


Abstract
Currently, there are no selective, well characterized inhibitors for CYP2A6. Therefore, the effects oftrans-(±)-2-phenylcyclopropylamine (tranylcypromine), a potent CYP2A6 inhibitor, on human liver microsomal cytochromes P450 (CYP) were studied to elucidate its selectivity. The IC50value of tranylcypromine in coumarin 7-hydroxylation (CYP2A6 model activity) was 0.42 ± 0.07 μM and in chlorzoxazone 6-hydroxylation (CYP2E1 model activity) 3.0 ± 1.1 μM. The IC50 values for CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 activities were >10 μM. Potency and selectivity of tranylcypromine were strongly dependent on the amine group, because its nonamine analog cyclopropylbenzene was much less potent inhibitor of CYP1A, CYP2A6, CYP2C19, and CYP2E1 activities and did not inhibit at all CYP2C9, CYP2D6, or CYP3A4 activities. In human liver microsomes tranylcypromine induced type II and cyclopropylbenzene type I difference spectrum. According to the double reciprocal analysis of these spectral responses both tranylcypromine and cyclopropylbenzene may have at least two P450-related binding sites in liver microsomes. TheKa values of tranylcypromine varied from 4.5 to 15.1 μM and −34.3 to 167 μM in microsomes derived from three different livers and of cyclopropylbenzene from −1.6 to 10.1 μM and −34.6 and 75.2 μM in the same liver microsomes. Based on these results, tranylcypromine seems an adequately selective CYP2A6 inhibitor for in vitro use.
The significance of characterizing CYP1isoform-selective inhibitor compounds lies in the fact that in screening of molecules during drug development, enzyme-specific inhibitors are convenient tools in delineating the metabolizing enzymes (Rodrigues, 1994; Pelkonen et al., 1998). Therefore, model inhibitors specific to every liver CYP enzyme are important when metabolic interactions are evaluated. In practice, an ideal CYP enzyme-specific reference inhibitor would be an easily obtainable, low-cost compound, which is thoroughly studied with respect to its in vitro inhibitory potential on CYP enzyme system. It is not so necessary for it to inhibit only one CYP if it inhibits the target CYP at clearly lower concentrations than other CYP enzymes.
CYP2A6, constituting about 5% of total hepatic CYPs, plays a crucial role in the bioactivation of some carcinogens such as nitrosamines and aflatoxin B1 (Pelkonen et al., 2000). Currently, there are no selective and well characterized chemical inhibitors for CYP2A6. Some potent inhibitors of CYP2A6 have been found (Mäenpää et al., 1994; Kimonen et al., 1995; Kinonen et al., 1995; Draper et al., 1997; Juvonen et al., 2000), but their specificity and potency against various CYP enzymes have not been thoroughly characterized. Methoxsalen is a widely used CYP2A6 reference inhibitor (Koenigs et al., 1997) but its selectivity is not clear. Likewise, R-(+)-menthofuran has recently been studied for this purpose (Khojasteh-Bakht et al., 1998).
trans-(±)-2-Phenylcyclopropylamine (tranylcypromine) is a nonhydrazine monoamine oxidase inhibitor used in psychiatry (Dollery et al., 1991). The structures of tranylcypromine (nonionized form) and its nonamine structural analog cyclopropylbenzene are presented in Fig.1. Tranylcypromine is also a potent CYP2A6 inhibitor (Draper et al., 1997). The purpose of this work is to characterize the inhibitory CYP selectivity of tranylcypromine in human liver microsomes and to study whether the amino group in the tranylcypromine molecule plays an important role in the inhibition of CYP-catalyzed activities. Therefore, several CYP-specific reactions were inhibited by tranylcypromine and cyclopropylbenzene. Furthermore, the spectral interactions of tranylcypromine and cyclopropylbenzene with microsomal P450 have been studied by measuring the chemical induced difference spectra in liver microsomes. In the present article we present evidence that at defined concentrations tranylcypromine is a suitable CYP2A6-selective inhibitor for in vitro metabolism studies.

