediately decreased after administration of sorivudine in both cases and that most of the DPD was inactivated 12 and 3 h after the initial dose when the parameters obtained by using human hepatic cytosol and rhDPD, respectively, were used in the simulation. The blood concentration of 5-FU was predicted to increase compared with control group, and the predicted AUC increase was 5.3- and 5.4-fold when the parameters obtained by using human hepatic cytosol and rhDPD, respectively, were used in the simulation.

Simulation of sorivudine effects on the hepatic DPD content and blood concentration of 5-FU in humans.
Time-courses of tegafur blood concentration (A), BVU blood concentration (B), active DPD content in the liver (C and E), and 5-FU blood concentration (D and F) were simulated according to eqs. 2, 3, and 9 through 21. C and D represent simulations based on parameters obtained with human hepatic cytosol. E and F represent simulations based on parameters obtained with rhDPD. Dashed lines, control; solid lines, coadministration of sorivudine.
Recovery of Active DPD in the Liver.
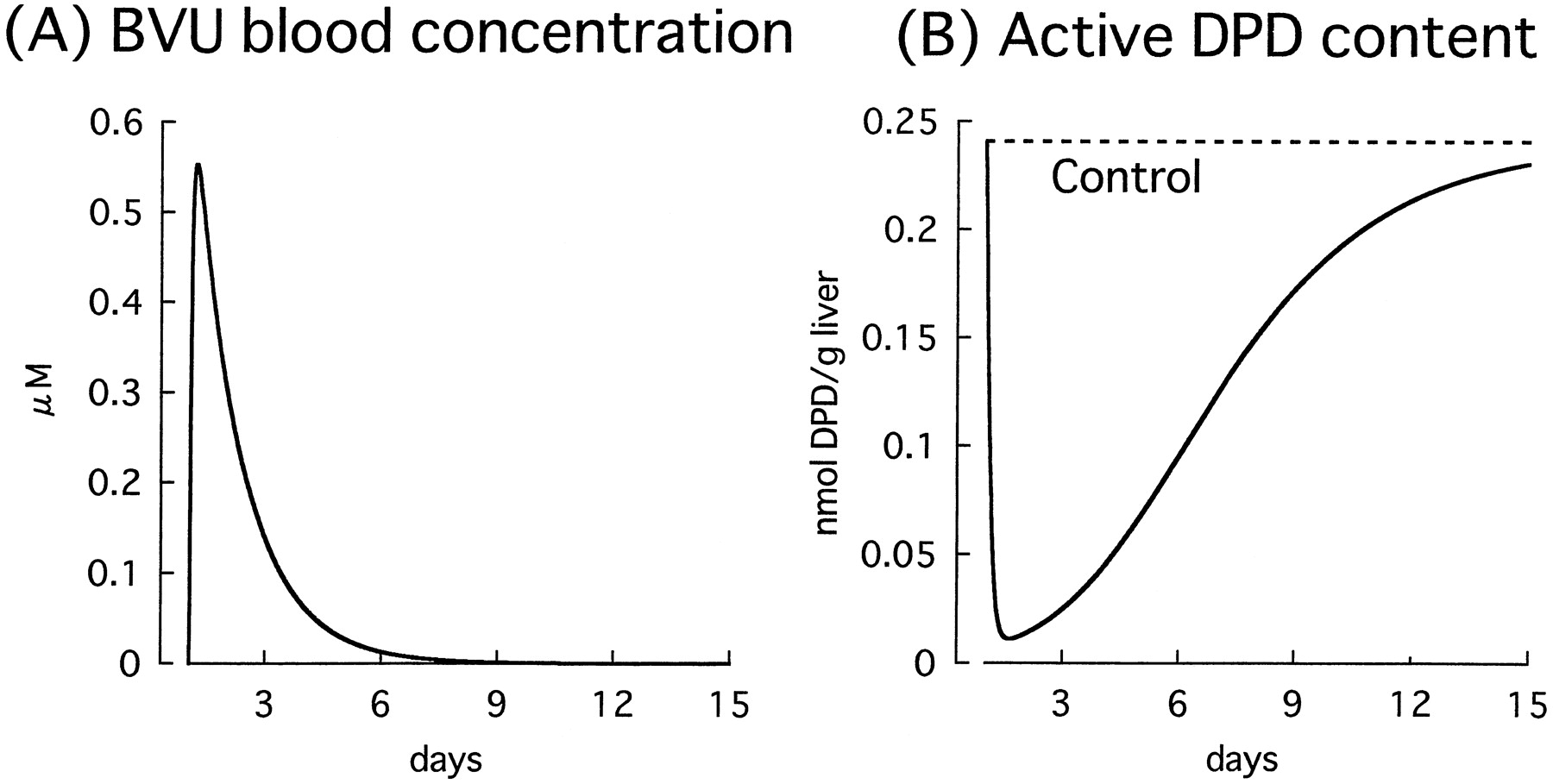
Figure 8 shows the time profiles of the BVU blood concentration and active DPD content in the liver after oral administration of a single tablet of sorivudine (50 mg) simulated by using kinetic parameters for DPD inactivation obtained from human hepatic cytosol studies. It was found that 95% of the DPD in the liver is inactivated 12 h after ingestion of only one tablet of sorivudine, and that about 2 weeks is required for the DPD content to return to normal.

Simulation of blood concentration of BVU (A) and the recovery of active DPD content in the liver (B) after administration of a single tablet of sorivudine (50 mg/dose).
Discussion
In the case of mechanism-based inhibition, the inhibitor is metabolically activated by an enzyme and irreversibly inactivates the same enzyme by covalent binding, exhibiting the following characteristics:
- 1.
