


Abstract
The most common clinical implication for the activation of the human pregnane X receptor (PXR) is the occurrence of drug-drug interactions mediated by up-regulated cytochromes P450 3A (CYP3A) isozymes. Typical rodent models do not predict drug-drug interactions mediated by human PXR because of species differences in response to PXR ligands. In the current study, a PXR-humanized mouse model was generated by bacterial artificial chromosome (BAC) transgenesis in Pxr-null mice using a BAC clone containing the complete human PXR gene and 5′- and 3′-flanking sequences. In this PXR-humanized mouse model, PXR is selectively expressed in the liver and intestine, the same tissue expression pattern as CYP3A. Treatment of PXR-humanized mice with the PXR ligands mimicked the human response, since both hepatic and intestinal CYP3As were strongly induced by rifampicin, a human-specific PXR ligand, but not by pregnenolone 16α-carbonitrile, a rodent-specific PXR ligand. In rifampicin-pretreated PXR-humanized mice, an ∼60% decrease was observed for both the maximal midazolam serum concentration (Cmax) and the area under the concentration-time curve, as a result of a 3-fold increase in midazolam 1′-hydroxylation. These results illustrate the potential utility of the PXR-humanized mice in the investigation of drug-drug interactions mediated by CYP3A and suggest that the PXR-humanized mouse model would be an appropriate in vivo tool for evaluation of the overall pharmacokinetic consequences of human PXR activation by drugs.
Multiple-therapy regimens are commonly used in patients with cancer, HIV, cardiovascular disease, and diabetes. According to recent population-based surveys on drug use in the United States, the highest overall prevalence of medication use was among patients aged over 65 years, of whom 12% took at least 10 medications and 50% took at least 5 medications (Kaufman et al., 2002). For patients with multiple drug exposure, drug-drug interactions (DDIs) have become an important issue in health care, which may decrease therapeutic efficacy and/or increase drug toxicity. A higher risk of adverse drug reactions was reported in patients receiving multiple drugs, which was proposed to be due to DDIs (May et al., 1977). Serious adverse drug reactions in patients require hospitalization, and some result in permanent disability or death (Lazarou et al., 1998). Therefore, understanding the mechanism of DDIs and the development of relevant animal models to predict DDIs in humans are both important goals for the improvement of drug safety.
Most DDIs are related to pharmacokinetics and result from interference of the metabolic clearance of one drug by a coadministered drug (Guengerich, 1997). Oral contraceptive failures are a typical example of DDIs that occur in women also receiving rifampicin (RIF) or phenobarbital (D'Arcy, 1986). Induction of metabolic enzymes and increase in oral contraceptive clearance have been reported as the cause for oral contraceptive failures (Back and Orme, 1990). The cytochromes P450 (P450) mainly involved in drug metabolism are found in the 1A, 2C, 2D, and 3A subfamilies (Gonzalez, 1992). Of these, CYP3A4 is the most abundant P450 in the liver and participates in the metabolism of over 50% of clinically used drugs (Guengerich, 1999). There exist three minor CYP3A enzymes, CYP3A5, CYP3A7, and CYP3A43, that also play a role in drug metabolism (Daly, 2006). Many common drugs, such as the glucocorticoid dexamethasone, the antibiotic rifampicin, and the antimycotic clotrimazole, increase the expression of CYP3A genes as a result of transcriptional activation mediated by the pregnane X receptor (PXR; NR1I2), a member of the nuclear receptor superfamily. PXR is the dominant activator of CYP3A transcription and also plays an important role in regulating other genes involved in drug metabolism and elimination (Kliewer et al., 1998; Kliewer, 2005). Human PXR undergoes ligand activation, and the resulting transcriptional activation of CYP3A involves the formation of a heterodimer with retinoid X receptor, which binds to PXR response elements in the 5′-flanking region of the CYP3A4 gene (Goodwin et al., 1999). Many clinical medicines have been identified as human PXR ligands (Honkakoski et al., 2003), including prescription drugs, herbal supplements, and vitamins. Thus, patients treated with combinations of drugs that are PXR ligands and are CYP3A substrates run the risk of DDIs.