The structures of tranylcypromine (1) and cyclopropylbenzene (2).
At physiological pH the amino group of tranylcypromine is approximately 96% protonated.
Materials and Methods
Chemicals.
rac-Mephenytoin was donated by Sandoz Pharma (Basel, Switzerland). Midazolam and 1′-hydroxymidazolam were donated by F. Hoffmann-La Roche (Basel, Switzerland). The reference substances dextrorphan, 6-hydroxychlorzoxazone, hydroxytolbutamide, and 4′-hydroxymephenytoin were purchased from Ultrafine Chemical Company (Manchester, UK) and trans-(±)-2-phenylcyclopropylamine hydrochloride and cyclopropylbenzene from Sigma Chemical Co. (St. Louis, MO). High-performance liquid chromatography (HPLC) grade solvents were obtained from Rathburn (Walkerburn, UK). Other chemicals were obtained from Sigma Chemical Co. and Boehringer (Ingelheim, Germany), and were of the highest purity available. The laboratory water was purified through Milli-Q system (Millipore S.A., Molsheim, France).
Preparation of Liver Microsomes.
Human liver samples used in this study were obtained from kidney transplantation donors. The collection of surplus tissue was approved by the Ethics Committee of the Medical Faculty of the University of Oulu, Finland, in accordance with the Helsinki declaration. Microsomes were prepared by differential ultracentrifugation (Pelkonen et al., 1974). The final microsomal pellet was suspended in 0.1 M phosphate buffer (pH 7.4) to achieve a concentration of approximately 20 mg of protein/ml. Protein content was measured according to the method ofBradford (1976). For the screening of the IC50values, microsomes from four livers were used. The livers were thoroughly characterized with CYP-specific substrates and reference inhibitors. The nomenclature for CYP enzymes is according toNelson et al. (1996). In enzyme kinetic studies a pool of microsomes from five well characterized livers was used. All the livers were screened for their CYP-specific model activities used in this study. Three livers contained all seven activities, two exhibited all exceptS-mephenytoin 4′-hydroxylation (or the level was not high enough for inhibition studies). CYP-specific activities in every liver sample were inhibitable by CYP-selective chemical inhibitors. These facts were considered a proof of the existence of specified CYP enzymes. For the determination of the IC50 values for CYP2C19 only livers exhibiting measurable and inhibitableS-mephenytoin 4′-hydroxylation levels were used.
CYP Isoform-Specific Enzyme Assays.
The following enzyme assays, which display at least some CYP isoform specificity, were used (the methods are modified from original publications): ethoxyresorufin O-deethylation (CYP1A1/2) (Burke et al., 1977); coumarin 7-hydroxylation (CYP2A6) [(Aitio, 1978), with slight modifications as described by Raunio et al. (1988,1990)]; tolbutamide hydroxylation (CYP2C9) [modified from Knodell et al. (1987) and Sullivan-Klose et al. (1996)]; S-mephenytoin 4′-hydroxylation (CYP2C19) (Wrighton et al., 1993); dextromethorphanO-demethylation (CYP2D6) (Park et al., 1984; Kronbach et al., 1987); chlorzoxazone 6-hydroxylation (CYP2E1) (Peter et al., 1990); and midazolam 1′-hydroxylation (CYP3A4/5) (Kronbach et al., 1989). The incubation and analysis conditions are summarized in Table1.
Incubation and analysis conditions to determine IC50 values in vitro in human liver microsomes for trans-(±)-2-phenylcyclopropylamine (tranylcypromine) and cyclopropylbenzene in seven CYP-specific activities
Determination of the Metabolites in CYP Isoform-Specific Assays.
