


Abstract
Time-dependent inhibition of CYP3A4 often results in clinically significant drug-drug interactions. In the current study, 37 in vivo cases of irreversible inhibition were collated, focusing on macrolides (erythromycin, clarithromycin, and azithromycin) and diltiazem as inhibitors. The interactions included 17 different CYP3A substrates showing up to a 7-fold increase in AUC (13.5% of studies were in the range of potent inhibition). A systematic analysis of the impact of CYP3A4 degradation half-life (mean t1/2deg = 3 days, ranging from 1 to 6 days) on the prediction of the extent of interaction for compounds with a differential contribution from CYP3A4 to the overall elimination (defined by fmCYP3A4) was performed. Although the prediction accuracy was very sensitive to the CYP3A4 degradation rate for substrates mainly eliminated by this enzyme (fmCYP3A4 ≥ 0.9), minimal effects are observed when CYP3A4 contributes less than 50% to the overall elimination in cases when the parallel elimination pathway is not subject to inhibition. Use of the mean CYP3A4 t1/2deg (3 days), average unbound systemic plasma concentration of the inhibitor, and the corresponding fmCYP3A4 resulted in 89% of studies predicted within 2-fold of the in vivo value. The impact of the interaction in the gut wall was assessed by assuming maximal intestinal inhibition of CYP3A4. Although a reduced number of false-negative predictions was observed, there was an increased number of overpredictions, and generally, a loss of prediction accuracy was observed. The impact of the possible interplay between CYP3A4 and efflux transporters on the intestinal interaction requires further evaluation.
As the most abundant human cytochrome P450 enzyme in both liver and intestine, CYP3A4 is susceptible to a number of reversible and irreversible metabolic drug-drug interactions. Irreversible, often referred to as mechanism-based, inhibition interactions involve the metabolism of an inhibitor by CYP3A4 to a reactive metabolite which inactivates the catalyzing enzyme in a concentration- and time-dependent manner (Silverman, 1995). A key characteristic of this type of inhibition is that inactivation occurs without the release of the reaction product from the catalytic site (Kent et al., 2001). The interaction between the inactivating species and enzyme can either be covalent or noncovalent, involving binding to protein or heme moiety, respectively (Zhou et al., 2005).
The two major kinetic parameters that characterize time-dependent inhibition interactions (TDIs) are kinact and Ki, the maximal inactivation rate constant and the inhibitor concentration leading to 50% of kinact, respectively (Silverman, 1995). The kinact/KI ratio is commonly taken as an indicator of the in vitro potency of a mechanism-based inhibitor. The methods used to obtain kinact and KI estimates generally vary across studies: the CYP3A4 probes used (midazolam, testosterone, or triazolam), their concentration (from below the Km to the concentrations equivalent to Vmax), preincubation to incubation time ratio, and data analysis method used, all of which may affect the estimates of inhibitor potency.
Quantitative in vitro-in vivo drug interaction prediction work has focused mainly on reversible interactions, whereas TDIs were assessed on an individual case-to-case basis (Ito et al., 2003; Wang et al., 2004). In contrast to reversible inhibition, CYP3A4 activity can only be restored by synthesis of new enzyme, often resulting in significant clinical interactions. General underprediction of an observed TDI using the simple [I]/Ki approach (Ito et al., 2004; Rostami-Hodjegan and Tucker, 2004) indicated a need for a more systematic analysis of this type of inhibition interaction.
The theoretical background for the quantitative prediction of TDI, incorporating the contribution of both hepatic and intestinal interaction, has been described previously (Wang et al., 2004) and was applied here (see Materials and Methods). The aim of the current study was to investigate systematically the significance of a number of parameters on the prediction of TDI, namely, enzyme degradation rate, the differential contribution of CYP3A4 to the victim drug elimination, and the effect of intestinal inhibition. Assessment was performed on 37 selected in vivo studies with macrolides (azithromycin, clarithromycin, and erythromycin) and diltiazem; the metric used was the AUCi/AUC ratio for the plasma concentration-time profiles in the presence and absence of the inhibitor, respectively, in agreement with previous analyses (Tucker et al., 2001; Galetin et al., 2005). The rationale for selection of these four inhibitors was the availability of both in vitro data and a wide range of in vivo interactions (including 17 different substrates with a differential contribution of CYP3A4 and renal excretion to their elimination). In addition, the prediction accuracy of this mechanistic approach was compared to the predictions based solely on reversible inhibition.
The rate of change of active enzyme concentration is determined by the equilibrium between the rates of de novo synthesis and degradation of the enzyme. Previous predictions of TDI (Ito et al., 2003; Wang et al., 2004) used CYP3A4 degradation half-life estimates (t1/2deg) obtained in either rat or Caco-2 cells (Correia, 1991; Malhotra et al., 2001), resulting in t1/2deg of 14 to 35 h. In the current study we have used estimates of human CYP3A4 kdeg from both induction studies or from in vitro investigations in liver slices. The impact of interindividual variability (estimated CYP3A4 t1/2deg varied from 1 to 6 days) on the prediction of TDI was assessed in addition to the fraction of the victim drug metabolized by CYP34 (defined by fmCYP3A4), as this parameter has been shown to have a significant impact on the drug-drug interaction prediction accuracy (Brown et al., 2005; Galetin et al., 2005; Ito et al., 2005).
Significant intestinal first-pass metabolism may contribute to the interindividual variability observed in bioavailability and the extent of drug-drug interactions for some CYP3A4 substrates. Therefore, the effect of assuming maximal intestinal inhibition on the precision and accuracy of the TDI predictions was investigated in conjugation with the detailed inhibitor- and substrate-related information (e.g., different inhibitor concentrations and parallel elimination pathways, respectively). The implications of maximal intestinal inhibition for drug elimination involving both metabolism and transporters are discussed.
Materials and Methods
In Vivo Studies. In the current analysis, 37 clinical studies of irreversible inhibition have been collated, focusing on macrolides (erythromycin, clarithromycin, and azithromycin) and diltiazem as time-dependent inhibitors of CYP3A4. The increase in the area under the plasma concentration-time profile in the presence of the inhibitor relative to control (AUCi/AUC) has been used as the metric to assess the degree of interaction. In the case of replicate studies (same dose of the inhibitor), the weighted mean of the AUC ratios was obtained for further analysis, as described previously (Galetin et al., 2005). Drug-drug interaction data resulting after the multiple dosing of an inhibitor and simultaneous oral administration of both drugs were used; studies selected involved 17 different CYP3A4 substrates (Table 1). In addition, clinical data on verapamil, mibefradil, saquinavir, and ritonavir interactions (n = 15) were collated for comparative reasons; no predictions were performed for these inhibitors.