An important concern is to identify whether drug candidates in development are PXR ligands and/or CYP3A substrates. Human liver microsomes and cDNA-expressed CYP3A4, together with CYP3A chemical inhibitors or CYP3A-inhibitory antibodies, are common approaches to identify CYP3A substrates (Newton et al., 1995; Mei et al., 1999). Unfortunately, it is difficult to identify human PXR ligands because of important deficiencies in laboratory rodent models (Jones et al., 2000; Honkakoski et al., 2003). For example, drugs such as rifampicin, clotrimazole, and troglitazone strongly activate human PXR but are weak activators of rodent PXR. In contrast, dexamethasone and pregnenolone 16α-carbonitrile (PCN) strongly activate rodent PXR but are weak activators of human PXR. Although cell-based reporter gene assays represent a simple and rapid in vitro tool for screening human PXR ligands and CYP3A inducers (Raucy et al., 2002a), there remains the problem of extrapolating from in vitro results to the in vivo situation. Therefore, a PXR-humanized mouse model would be an ideal in vivo tool to study DDIs triggered by human PXR ligands. In previous studies, two humanized PXR mouse models, Alb-VP-hPXR and FABP-VP-hPXR transgenic mice, were generated by cDNA transgenesis (Xie et al., 2000; Gong et al., 2006). In the present study, a PXR-humanized mouse model was produced by bacterial artificial chromosome (BAC) transgenesis, in which the transgene contains the complete human PXR gene and the 5′- and 3′-flanking sequences. In this PXR-humanized mouse model, the role of human PXR on CYP3A induction was determined by pretreatment of mice with rifampicin and the investigation of the pharmacokinetics of midazolam (MDZ), a prototypical CYP3A4 drug substrate. This rifampicin/midazolam interaction illustrates the potential effectiveness of using this PXR-humanized mouse model in the study of DDIs mediated by CYP3A.
Materials and Methods
Chemicals. RIF, PCN, MDZ, ketoconazole (KCZ), and NADPH were obtained from Sigma-Aldrich (St. Louis, MO). 1′-Hydroxymidazolam (1′-OH-MDZ) was purchased from BD Gentest (Woburn, MA). All other chemicals were of the highest grade commercially available.
Generation of PXR-Humanized Transgenic Mice. The BAC clone RP11-169N13 (Resgen/Invitrogen Corporation, Huntsville, AL) containing the complete human PXR gene sequence (Zhang et al., 2001), including 5′- and 3′-flanking sequences, was purified by using a Maxi Prep kit (QIAGEN, Valencia, CA). The BAC clone was verified initially by PCR using primers designed to amplify specific regions within exons 2 and 9, and the 5′ UTR, as described previously (Zhang et al., 2001). Additional verification of the BAC clone was carried out by Southern blot analysis with 32P-end-labeled human PXR cDNA (Lehmann et al., 1998) and DNA oligonucleotide probes recognizing specific regions (exons 1 and 9, and –12.5 kilobases upstream) of the human PXR gene. The BAC clone was linearized by restriction enzyme digestion (P1-Sce) and purified before microinjection into fertilized FVB/N mouse eggs. Mice resulting from this breeding step that were positive for the human PXR transgene by PCR analysis were bred further with Pxr-null mice (Staudinger et al., 2001) kindly provided by GlaxoSmithKline Inc. (Uxbridge, Middlesex, UK) and Dr. Steven A. Kliewer (University of Texas Southwestern Medical Center, Dallas, TX). Mice positive for the human PXR transgene and the Pxr null allele, as determined by PCR genotyping, were designated as PXR-humanized transgenic (hPXR) mice. The hPXR mice were further bred with Pxr-null mice for at least four generations onto a C57BL/6 genetic background. Mice heterozygous for the hPXR transgene were then interbred to generate a homozygous line that was confirmed to be homozygous by breeding wild-type mice; all litters were positive for hPXR.
Animals and Treatments. Male, 2- to 4-month-old hPXR, Pxr-null, and wild-type (WT) mice, were maintained under a standard 12-h light/12-h dark cycle with water and chow provided ad libitum. The WT mouse background is C57BL/6. Handling was in accordance with animal study protocols approved by the National Cancer Institute Animal Care and Use Committee. For CYP3A induction, WT and Pxr-null and hPXR mice were injected i.p. with PCN, 10 mg/kg/day, or p.o. with RIF, 10 mg/kg/day, daily for 3 days. All mice were sacrificed by CO2 asphyxiation 24 h after the last dose. Liver and small intestine were collected and frozen at –80°C for further analysis. To detect CYP3A expression after the withdrawal of PXR ligand, the hPXR mice were treated with RIF (10 mg/kg/day) for 3 days. Then, 1, 2, 4, 6, 8, and 10 days after the last dose, mouse liver and small intestine were collected and frozen at –80°C for CYP3A expression analysis.