For the HPLC method of the metabolites of chlorzoxazone, dextromethorphan, S-mephenytoin, midazolam, and tolbutamide Symmetry C18 column was used (3.9 × 150 mm, 5-μm particle size; Waters Corporation, Milford, MA). Column temperature was ambient. A Lichrospher 100 RP-18 guard column (4 × 4 mm; E. Merck, Darmstadt, Germany) was used to protect the analytical column. For UV-HPLC analysis samples were centrifuged at 10,000g for 15 min before injection into HPLC. The apparatus used was a Shimadzu VP series HPLC with autoinjector.
In Vitro Inhibition of CYP Isoform-Specific Assays by Tranylcypromine and Cyclopropylbenzene.
The studied compounds were added from stock solutions in different concentrations (final concentrations in the incubation mixture were usually 0.01, 0.1, 1.0, 10, 100, and 1000 μM) into the incubation mixture in a small volume of solvent, 0.1 M NaOH or ethanol, respectively. Ethanol concentration was <1% in incubation mixture. For chlorzoxazone 6-hydroxylation inhibition study the solvent of cyclopropylbenzene was acetonitrile. For each assay fresh solutions of tranylcypromine and for cyclopropylbenzene new dilutions from stock solution (100 mM in ethanol or acetonitrile, stored at −20°C) were used. The enzyme activities in the presence of tranylcypromine or cyclopropylbenzene were compared with the control incubations into which only solvent was added instead of tranylcypromine or cyclopropylbenzene. The IC50 values for inhibitors (concentration causing 50% reduction of control activity) were determined by linear regression analysis from the plot of the logarithm of inhibitor concentration versus percentage of the activity remaining after inhibition using MicroCal Origin software, version 4.10 (MicroCal Software, Inc., Northampton, MA).
Analysis of Data for Determining ApparentKm, Vmax, andKi.
The enzyme kinetic studies for determining apparentKi values for tranylcypromine in coumarin 7-hydroxylation were performed by using a pool of microsomes from five different livers and chlorzoxazone 6-hydroxylation reactions were performed by using one well characterized human liver sample containing all drug-metabolizing CYPs. The existence of CYPs was determined by activity screening and by inhibition with reference chemical inhibitors (data not shown). Incubations for the determination of kinetic parameters were conducted under initial velocity conditions. The formation of metabolites in both coumarin 7-hydroxylation and chlorzoxazone 6-hydroxylation was linear up to 1.0 mg of protein/ml and 60 min of incubation time in the used microsomes. The Eadie-Hofstee plot exhibited single enzyme kinetics for both activities. Protein amounts in these assays were 0.2 and 0.5 mg/ml, and incubation times were 10 and 15 min, respectively. The formation of metabolites was calculated and expressed as picomoles per minute per milligram of protein. For the determination of apparentKm and Vmaxplots of metabolite formation rate versus substrate concentration and Lineweaver-Burk plots were constructed. The determination of apparentKi in activities used was conducted from the respective Dixon plots. The lines in the plot were fitted by linear regression analysis (MicroCal Origin, version 4.10). The intersection point of lines (Ki value) was determined graphically.
Determination of Difference Spectra Induced by Tranylcypromine and Cyclopropylbenzene.
For studying tranylcypromine- and cyclopropylbenzene-induced difference spectra the microsomes (3 mg of protein/ml) were first solubilized for 30 min in 0.6% cholate solution containing 100 mM phosphate buffer, pH 7.4, 20% glycerol, 0.1 mM EDTA, and 0.1 mM dithiothreitol and the samples were centrifuged for 1 h at 105,000g. The supernatant was aliquoted into two spectrophotometer cells (1-cm path length), tranylcypromine or cyclopropylbenzene was added from stock solution (1000 times dilution) in ethanol to the sample cell and an equal volume of ethanol to the reference cell, and the spectrum was recorded from 460 to 360 nm (Jefcoate, 1978). The total added volume of ethanol was less than 20 μl (2%) and the concentrations of tranylcypromine and cyclopropylbenzene varied between 1 and 900 μM.
Analysis of Substrate-Induced Difference Spectra.