Observed in vivo AUCi/AUC ratios for 37 CYP3A4 time-dependent interactions
In Vitro Data. Critical assessment of the available in vitro data were performed, and the estimates obtained in studies with the most appropriate experimental design (F. Ghanbari, K. Rowland-Yeo, J. Bloomer, S. Clarke, M. S. Lennard, G. T. Tucker, and A. Rostami-Hodjegan, submitted for publication) were used for prediction. The main focus was on the preincubation to incubation time ratio (>1), use of the dilution factor from the preincubation to the incubation stage (ideally, 1:10 to reduce the occurrence of competitive inhibition between the inhibitor and marker substrate in the second incubation), use of high substrate concentrations (equivalent to Vmax), and nonlinear regression analysis for obtaining the kinact and KI parameters. The kinact and KI estimates selected (Table 2) were obtained in the pooled human liver microsomes using either midazolam (macrolides; Ito et al., 2003) or testosterone (diltiazem; Rowland Yeo and Yeo, 2001) as the in vitro probe, whereas the estimates for N-desmethyldiltiazem were obtained by spectroscopic measurements of the metabolite-intermediate complex formation (Mayhew et al., 2000). No correction for microsomal binding was used for macrolides and N-desmethyldiltiazem, whereas a microsomal fraction unbound of 0.61 was used for diltiazem [calculated using the equation defined by Austin et al. (2002) and the logP value of 2.7 obtained by Wang et al. (1997)].
List of inhibitor concentrations and in vitro estimates for macrolides and diltiazem used for the prediction of 37 time-dependent interactions
Prediction of Time-Dependent Drug Interactions. The extent of time-dependent inhibition interaction incorporating the contribution of parallel elimination pathways (other cytochrome P450 enzymes or renal clearance, defined by 1 – fmCYP3A4) was predicted using the following equation (Wang et al., 2004):  where kinact represents the maximal inactivation rate constant, KI is the inhibitor concentration at 50% of kinact, Iu is the unbound inhibitor concentration (either the average systemic plasma concentration after repeated oral administration ([I]av) or the maximum hepatic input concentration ([I]in) (taken from Ito et al., 2004; range shown in Table 2), fmCYP3A4 is the fraction of victim drug metabolized by CYP3A4, kdeg is the endogenous degradation rate constant of the enzyme, and FG′ and FG are the intestinal wall availability in the presence and absence of inhibitor, respectively. Absorption rate constants used to obtain the [I]in values for erythromycin, clarithromycin, azithromycin, and diltiazem were 0.011, 0.018,0.00175, and 0.039 min–1, whereas the unbound fractions to the plasma protein were 0.16, 0.7, 0.28, and 0.175, respectively (Echizen and Eichelbaum, 1986; Ito et al., 2003). The impact of N-desmethyldiltiazem, an active metabolite of diltiazem, on the predicted degree of interaction was investigated assuming the same plasma protein binding as for diltiazem, and the relative plasma concentration is 38% of the parent (Echizen and Eichelbaum, 1986).
where kinact represents the maximal inactivation rate constant, KI is the inhibitor concentration at 50% of kinact, Iu is the unbound inhibitor concentration (either the average systemic plasma concentration after repeated oral administration ([I]av) or the maximum hepatic input concentration ([I]in) (taken from Ito et al., 2004; range shown in Table 2), fmCYP3A4 is the fraction of victim drug metabolized by CYP3A4, kdeg is the endogenous degradation rate constant of the enzyme, and FG′ and FG are the intestinal wall availability in the presence and absence of inhibitor, respectively. Absorption rate constants used to obtain the [I]in values for erythromycin, clarithromycin, azithromycin, and diltiazem were 0.011, 0.018,0.00175, and 0.039 min–1, whereas the unbound fractions to the plasma protein were 0.16, 0.7, 0.28, and 0.175, respectively (Echizen and Eichelbaum, 1986; Ito et al., 2003). The impact of N-desmethyldiltiazem, an active metabolite of diltiazem, on the predicted degree of interaction was investigated assuming the same plasma protein binding as for diltiazem, and the relative plasma concentration is 38% of the parent (Echizen and Eichelbaum, 1986).
Values for fmCYP3A4 were taken from Brown et al. (2005), Galetin et al. (2005), and Egnell et al. (2003) (carbamazepine); cyclosporine values were generated by regression analysis using eq. 1, as described previously (Ito et al., 2005). For loratadine, terfenadine, and nitrazepam, the number of studies was limited and the fmCYP3A4 was obtained by the rank order approach using studies with midazolam and the same dose of erythromycin as applied previously (Brown et al., 2005).
Human CYP3A degradation constants were collated from induction studies with carbamazepine, ritonavir, rifampin, and in vitro studies, as shown in Table 3. The impact of the different kdeg values (0.0005–0.00008 min–1, corresponding to CYP3A4 t1/2deg = 1–6 days) on the predicted AUC ratio was assessed for cases with an increasing contribution of CYP3A4 to the overall elimination (fmCYP3A4 = 0.4–1). This was carried out both by simulation at a constant or a range of inhibitor concentrations and by assessing the precision and accuracy of the predicted AUCi/AUC (see below). In addition, simulations of the effects of the multiple time-dependent inhibitors were performed assuming that the inhibitors inhibit the same enzyme, i.e., act via the same mechanism. The simulations investigated the scenarios in which the second inhibitor is either equipotent or 5-, 10-, and 50-fold more potent (greater kinact/KI ratio) than the first inhibitor, for different fmCYP3A4 values.
CYP3A4 degradation rate constant and the corresponding half-lives in humans
The contribution of the enzyme interaction in the intestinal wall was evaluated by incorporating the ratio of intestinal wall availability in the presence and absence of inhibitor, FG′ and FG, respectively, into the hepatic prediction (Wang et al., 2004), as shown in eq. 1. The impact of the maximal inhibition of intestinal metabolism in the presence of the inhibitor (FG′ = 1) on the prediction of TDI was assessed. The FG control values for the corresponding substrates were obtained indirectly from oral and i.v. administration data assuming complete absorption, negligible intestinal metabolism after i.v. administration, and that the systemic clearance of drug after i.v. dose reflects only hepatic elimination (eqs. 2–3) (Hall et al., 1999): 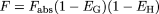
 where F represents the overall oral bioavailability, Fabs is the fraction of the oral dose absorbed intact across the apical membrane of the epithelial layer, EG and EH are the intestinal and hepatic extraction ratios, respectively, CLH is the hepatic blood clearance, and QH is the hepatic blood flow.
where F represents the overall oral bioavailability, Fabs is the fraction of the oral dose absorbed intact across the apical membrane of the epithelial layer, EG and EH are the intestinal and hepatic extraction ratios, respectively, CLH is the hepatic blood clearance, and QH is the hepatic blood flow.