PCR Genotyping. The presence of the human PXR transgene was determined using the following primers: hPXR 5′ UTR Fwd, 5′-GCACCTGCTGCTAGGGAATA-3′ and hPXR 5′ UTR Rev, 5′-CTCCATTGCCCCTCCTAAGT-3′ amplifying a PCR product of 576 bp in only the samples positive for this transgene. Mouse epoxide hydrolase 1 gene (Ephx1) primers served as an internal positive control for amplification, yielding a fragment of 341 bp in all samples (Miyata et al., 1999). The following primers were used to identify the mouse PXR WT and null alleles: PXR-Fwd1, 5′-CTGGTCATCACTGTTGCTGTACCA-3′; PXR-Rev2, 5′-GCAGCATAGGACAAGTTATTCTAGAG-3′; and PXR-Rev3, 5′-CTAAAGCGCATGCTCCAGACTGC-3′ amplifying a PCR product of 348 bp for WT allele and 265 bp for Pxr-null allele (Guo et al., 2003).
Southern Blot Analysis. To analyze the BAC clone, DNA was digested with EcoRI, SacI, BglII, NcoI, or AflII. Electrophoresis and Southern hybridization conditions were described previously (Granvil et al., 2003). Random-primer 32P-labeled human PXR cDNA and DNA oligonucleotide probes recognizing specific regions of the human PXR gene were used for analysis of the BAC clone.
RNA Analysis. Human PXR tissue distribution in hPXR mice was analyzed by quantitative real-time PCR (qPCR). RNA was extracted from different tissues using TRIzol reagent (Invitrogen, Carlsbad, CA). qPCR was performed using cDNA generated from 1 μg of total RNA with a SuperScript II Reverse Transcriptase kit (Invitrogen). Primers were designed for qPCR using the Primer Express software (Applied Biosystems, Foster City, CA). hPXR: Fwd, 5′-GGCCACTGGCTATCACTTCAA-3′; Rev, 5′-TTCATGGCCCTCCTGAAAA-3′. mPXR: Fwd, 5′-AAGAAGCAGACTCTGCCTTGGA-3′; Rev, 5′-GTGGTAGCCATTGGCCTTGT-3′. CYP3A11: Fwd, 5′-AGCAGGGATGGACCTGG-3′; Rev, 5′-CGGTAGAGGAGCACCAA-3′. qPCR reactions were carried out using SYBR Green PCR master mix (SuperArray) in an ABI Prism 7900HT Sequence Detection System (Applied Biosystems). Values were quantified using the comparative cycle threshold (Ct) method, and samples were normalized to β-actin.
Microsome Preparation and Western Blot Analysis. Liver and small intestine were homogenized in ice-cold buffer (50 mM Tris-HCl, 150 mM KCl, 1 mM EDTA, and 20% glycerol). Microsomes were prepared by centrifugation at 10,000g for 20 min at 4°C, and the resulting supernatant was spun at 100,000g for 1 h at 4°C. Microsomal pellets were resuspended in the same ice-cold buffer used for homogenization. For Western blot analysis, microsomal protein (10 μg) from each sample was separated by SDS-polyacrylamide gel electrophoresis, electrophoretically transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH), and probed using anti-rat CYP1A2 mAb (clone 22-341), anti-rat CYP2C polyclonal Ab, anti-rat CYP2D polyclonal Ab, anti-rat CYP2E1 mAb (clone 1-98-1), anti-rat CYP3A1/2 mAb (clone 2-13-1), and anti-GAPDH polyclonal Ab (Chemicon International, Temecula, CA). Secondary antibodies, goat anti-mouse IgG (catalog number 115-036-071) and goat anti-rabbit IgG (catalog number 111-036-045), were purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA). Detection of immunoreactive proteins was done by an enhanced chemiluminescence blot detection system (GE Healthcare Bio Sciences, Little Chalfont, Buckinghamshire, UK).
CYP3A Activity Analysis. Midazolam 1′-hydroxylation was used as a probe for CYP3A activity (Thummel et al., 1994). The incubation was performed in 100 mM sodium phosphate buffer (pH 7.4) containing microsomes with 50 to 100 μg of protein and 50 μM MDZ in a final volume of 200 μl. For the inhibitory analysis, 2 μM ketoconazole (final concentration) was added in the incubation system and preincubated at 37°C for 5 min. The reaction was initiated by the addition of 20 μl of 20 mM NADPH, 37°C for 10 min, and terminated by the addition of 1 ml of ethyl acetate and 1 ml of methyl t-butyl ether mixture. Samples were centrifuged at 3000 rpm for 5 min at 4°C. The organic layer was then transferred to a new tube, dried with N2, and reconstituted in 100 μl of 70% methanol and 30% H2O containing 0.1% formic acid. All reactions were performed in duplicate. 1′-OH-MDZ was detected by liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS).