The data of substrate-induced difference spectra were analyzed by the double reciprocal plot of the changes produced by different concentrations of tranylcypromine or cyclopropylbenzene (Morgan et al., 1982; Vaz et al., 1992) (GraphPad Prism, version 2.0).
Results
In Vitro Inhibition of CYP-Specific Activities by Tranylcypromine and Cyclopropylbenzene.
The IC50 values of tranylcypromine and cyclopropylbenzene were determined for CYP-specific ethoxyresorufinO-deethylation (CYP1A2), coumarin 7-hydroxylation (CYP2A6) and tolbutamide hydroxylation (CYP2C9), S-mephenytoin 4′-hydroxylation (CYP2C19), dextromethorphan O-demethylation (CYP2D6), chlorzoxazone 6-hydroxylation (CYP2E1), and midazolam 1′-hydroxylation (CYP3A4) activities (Table2). Tranylcypromine inhibited all activities except midazolam 1′-hydroxylation, whereas cyclopropylbenzene did not inhibit tolbutamide hydroxylation, dextromethorphan O-demethylation, and midazolam 1′-hydroxylation. Tranylcypromine inhibited all these monooxygenase activities at 40- to 1000-fold lower concentrations than cyclopropylbenzene (Table 2).
IC50 values of tranylcypromine and cyclopropylbenzene against human CYP isoforms
Because the IC50 values of tranylcypromine were the lowest against coumarin 7-hydroxylation and chlorzoxazone 6-hydroxylation, the inhibition kinetics were studied for these enzyme activities. Tranylcypromine appeared to be a mixed type inhibitor for both enzyme activities in the studied microsomes. For comparative reasons, we performed calculations supposing that the activities studied were inhibited competitively. TheKi values were calculated according toTornheim (1994) (Table 2). The Ki value of tranylcypromine for coumarin 7-hydroxylation was the lowest, 0.1 to 0.17 μM, and it was 16 to 17 times lower than the next most potently inhibited chlorzoxazone 6-hydroxylation or S-mephenytoin 4′-hydroxylation. Calculated Ki values did not markedly differ from experimental values for coumarin 7-hydroxylation and chlorzoxazone 6-hydroxylation.
Spectral Interactions and Affinities of Tranylcypromine and Cyclopropylbenzene with Human Liver Microsomes.
Tranylcypromine induced a type II difference spectrum, i.e., the absorbance maximum at 431 nm and the minimum at 410 nm (Fig.2A). This means that spin equilibria between CYPs are changed mainly from low-spin state to altered low-spin state by tranylcypromine. Furthermore, the spectrum suggests that the amino group of tranylcypromine is coordinated with heme iron of CYP because the altered spin state of CYP is detected in the spectrum. Cyclopropylbenzene induced a type I difference spectrum with absorbance maximum at around 385 nm and minimum at 416 to 418 nm (Fig. 2B). Therefore, cyclopropylbenzene altered spin equilibrium of CYPs so that the relative amount of low-spin CYPs decreased and high-spin CYPs increased.

Tranylcypromine- and cyclopropylbenzene-induced difference spectra.
In the experiment tranylcypromine (A) or cyclopropylbenzene (B) was added to the solubilized human liver microsomes (sample cell) and the equal volume of solvent was added into the otherwise similar reference cell. Concentrations of tranylcypromine were 87 and 89 μM for cyclopropylbenzene, whereas the cytochrome P450 concentrations were 0.35 and 0.48 μM, respectively. The spectra shown are from representative examples.