The bias of TDI prediction was assessed from the geometric mean of the ratio of predicted and actual value [average-fold error (afe); eq. 4]. The mean squared prediction error (mse; eq. 5) and the root mean squared prediction error (rmse; eq. 6) provided a measure of precision for the predictions of 37 in vivo TDIs assessing the contribution of kdeg and FG ratio applying various inhibitor concentrations (Sheiner and Beal, 1981; Obach et al., 1997): 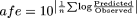


A 2-fold threshold value was selected on the basis of previous consensus reports (Tucker et al., 2001; Bjornsson et al., 2003) for a significant increase in AUC ratio. Thus, predicted areas <2 for interactions observed to be >2 were classed as false-negative and, similarly, predicted areas >2 for observed areas <2 were classed as false-positive interactions.
Results
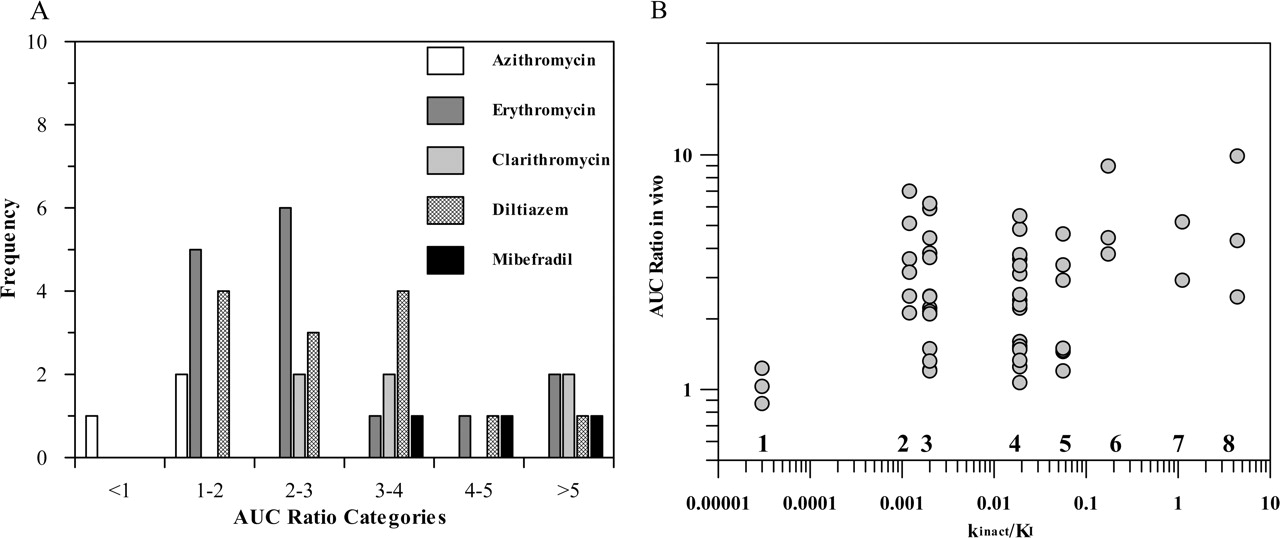
In Vivo Data. Previously reported time-dependent interactions (Table 1) have been classified using the recommendations of Bjornsson et al. (2003) for CYP3A4 inhibition interactions. Figure 1A shows the frequency distribution of the erythromycin, clarithromycin, azithromycin, and diltiazem interactions observed in comparison to mibefradil, a well known potent irreversible inhibitor of CYP3A4. This classification indicated that 54% of the interactions investigated were moderate (increase in AUC 2- to 5-fold), 14% were in the range of potent inhibition (AUCi/AUC > 5), and the remainder (32%) were weak (AUCi/AUC < 2).
In Vitro Data. TDI in vitro risk assessment is commonly based on the kinact/Ki ratio as an indicator of in vitro potency. The kinact values obtained for three macrolides and diltiazem ranged from 0.020 to 0.07 min–1 (Table 2), resulting in a maximum inactivation t1/2 of 10 to 35 min for diltiazem and azithromycin, respectively, significantly longer than those for mibefradil (inactivation t1/2 = 1.7 min) (Prueksaritanont et al., 1999). The kinact/KI ratio reported for N-desmethyldiltiazem (0.035) was equivalent to that for diltiazem. Literature-reported estimates obtained in both human liver and recombinant enzymes show large variability (0.001–0.008 and 0.005–0.023, respectively, in the case of erythromycin), with a general trend of greater estimated potency using recombinant enzymes in comparison to the liver microsomes. However, no direct relationship between the in vitro potency parameter and in vivo AUCi/AUC ratios was observed, as shown in Fig. 1B for eight time-dependent inhibitors, namely, azithromycin, clarithromycin, erythromycin, diltiazem, verapamil, mibefradil, saquinavir, and ritonavir. These findings are only partially dose-related as the same dose of erythromycin (1500 mg/day) results in either no effect (carbamazepine) or a 6-fold increase in AUC ratio (simvastatin) confirming that the in vitro parameters, although good indicators of potency, are not sufficient to predict the extent of TDI; additional substrate and inhibitor-related parameters are required.

Assessment of inhibitory potential of macrolides, diltiazem, and other compounds in vivo and in vitro. A, classification of azithromycin, erythromycin, clarithromycin, diltiazem, and mibefradil interactions according to Bjornsson et al. (2003), where AUCi/AUC >5 indicates potent, 2 < AUCi/AUC < 5 indicates moderate, and AUCi/AUC <2 indicates weak interactions. B, relationship between obtained kinact/KI ratio and the degree of interaction observed in vivo for azithromycin (1), erythromycin (2), clarithromycin (3), diltiazem (4), verapamil (5), mibefradil (6), saquinavir (7), and ritonavir (8) (inhibitors are listed in the order of increasing kinact/KI).