- Preincubation time-dependent inhibition of the enzyme (time dependence).
- 2.
- No inhibition if the cofactors necessary to produce the activated inhibitor (e.g., NADPH for P450 metabolism) are not present in the preincubation medium.
- 3.
- Potentiation of the inhibition depending on the inhibitor concentration (saturation kinetics).
- 4.
- Slower inactivation rate of the enzyme in the presence of substrate compared with its absence (substrate protection).
- 5.
- Enzyme activity not recovered after gel filtration or dialysis (irreversibility).
- 6.
- 1 : 1 Stoichiometry of the inhibitor and the active site of the enzyme (stoichiometry of inactivation).
Mechanism-based inhibitors should satisfy these criteria (Silverman, 1988).
Desgranges et al. (1986) reported that when rat hepatic cytosol was preincubated with BVU in the presence of NADPH, DPD was irreversibly inactivated and the activity was not recovered after dialysis. Furthermore, when purified rat hepatic DPD was preincubated with radioactive BVU in the presence of NADPH, DPD was inactivated depending on the preincubation time and was irreversibly bound to BVU (Okuda et al., 1997, 1998; Watabe et al., 1997). These findings show that BVU is a mechanism-based inhibitor of DPD. The aim of the present study was to apply the methodology for predicting drug-drug interactions in vivo, based on the mechanism-based inhibition, from in vitro data to the interaction between 5-FU and BVU.
As shown in Fig. 5, an 11-fold increase in 5-FU AUC (from 78 to 874 μM · h) was predicted based on the parameters obtained by using rat hepatic cytosol. This increase was comparable with the reported 8.1-fold increase (from 66 to 534 μM · h; Desgranges et al., 1986), suggesting that the present prediction methodology is appropriate for this interaction.
Different values of kinact andK′app were obtained in the in vitro studies that used rhDPD and human hepatic cytosol (Table 5). To clarify if this was attributable to endogenous substances in the cytosol fraction, the inhibitory effect of BVU was examined by changing the concentration of human hepatic cytosol and by adding ultra-filtrate of human hepatic cytosol to rhDPD (data not shown). No clear difference was observed in either study, suggesting that the difference in the parameters could be attributable to a difference in the enzyme itself (Ogura et al., 1998).
Coadministration of sorivudine was predicted to increase the AUC of 5-FU more than 5-fold compared with control, based on the parameters obtained by using human hepatic cytosol or rhDPD. Thus, it could be predicted that combination therapy of fluorouracil anticancer drugs and sorivudine is absolutely to be avoided. Furthermore, it was predicted that 95% of DPD in the liver was inactivated 12 h after ingestion of only one tablet of sorivudine and that 2 weeks is required for the DPD content to return to normal (Fig. 8) (Yan et al., 1997). In the case of mechanism-based inhibition, the inhibitory effect remains even after the inhibitor is eliminated from the body. The present simulation study indicates that administration of a mechanism-based inhibitor is very dangerous because waiting for the enzyme to recover due to natural turnover is the only way for the inhibitory effect to disappear.
As shown in Fig. 3, about a 3-fold interindividual difference was observed in 5-FU-metabolizing activity and DPD content in the liver. This means that, if the metabolic clearance associated with DPD is 80% of the total body clearance of 5-FU (Diasio and Harris, 1989), the total body clearance of a person with the lowest DPD activity is only half that of a person with the highest activity. In other words, when 5-FU is administered to both persons, theoretically there should be twice the difference in the AUC of 5-FU. Such interindividual differences in metabolic activity may have to be taken into consideration in planning the dosing schedule.
It is important in the present prediction to precisely estimate the unbound concentration of inhibitor in the liver, and this cannot be measured, especially in humans. However, some of the pharmacokinetic parameters can be determined to fit the blood concentration profile of the inhibitor, which can be measured in many cases. Other uncertain parameters which are not measured [e.g., liver-to-blood concentration ratio (Kp)] may have to be changed to some extent in the simulation study to predict the Eact and delay in substrate elimination with a range.
Okuda et al. (1998) reported that the DPD activity in the liver fell to 14% of the control after repeated oral administration of BVU (3.7 mg/kg) to rats. However, the simulation that used the parameters obtained in the present study showed that DPD activity had fallen to 1.2% 4 h after administration of BVU (data not shown). The effect of endogenous substances may be one of the reasons for the overestimation of the effect of BVU by the present prediction method. Because DPD was inhibited by BVU, the level of pyrimidines such as uracil may have risen and it may have functioned as a competitive inhibitor in vivo; it is unlikely that such effects of endogenous substances could be predicted from the in vitro studies.
In the future, it is important to confirm the validity of the present prediction method in animal studies, where inhibition studies can be performed both in vitro (by using e.g., hepatic cytosol for DPD and hepatic microsomes for P450) and in vivo. Because invasive experiments are possible in this case, including measurements of theKp of the inhibitor in the liver and enzyme activity in the liver, this may allow more accurate predictions to be made.
Footnotes
-
Send reprint requests to: Yuichi Sugiyama, Graduate School of Pharmaceutical Sciences, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail:sugiyama{at}seizai.f.u-tokyo.ac.jp
- Abbreviations used are::
- 5-FU
- 5-fluorouracil
- BVU
- (E)-5-(2-bromovinyl)uracil
- DPD
- dihydropyrimidine dehydrogenase
- rhDPD
- human recombinant DPD
- ECL
- enhanced chemiluminescence
- FBAL
- α-fluoro-β-alanine
- AUC
- area under the curve
- Received June 26, 1999.
- Accepted December 14, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}