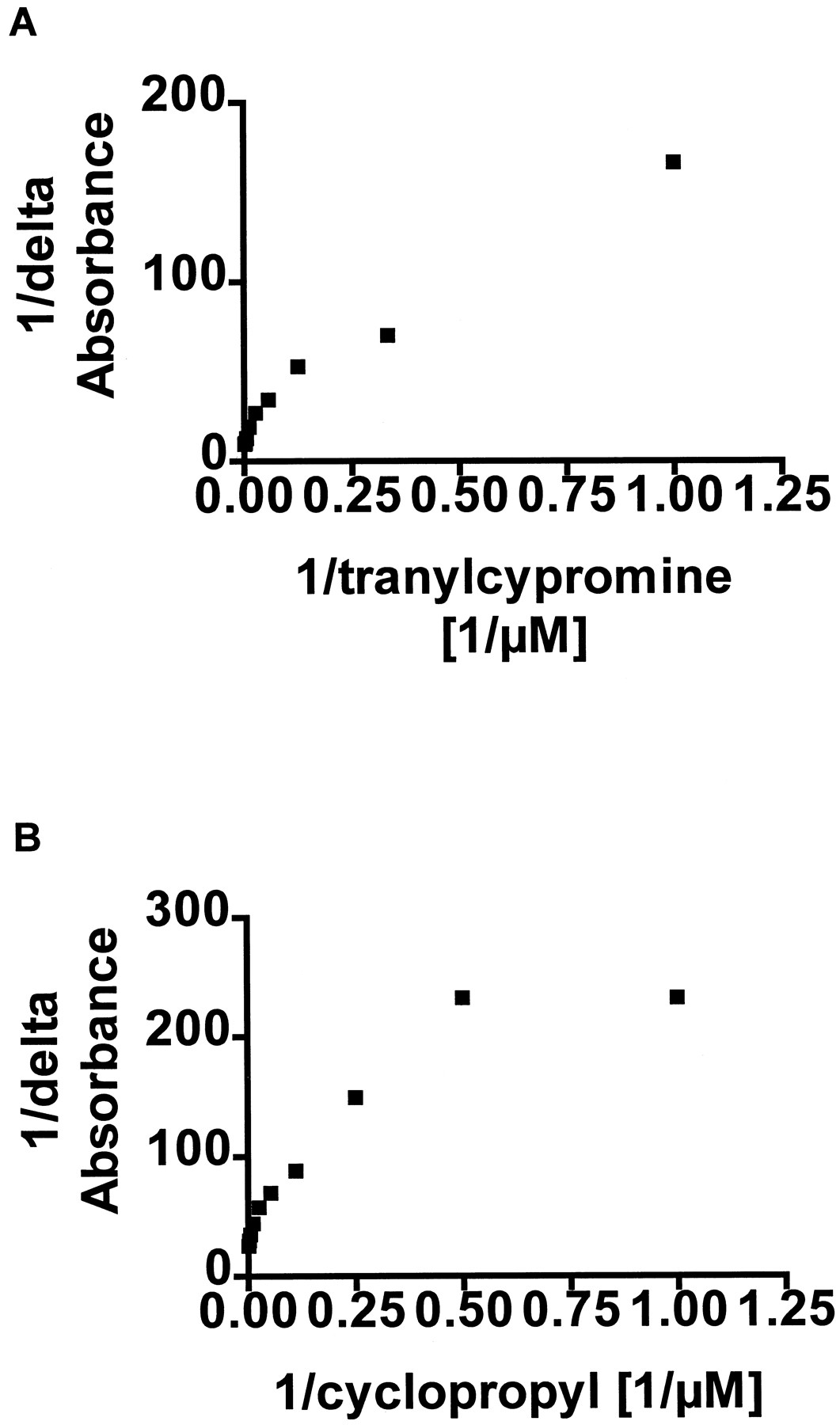
According to both the double reciprocal analysis (Fig.3, A and B) and the Scatchard analysis (data not shown) there may be at least two binding sites for both tranylcypromine and cyclopropylbenzene with liver microsomal P450, because two linear lines seems to be formed. The lowerKa value of tranylcypromine varied between 4.5 and 15.1 μM and the higher Ka value between −34.3 and 163 μM in different solubilized human liver microsomes. The lower Ka value of cyclopropylbenzene varied between −1.6 and 10.1 μM and the higherKa value between 57 and 154 μM in different solubilized human liver microsomes. According to theoretical calculations for protonation of tranylcypromine at pH 7.4, approximately 4% is in unprotonated form and therefore capable of coordination to heme ferric ion of P450 (theoretical calculation based on the calculated pKa of 8.78 for tranylcypromine, data not presented, ACD/pKa software, version 4.56).