Impact of Enzyme Degradation and Parallel Elimination Pathways on TDI Prediction. Human kdeg estimates from induction studies with carbamazepine or ritonavir are shown in Table 3. The small number of livers and the 7-fold range in CYP3A4 t1/2deg observed represent a limitation of these estimates. The mean value of 3 days (corresponding to a kdeg = 0.00016 min–1) is consistent with in vitro findings in liver slices (Renwick et al., 2000) and was applied in all our TDI predictions.
The simulation in Fig. 2 shows the combined effect of substrate-related (fmCYP3A4 = 0.4–1) and enzyme-related (kdeg = 0.00008–0.0005 min–1) parameters on the predicted AUC ratio, taking erythromycin as an example (kinact and KI values of 0.025 min–1 and 13 μM, respectively, for an [I] of 0.5 μM, assuming no inhibition of the parallel pathway). The weak interaction potential predicted (2-fold increase in the AUC) refers to the substrates with low CYP3A4 contribution to the overall elimination (e.g., cerivastatin) in patients with a short CYP3A4 t1/2deg, whereas the compounds with fmCYP3A4 >0.9 in patients with CYP3A4 t1/2deg = 6 days represent the highest interaction risk (AUC increase up to 7-fold). The sensitivity of the predicted AUC ratios to different kdeg values at high and low fmCYP3A4 (0.9 and 0.5, respectively) over a range of inhibitor concentrations is illustrated in Fig. 3, A and B, for a weak and potent time-dependent inhibitor (e.g., azithromycin and mibefradil, respectively). Different CYP3A4 t1/2deg estimates have the greatest impact at higher fmCYP3A4 values, a trend observed regardless of the inhibitor potency; the major difference is the inhibitor concentration required to achieve the maximum effect.
The effect of various kdeg values (CYP3A4 t1/2deg = 1–6 days) on the prediction accuracy of 15 erythromycin, 6 clarithromycin, 3 azithromycin, and 13 diltiazem studies was investigated. Comparison of the predicted and observed AUCi/AUC for 37 TDI studies using the mean kdeg value of 0.00016 min–1 (CYP3A4 t1/2deg = 3 days), [I]av,u, and the corresponding fmCYP3A4 is shown in Fig. 4A. Of 37 studies, 89% were predicted within 2-fold of in vivo value; the use of extreme values (1 and 6 days) resulted in 22 and 25% of the studies under- or overpredicted, respectively. The prediction precision using the mean kdeg was improved by 10- and 21-fold (lower rmse) in comparison to the CYP3A4 t1/2deg of 1 and 6 days, respectively. In comparison to [I]av,u, the use of [I]in,u resulted in an increased number of overpredictions (up to 41% for t1/2 deg of 6 days) and a generally lower prediction accuracy since 59 to 68% of the studies predicted were within 2-fold limits over the range of kdeg.
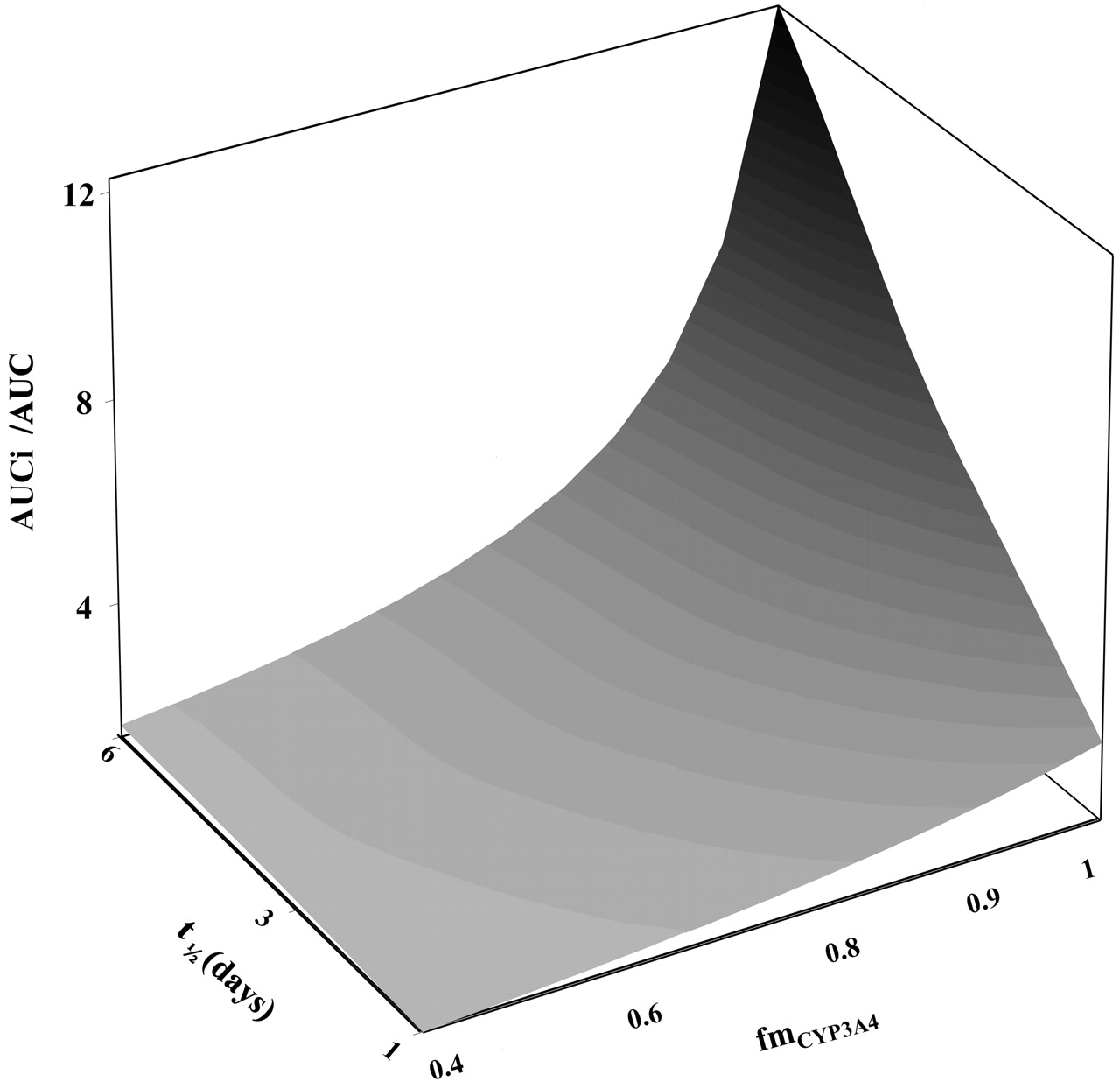
Effect of fmCYP3A4 and CYP3A4 t1/2deg on the predicted AUC ratio. Surface simulated for erythromycin kinact and KI values (0.025 min–1 and 13 μM, respectively) for inhibitor concentration of 0.5 μM over the range of fmCYP3A4 = 0.4 to 1 and CYP3A4 t1/2deg = 1 to 6 days; eq. 1 was used with FG′/FG = 1.