The double reciprocal analysis of tranylcypromine- (A) and cyclopropylbenzene (B)-induced difference spectra.
Tranylcypromine- and cyclopropylbenzene-induced difference spectra were analyzed as shown in Fig. 2 and in greater detail underMaterials and Methods. Representative analysis of both tranylcypromine and cyclopropylbenzene are shown with results of one well characterized liver.
Discussion
Tranylcypromine as a Selective CYP2A6 Inhibitor.
The main results of this study are that 1) tranylcypromine is a more potent inhibitor of CYP-catalyzed activities than cyclopropylbenzene; 2) tranylcypromine is a selective, mixed type inhibitor for CYP2A6 in the human liver microsomes; and 3) the amine-group of tranylcypromine is essential for potency and selectivity toward CYP enzymes in microsomes; however, 4) spectrally detectable affinities were rather similar for both compounds. This could be explained by the protonation of the amine group of tranylcypromine. Considering the protonation of tranylcypromine being about 96% there is actually a large difference in affinity between the two compounds (see below).
As seen in Table 2, tranylcypromine most potently inhibits CYP2A6, the apparent Ki being 0.17 μM. The apparentKm of coumarin in 7-hydroxylation was 2.0 in the pool of microsomes from five different livers. Draper et al. (1997) determined Ki of tranylcypromine to be 0.04 μM and the reaction type to be competitive. Because IC50 value is dependent on the substrate used and its concentration, we have derived Kivalues from determined IC50 values according toTornheim (1994), assuming competitive inhibition. This approach has been used for comparison purposes, although according to performed enzyme kinetic experiments inhibition by tranylcypromine of CYP2A6- and CYP2E1-catalyzed activities was of mixed type. As seen in Table 2, the calculated values are similar to respective apparent values. However, it must be kept in mind that the type of inhibition of activities catalyzed by other P450 is not known. If the calculatedKi values of tranylcypromine in other CYP-specific activities are divided by the apparentKi value of tranylcypromine in coumarin 7-hydroxylation, we will get a rank of inhibitory potencies (13.5 for CYP2C19, 14.7 for CYP2E1, 25.3 for CYP2D6, 27.6 for CYP1A2, and 92.9 for CYP2C9, no inhibition for CYP3A4) showing that tranylcypromine is over 13 times more potent inhibitor of CYP2A6 than any of the other CYP enzymes studied here.
There is a clear difference in apparent Kivalues of tranylcypromine between coumarin 7-hydroxylation and chlorzoxazone 6-hydroxylation. Apparent Kifor CYP2E1 mediated reaction is over 14 times theKi for CYP2A6-catalyzed activity. When using over 1 μM concentrations of tranylcypromine one should be aware of its effect on CYP2E1. The involvement of CYP2E1 can, on the other hand, be evaluated by inhibition with pyridine, which is a highly selective inhibitor of CYP2E1 in human liver microsomes (unpublished results). It could be used as a tool in differentiating between CYP2A6 and CYP2E1, if there is suspicion about CYP2E1 being involved in the metabolism of an unknown compound.
Tranylcypromine has been used as a CYP2C19-specific reference inhibitor (Chauret et al., 1997). Based on our results, tranylcypromine is not a good reference inhibitor for CYP2C19, because it inhibits CYP2A6 and CYP2E1 more potently. Chauret et al. (1997) used tranylcypromine in a mechanism-based manner with 15-min preincubation, whereas we used an ordinary 2-min preincubation period. We have not studied the inhibitory effect of tranylcypromine, or cyclopropylbenzene, in a mechanism-based manner at this time.
Comparison of Tranylcypromine with Methoxsalen and (R)-(+)-Menthofuran as a Specific Inhibitor of CYP2A6.
Methoxalen is known to inhibit other CYPs than merely CYP2A6 with nearly the same potency. CYP1A2 and CYP2E1 at least are sensitive to its inhibitory effect (Mäenpää et al., 1994; unpublished results). For CYP2E1, aKi value of 2 μM for methoxsalen has been presented (Yamazaki et al., 1992; Mäenpää et al., 1994), which is of the same order of magnitude asKi for diethyldithiocarbamate, a commonly used CYP2E1 inhibitor (Yamazaki et al., 1992). For methoxsalen,Ki in coumarin 7-hydroxylation is around 1 μM (Yamazaki et al., 1992; Mäenpää et al., 1994).Koenigs et al. (1997) did not find any inhibition toward other CYPs by methoxsalen.
We have studied the effect of methoxsalen as an inhibitor to CYP1A2-catalyzed ethoxyresorufin O-deethylation and found it to be a mixed type inhibitor with Ki value of 2.9 μM in nonmechanism-based incubation conditions. In other studies on this topic by Mäenpää et al. (1994) andYamazaki et al. (1992) different substrates were used to determine a specific CYP enzyme. For example, in chlorzoxazone 6-hydroxylation,Mäenpää et al. (1994) and Yamazaki et al. (1992) used a substrate concentration of 500 μM, which is 10 times theKm of the reaction and inS-mephenytoin 4′-hydroxylation they used 200 μMS-mephenytoin, which is approximately 4 times theKm of the reaction. Concerning CYP2E1, inhibition by methoxsalen can certainly be masked by the substrate concentration. In the current study, there is one activity for which too high a substrate concentration was used, namely, 100 μM in CYP2D6 catalyzed dextromethorphan O-demethylation. This is about 10 times the Km determined for dextromethorphan in this reaction. However, in agreement with our findings, Draper et al. (1997) have demonstrated that CYP2D6 is not inhibited by tranylcypromine. Hence, compared with methoxsalen as a mechanism-based inhibitor, it is a viable conclusion that tranylcypromine as a competitive/mixed type inhibitor is more convenient to use than methoxsalen, and the difference betweenKi values for CYP2A6 and other CYPs seems wider with tranylcypromine than with methoxsalen.
Khojasteh-Bakht et al. (1998) have shown that (R)-(+)-menthofuran is a potent, mechanism-based inactivator of human liver CYP2A6 with a Ki of 2.5 μM. (R)-(+)-menthofuran did not inhibit CYP1A2, CYP2D6, CYP2E1, or CYP3A4. There is no information about inhibitory potency of menthofuran against CYP2C enzymes. In the Khojasteh-Bakht et al. (1998)study, substrate concentrations for marker activities have not been reported, and therefore it is difficult to evaluate the inhibition on the basis of the Khojasteh-Bakht et al. (1998) work. According to our unpublished results, the IC50 value for (R)-(+)-menthofuran for CYP2A6-catalyzed activity was 2.4 μM, for CYP1A2 it was 7.1 μM, CYP2C9-catalyzed tolbutamide hydroxylation was slightly activated, CYP2C19 was inhibited by the IC50 value of 338 μM, for CYP2E1 the value was 510 μM, and for CYP3A4 479 μM. Although this substance inhibits CYP1A2 at very low concentrations, we have considered (R)-(+)-menthofuran as a reference inhibitor for CYP2A6 in our metabolic studies with new chemical compounds. However, we have found (R)-(+)-menthofuran very uncomfortable to use because of the pure compounds sensitivity to light, air, and humidity under storage, not to mention the fact that it evaporates readily, and the dilution process needs to be conducted under a hood for safety reasons.