In all the scenarios investigated, the contribution of N-desmethyldiltiazem, an equipotent time-dependent inhibitor to diltiazem, was incorporated in the prediction. The mechanism of inhibition is the same for both parent and metabolite, and therefore, the maximal inhibition effect is not significantly increased by incorporating the metabolite in the prediction. The net effect is fmCYP3A4-dependent and decreases progressively with the decreasing contribution of CYP3A4 to the overall elimination of a substrate (from 19 to 0.5% for fmCYP3A4 = 0.9 and 0.1, respectively). A simulated interaction scenario with two inhibitors acting by the same mechanism, in which the second inhibitor is either equipotent (e.g., N-desmethyldiltiazem) or 5-, 10-, and 50-fold more potent (greater kinact/KI ratio) than the first inhibitor (erythromycin used as an example) is shown in Fig. 5 for the case when fmCYP3A4 is 0.9. The increased potency of the second inhibitor causes the maximal effect to be achieved at lower inhibitor concentrations, the level of which is dependent on the contribution of CYP3A4 to the elimination.

Effect of CYP3A4 degradation half-life on AUC ratio for inhibitors with different potency. The sensitivity of the predicted AUC ratios on t1/2deg of 1 (– – –), 3 (—), and 6 days (—) at high and low fmCYP3A4 (0.9 and 0.5, respectively) over a range of inhibitor concentrations for azithromycin (A) and mibefradil (B); eq. 1 was used with FG′/FG = 1.
Misspecification of the Inhibition Mechanism. Prediction of TDI using the [I]/Ki approach with the assumption of reversible inhibition resulted in 70% of predictions being classified as false-negatives and only 30% within 2-fold range of in vivo values (Table 4). In comparison, predictions made using the time-dependent model resulted in a marked reduction in false-negatives and a significant increase in the number of studies predicted within 2-fold (89% when no corrections for the intestinal interaction were incorporated).
Comparison of predictions applying reversible and time-dependent inhibition mechanism (with or without the correction for intestinal interaction)
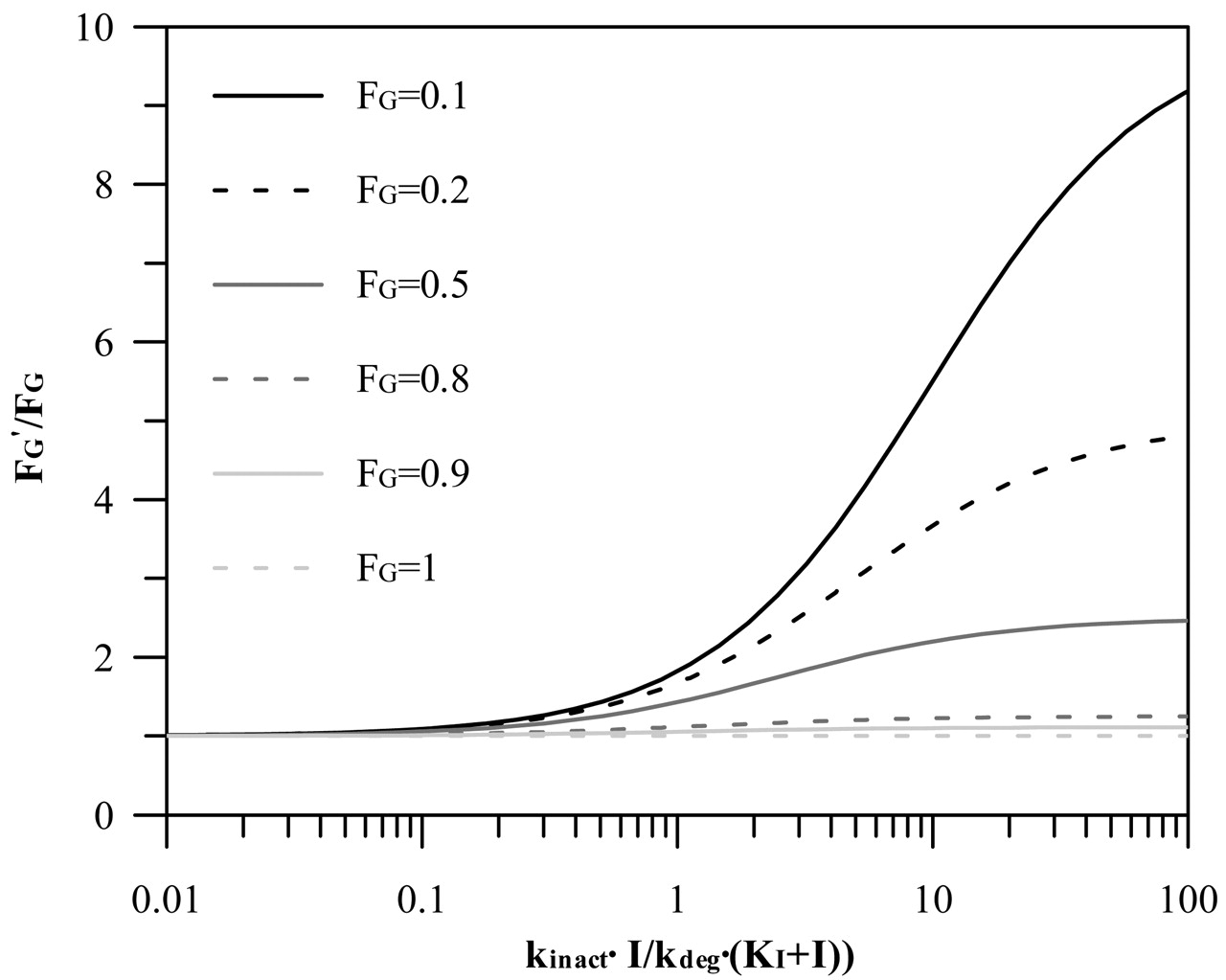
Maximal Intestinal Inhibition. To assess the contribution of the gut wall to the interaction, the ratio of the intestinal wall availability in the presence and absence of the inhibitor (FG′ and FG, respectively) is incorporated in the prediction equation based on hepatic estimates. Figure 6 shows the general relationship between FG′/FG ratio and the relative change in the intestinal clearance caused by an inhibitor [defined by the inactivation index, kinact · I/kdeg · (KI + I)]. For the case when this inactivation index is >1 (resulting in more than 50% decrease in the intestinal clearance), the effect of the FG ratio is most pronounced for compounds with high intestinal extraction (e.g., buspirone, tacrolimus), whereas it is minimal for the drugs with intestinal extraction approximately and below 50% (e.g., indinavir, alprazolam). The list of the FG values collated from i.v. and oral studies for nine CYP3A4 substrates analyzed in the current study is presented in Table 5. For the remaining substrates (loratadine, cerivastatin, nitrazepam, terfenadine, carbamazepine, pimozide, cisapride, and lovastatin, representing 24% of the interactions investigated), FG values were not available and only hepatic inhibition was taken into consideration (FG′/FG assumed to be 1).