Studies on the difference spectra of tranylcypromine and cyclopropylbenzene with microsomes indicated that tranylcypromine elicited a type II (Fig. 2A) and cyclopropylbenzene a type I spectrum (Fig. 2B). Type II difference spectrum is typical for chemicals containing a nitrogen atom, which is coordinated toward the P450 heme iron (Jefcoate, 1978; Schenkman et al., 1981; Gibson and Tamburini, 1984). When tranylcypromine binds to the P450 enzymes, it is probable that the amine group of tranylcypromine is toward heme iron in the enzyme and this is undoubtedly a factor in increasing its potency and selectivity to inhibit CYP2A6 compared with cyclopropylbenzene. Another example of this phenomenon is pilocarpine, which induces a type II difference spectrum in both mouse and human liver microsomes (Kinonen et al., 1995). In the study by Kinonen et al. (1995) the authors suggested nitrogen N(3) in pilocarpine to be coordinated toward the heme iron of CYP2A6 and CYP2A5 active site.
Comparison of Inhibitory Potencies and Affinities of Tranylcypromine and Cyclopropylbenzene.
Although tranylcypromine and cyclopropylbenzene are structural analogs differing from each other only by one amine group, tranylcypromine is a much more potent CYP inhibitor than cyclopropylbenzene. However, both substances elicit spectral interactions roughly at the same concentrations. There are five aspects that need to be taken into consideration when reconciling these above-mentioned differences between inhibition potency and spectral interaction and similarities in affinities of both substances according to spectral interactions. First, substrate-induced spectral change is not as sensitive as the inhibition to detect interaction with specific CYP enzymes. Second, inhibition studies indicate that both compounds interact with several CYP enzymes and the difference in inhibition potency between tranylcypromine and cyclopropylbenzene is less with other CYP enzymes than with CYP2A6 and CYP2E1. Substrate-induced spectral change is dependent on the amount of specified CYP enzyme in microsomes. Relative amount of CYP2A6 in human liver microsomes is low and it is possible that its interaction with tranylcypromine has not been detected as a spectral change. Third, spectral interaction indicates that the amine group of tranylcypromine is interacting with heme iron of CYPs, whereas this kind of interaction is not possible with cyclopropylbenzene and therefore its interaction is stronger and more stable. This is probably an important factor that affects the inhibition potency because there is competition between the substrate and inhibitor for access to the active site of the CYP. However, this kind of experimental condition does not exist when spectral output of interaction is measured. Fourth, the most probable reason for very similar affinities may be the protonation of the amino group in tranylcypromine at pH 7.4. According to theoretical calculations for protonation of tranylcypromine at pH 7.4, about 4% is in unprotonated form and therefore capable of binding to P450. This means that the Ka values for unprotonated tranylcypromine actually are about 4% of the determined, i.e., about 0.18 to 0.6 μM and 1.4 to 6.5 μM. Fifth, it is still possible that the high-affinity component is due to a CYP enzyme that was not measured here in the inhibition panel.
Acknowledgments
The skillful technical assistance of Anne Hyry and Ritva Tauriainen is gratefully acknowledged. We thank Dr. Antti Poso for theoretical calculations of pKa for tranylcypromine.
Footnotes
-
Send reprint requests to: Päivi Taavitsainen, Department of Pharmacology and Toxicology, P.O. Box 5000, University of Oulu, FIN-90014 Finland. E-mail: paivi.taavitsainen{at}oulu.fi
-
This study was partially supported by the European Union Framework 4 Biomed2 project EUROCYP, by Orion Pharma, and by the Farmos Science and Research Foundation. This work was partially presented as an abstract in DMW/ISSX 2000 Congress, St. Andrews, Fife, Scotland, UK. Abstract appears in Drug Metab Rev (2000)32 (Suppl 1):111.
- Abbreviations used are::
- CYP
- P450, cytochrome P450
- HPLC
- high-performance liquid chromatography
- rac
- racemic
- Received August 14, 2000.
- Accepted November 16, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}