FG and maximum FG ratio values for nine CYP3A4 substrates
The FG′/FG ratio can be determined experimentally from the relative change in the intestinal clearance of a compound in the presence of an inhibitor. However, when this is not available, an initial estimate of the extent of intestinal inhibition can be obtained by assuming the “worst case scenario,” i.e., the maximum inhibition of intestinal CYP3A4 (FG′ = 1). Table 5 indicates a 5-fold range of maximum FG′/FG ratios for the nine CYP3A4 substrates investigated. An issue of concern is the extent of interindividual variability in the estimates of FG, exemplified by a range of values for midazolam intestinal extraction (43–63%) (Olkkola et al., 1996; Thummel et al., 1996). For the majority of TDI studies involving erythromycin and clarithromycin, inclusion of the maximal FG′/FG ratios improved the prediction, in contrast to the diltiazem studies, which were mainly overpredicted. In addition, not surprisingly, this correction was not advantageous to the minimal interactions observed with azithromycin.
Overall, the inclusion of the maximal FG′/FG ratio for the respective substrates increased the number of overpredictions and reduced the prediction accuracy (70% estimates were within 2-fold of the in vivo value using the mean kdeg) (Fig. 4B; Table 4). The number of overpredictions was related to the CYP3A4 t1/2deg used, increasing from 0 to 6 and from 9 to 16 for the t1/2deg of 1 to 6 days, respectively. When [I]in,u rather than [I]av,u was applied in the prediction, the inclusion of the maximal FG ratio resulted in the overprediction of more than 50% of the studies, irrespective of the CYP3A4 t1/2deg used.
In addition to inhibitors, the predictions of 28 TDIs (based on eq. 1, CYP3A4 t1/2deg = 3 days; corresponding fmCYP3A4 and [I]av,u values) before and after the FG ratio correction (Fig. 4, C and D, respectively) are identified according to the particular substrates. In contrast to midazolam, triazolam, and nifedipine interactions, which were generally improved by the gut correction, pronounced overpredictions were observed for the interactions with cyclosporine and buspirone, in particular, with diltiazem as an inhibitor. In contrast to assuming FG′ = 1, the assumption that diltiazem reduces intestinal CYP3A4 activity by 62% (assessed by midazolam 1′-hydroxylation; Pinto et al., 2005) reduced this ratio and the extent of overpredictions, mainly for cyclosporine and buspirone interactions, respectively, by 54 to 190%; however, the predictions were still outside the 2-fold limits of in vivo values, in particular, for cyclosporine.

Relationship between predicted and observed AUC ratios for time-dependent interactions. Impact of gut interaction identifying either inhibitors or substractes. A, prediction of 37 TDIs using eq. 1, applying the average unbound plasma concentration of the inhibitor, corresponding fmCYP3A4, and CYP3A4 t1/2deg of 3 days (FG ratio = 1). Interactions are identified according to the inhibitors: ▪, azithromycin (3); ▵, erythromycin (15); •, clarithromycin (6); and *, diltiazem (13). B, effect of incorporating the maximal intestinal inhibition (FG′ = 1) on the prediction; all other conditions and symbols are as in panel A. C, predictions of 28 TDIs using eq. 1 (CYP3A4 t1/2deg, fmCYP3A4, and [I]av,u as in panel A) using the nine substrates with the available FG value. Interactions are identified according to the substrates: ▵, midazolam; ○, triazolam; □, alprazolam; ▴, buspirone; ▾, quinidine; ⋄, simvastatin; ▪, cyclosporine; •, felodipine; and *, nifedipine. D, effect of incorporating the maximal intestinal inhibition (FG′ = 1) on the predictions; all other conditions and symbols are as in panel C. In all the panels, the solid line represents line of unity, whereas dashed lines represent the 2-fold limit in prediction accuracy. The shaded areas correspond to the true negative and positive time-dependent interactions defined by the 2-fold increase in the AUC.
Discussion
The general approach to TDI in vitro risk assessment is based on the use of the kinact/KI ratio as an indicator of in vitro potency. However, there is a great variation in the methods used to generate this estimate, consistent with the 8-fold range observed in the literature-reported estimates for erythromycin in human liver microsomes and recombinant enzymes. The large variability observed can only be partially related to the use of different in vitro probes to assess the remaining CYP3A4 activity (triazolam, midazolam, or testosterone). The highly variable substrate concentrations used (e.g., midazolam at 8–200 μM) and the differential methods applied for obtaining parameter estimates (linear or nonlinear regression analysis) observed here and in other studies (F. Ghanbari, K. Rowland-Yeo, J. Bloomer, S. Clarke, M. S. Lennard, G. T. Tucker, and A. Rostami-Hodjegan, submitted for publication) imply a need for a more consistent in vitro experimental design for quantitative TDI assessment. A recent correlation analysis of kinact/KI estimates for five HIV-protease inhibitors by Ernest et al. (2005) indicated higher potency estimates in recombinant enzymes in comparison to the liver microsomes, a trend also seen in the current analysis with macrolides. However, standardization of in vitro methodology and the use of the CYP3A4 probe at high substrate concentration (equivalent to the Vmax) in the study by Ernest et al. (2005) reduced the difference between the in vitro systems (with the exception of saquinavir) to only 2-fold. The extrapolation of the in vitro potency (kinact/KI) to the in vivo situation depends on the relative magnitude of this parameter to the enzyme degradation; in the case where the inactivation index kinact · I/kdeg · (KI + I) is ≪kdeg (<10%, e.g., azithromycin), a weak inhibitory effect can be expected, with the AUC ratio ≤2. The opposite applies for moderate to potent inhibition; however, the relationship is more sensitive to the inhibitor concentration.

Simulations of interactions with multiple time-dependent inhibitors. The mechanism of inhibition is assumed to be the same and the second inhibitor is either equipotent or 5-, 10-, and 50-fold more potent (greater kinact/KI ratio) than the first inhibitor for the case when fmCYP3A4 is 0.9; eq. 1 was used with FG′/FG = 1.
The current study investigated the impact of CYP3A t1/2deg and the effect of maximal inhibition of intestinal metabolism (assessed by the FG ratio) on the prediction of 37 TDIs showing increases in AUC ratio of up to 7-fold. The substrates investigated cover a wide spectrum of interaction risk as they vary in the relative importance of CYP3A4 and renal clearance to their elimination, the significance of intestinal first-pass, the possible interplay with P-glycoprotein (Benet et al., 2004), and the contribution of CYP3A5 to the overall clearance (Galetin et al., 2004).
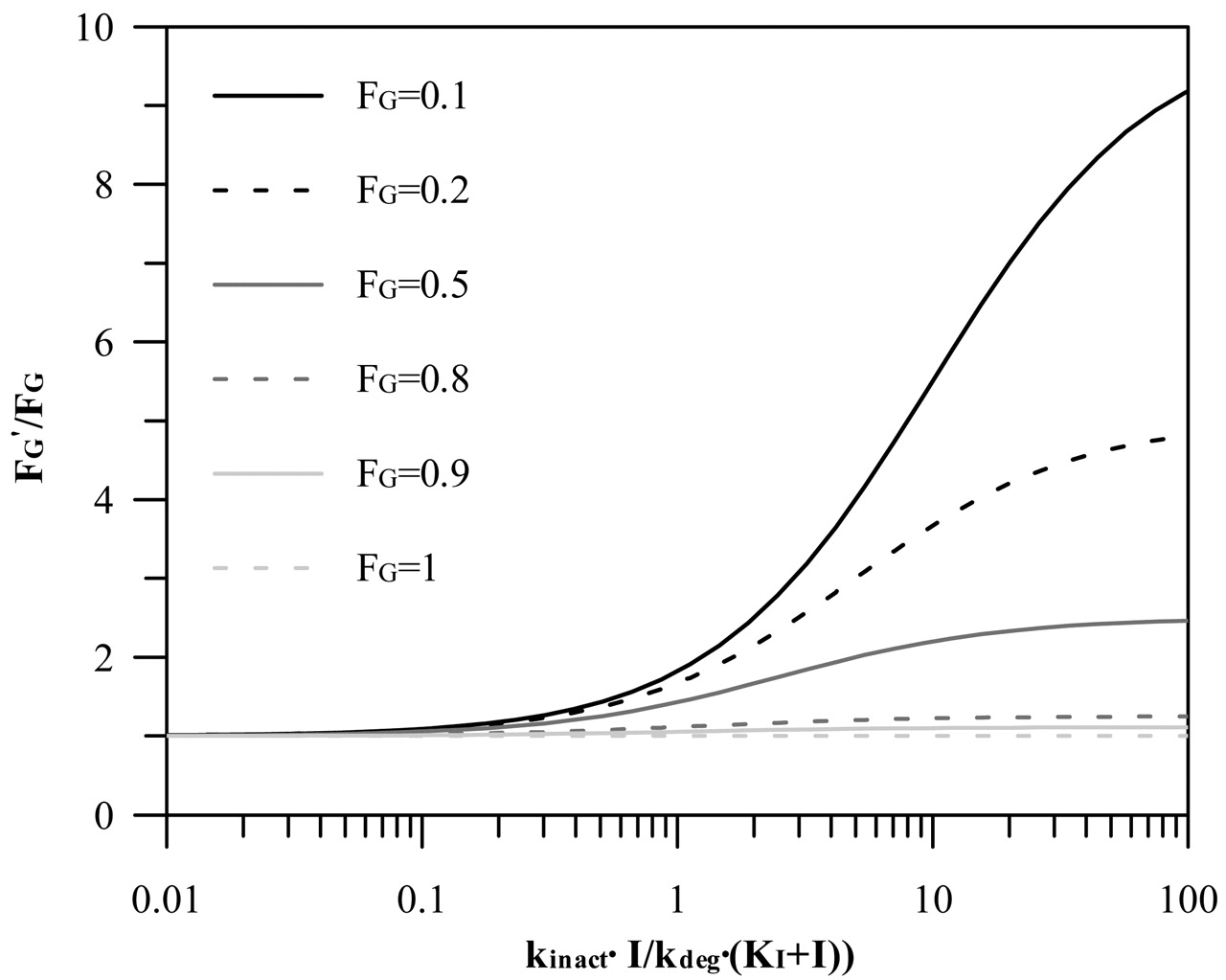
Relationship between FG′/FG and inactivation index, kinact · I/kdeg · (KI + I) for varying degrees of intestinal extraction (0–1). FG′/FG was simulated from the relative change in the intestinal intrinsic clearance caused by the inhibitor, which is estimated from the inactivation index parameters (Wang et al., 2004).
Venkatakrishnan and Obach (2005) have indicated the importance of accurate enzyme degradation estimates for the prediction of CYP2D6 inactivation by paroxetine; however, there is no comparable study performed for CYP3A4. Most of the prediction work in the area of time-dependent interactions is based on the use of CYP3A4 t1/2deg estimates obtained in either rat or Caco-2 cells (Ito et al., 2003; Wang et al., 2004). A number of indirect methods (Table 3) have been used to assess the CYP3A4 t1/2deg, all showing a great extent of interindividual variability. Therefore, the current study examines the impact of a range of human CYP3A4 t1/2deg values (20–146 h used as the extreme values) on the assessment of TDI potential. The mean estimated decay of CYP3A4 activity is, in general, longer in comparison to most other cytochrome P450 enzymes (F. Ghanbari, K. Rowland-Yeo, J. Bloomer, S. Clarke, M. S. Lennard, G. T. Tucker, and A. Rostami-Hodjegan, submitted for publication). The differential degradation half-lives reported for CYP3A4 and CYP3A5 in vitro (79 versus 35 h, respectively; Renwick et al., 2000) and the lower susceptibility to inhibition observed for CYP3A5 (McConn et al., 2004) may contribute to the extent of interindividual variability observed in the magnitude of interactions.
The effect of the CYP3A kdeg on the predicted AUC ratios for 37 interactions with macrolides and diltiazem was dependent on the inhibitor concentration used in the prediction. Use of the mean kdeg (0.00016 min–1) and [I]av,u, with no corrections for the intestinal interaction, provided low bias and the highest precision of TDI predictions (Table 4), with 33 of 37 studies predicted within 2-fold (Fig. 4A), whereas use of [I]in,u gave generally poorer results. For all the scenarios investigated (t1/2deg = 1–6 days), the use of total inhibitor concentration, in particular, hepatic input values, resulted in significant overprediction regardless of the CYP3A4 t1/2deg estimates used. As indicated previously for CYP2D6 (Venkatakrishnan and Obach, 2005), the sensitivity of the predicted extent of interaction to the differential CYP3A4 degradation rate was dependent on fmCYP3A4, as illustrated in Figs. 2 and 3. The current analysis indicates the suitability of the mean CYP3A4 t1/2deg of 3 days in the assessment of time-dependent interaction potential.
For CYP3A4 substrates, a certain degree of interaction at the level of the gut wall is to be expected because the small intestine represents the first in the series of first-pass eliminating organs and is exposed to orally administered inhibitors or inducers. A number of studies indicated a more pronounced inhibition (and induction) of intestinal CYP3A4 in comparison to the liver (Gomez et al., 1995; Floren et al., 1997; Tsunoda et al., 1999). The interpretation of such events has to be cautious for compounds that are mutual substrates for P-glycoprotein and CYP3A4 (e.g., cyclosporine, tacrolimus) due to their interactive nature and differing interrelationship in the intestine and liver (Benet et al., 2004). Intestinal interactions are rarely incorporated in the assessment of interaction potential, either for reversible or irreversible types (Rostami-Hodjegan and Tucker, 2004), which may represent a limitation for some CYP3A4 compounds. For example, a 2.6-fold greater increase in the AUCi/AUC ratio after an oral dose of midazolam in comparison to i.v. administration was observed in the presence of clarithromycin, indicating the contribution of intestinal interaction (Gorski et al., 1998). This observed difference is in good agreement with the predicted maximal intestinal FG ratio (Table 5). Predicted values of the maximal FG′/FG ratio are directly related to the accuracy of the initial estimates of the intestinal extraction and are based on the assumption that the extent of intestinal metabolism after i.v. administration is negligible and that absorption is complete, which may not be correct. The sensitivity of TDI prediction to the interindividual variability observed in the estimates of FG is greatest for substrates with FG < 0.5 (e.g., buspirone), as illustrated in Fig. 6.
The current study assessed the impact of assuming maximal intestinal inhibition (FG′ = 1), and despite the fact that some of the inhibitors investigated show moderate/weak potency, this assumption was satisfactory. The highest concentrations of the inhibitor are expected during its absorption phase, and the number of weak interactions (azithromycin being the weakest inhibitor investigated) is small in comparison to the overall number of studies. Of 28 studies in which the corrections for the intestinal interaction were applied, 18 were predicted within 2-fold of the in vivo value, with a low number of false-negative predictions. However, in 10 cases, incorporation of the FG ratio (notably for diltiazem studies) resulted in an overprediction (Fig. 4, B and D). In addition to the inhibitor, this observed overprediction trend can also be associated with the substrate-related characteristics as illustrated in Fig. 4, C and D. The overestimate of the magnitude of interaction observed in the case of cyclosporine is anticipated due to the possible contribution of efflux transporters and initial overprediction of the significance of the intestinal metabolism. For this and other high permeability/low solubility compounds like tacrolimus and lovastatin (Wu and Benet, 2005) the relationship between the FG ratio and the inhibitor potency will be more complex in comparison to the substrates mainly cleared by metabolism (e.g., midazolam, nifedipine). In the case of midazolam, maximal predicted and observed FG ratios were in good agreement in the presence of a number of irreversible and reversible inhibitors (data not shown); however, the general lack of i.v. studies undertaken prevents a more comprehensive comparison.
Prediction of TDI using the [I]/Ki approach and the assumption of a reversible inhibition process generally results in the false-negative predictions (Ito et al., 2004; Venkatakrishnan and Obach, 2005). When this model was used for the prediction of interactions with macrolides and diltiazem, 70% of the studies investigated were underpredicted (by 2- to 7-fold), whereas the time-dependent prediction model described here decreased the percentage of false-negatives to 19 and 8% before and after the FG ratio correction, respectively (Table 4).
In conclusion, the current study provides a systematic analysis of the impact of CYP3A4 degradation rate, parallel elimination pathways, and intestinal inhibition on the prediction of a range of TDIs. The impact of a differential CYP3A4 degradation rate on prediction accuracy and precision is important for substrates of which CYP3A4 contributes more than 50% to the overall elimination. Maximal FG ratio is proved to be a useful initial indicator of the extent of intestinal inhibition and minimized the number of false-negative predictions. However, the increased number of overpredictions observed for true positive TDIs when the maximal intestinal inhibition was assumed indicated a need for further evaluation of this approach and assessment of the interplay with the efflux transporters and variability in FG estimates.
Acknowledgments
We are grateful to Dr. Amin Rostami-Hodjegan (University of Sheffield and Simcyp Ltd.) for useful discussions.
Footnotes
-
Financial support for this project was provided by the following Centre for Applied Pharmacokinetic Research Consortium members: Eli Lilly, GlaxoSmithKline, Novartis, Pfizer, and Servier.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.006874.
-
ABBREVIATIONS: TDI, time-dependent drug-drug interaction; AUC, area under the plasma concentration-time curve; AUCi, area under the curve in the presence of the inhibitor; FG, intestinal wall availability; FG′, intestinal wall availability in the presence of an inhibitor; kinact, maximal inactivation rate constant; KI, inhibitor concentration at 50% of kinact; fmCYP3A4, fraction of victim drug metabolized by CYP3A4; kdeg, enzyme degradation rate constant; t1/2deg, degradation half-life.
- Received August 12, 2005.
- Accepted October 7, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}