Pharmacokinetic Study of Midazolam in hPXR Mice. WT and hPXR mice were pretreated with or without 10 mg/kg RIF, once daily for 3 days. Twenty-four hours after the last dose of RIF, mice were administered with 5 mg/kg MDZ orally by gavage. Blood samples were collected from suborbital veins using heparinized tubes at 0, 5, 10, 20, 30, 60, 90, 120, and 180 min after administration of MDZ. Serum was separated by centrifugation at 8000g for 10 min. For MDZ extraction, 50 μl of serum was mixed with 150 μl of phosphate-buffered saline, 200 μl of ethyl acetate, and 200 μl of methyl t-butyl ether. The mixture was centrifuged at 3000 rpm for 5 min at 4°C. The organic layer was then transferred to a new tube, dried with N2, and reconstituted in 100 μl of 70% methanol and 30% H2O containing 0.1% formic acid. MDZ and 1′-OH-MDZ were detected by LC-MS/MS. Pharmacokinetic parameters for MDZ were estimated from the plasma concentration-time data by a noncompartmental approach using WinNonlin (Pharsight, Mountain View, CA). The maximum concentration in serum (Cmax) was obtained from the original data. The area under the serum concentration-time curve (AUC0–180 min) was calculated by the trapezoidal rule.
Analysis of Midazolam and 1′-OH-Midazolam by LC-MS/MS. MDZ and its metabolites 1′-OH-MDZ were determined by LC-MS/MS based on a previous method (Granvil et al., 2003) with minor modifications. LC-MS/MS analysis was carried out using a high-performance liquid chromatography system consisting of a PerkinElmer Series 200 quaternary pump, vacuum degasser, and autosampler (PerkinElmer Life and Analytical Sciences, Boston, MA) with a 100-μl loop interfaced to an API2000 SCIEX triple-quadrupole tandem mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA). MDZ, 1′-OH-MDZ, and 6-chloromelatonin (internal standard) were separated on a Luna C18 50 mm × 4.6 mm i.d. column (Phenomenex, Torrance, CA). The flow rate through the column at ambient temperature was 0.25 ml/min with 70% methanol and 30% H2O containing 0.1% formic acid. Each analysis lasted for 5.0 min. The mass spectrometer was operated in the turbo ion spray mode with positive ion detection. The turbo ion spray temperature was maintained at 300°C, and a voltage of 4.8 kV was applied to the sprayer needle. N2 was used as the turbo ion spray and nebulizing gas. The detection and quantification of analysts were performed using the multiple-reaction monitoring mode, with m/z 326/291 for MDZ, m/z 342/203 for 1′-OH-MDZ, and m/z 267/208 for 6-chloromelatonin.
Statistical Analysis. All values are expressed as the means ± S.D. and group differences analyzed by unpaired Student's t test.
Results
Generation ofPXR-Humanized Mice. A transgenic mouse line was created using a BAC clone containing the complete human PXR gene sequence and including 5′- and 3′-flanking sequences (Fig. 1A). The BAC clone was verified initially by PCR using primers designed to amplify specific regions within exons 2 and 9, and the 5′ UTR (Fig. 1B). Additional verification of the BAC clone was carried out by Southern blot analysis with 32P-end-labeled human PXR cDNA and DNA oligonucleotide probes recognizing specific regions (exons 1 and 9, and –12.5 kilobases upstream) of the human PXR gene. The size of each hybridized band corresponded well with the size predicted from the map of the human PXR gene sequence (data not shown). The BAC clone was linearized by restriction enzyme digestion and purified before microinjection into fertilized FVB/N mouse eggs. Several transgenic founders were identified by Southern blot analysis and bred with Pxr-null mice. Mice that were positive for the human PXR transgene and containing the mouse Pxr-null allele, as determined by PCR genotyping (Fig. 1C), were bred to generate homozygous mice and designated as PXR-humanized transgenic (hPXR) mice.

Generation and genetic characterization of PXR-humanized mice. A, structure of the BAC clone containing the complete human PXR gene sequence (exons 1–9) and the 5′- and 3′-flanking sequences. B, verification of the BAC clone by PCR using primers designed to amplify specific regions within exons 2 and 9, and the 5′ UTR. C, a representative genotyping result for hPXR mice. Mouse epoxide hydrolase 1 gene (Ephx1) primers served as an internal positive control. Mouse lines 4 and 5 were positive for the human PXR transgene and containing the mouse Pxr-null allele.
PXR Tissue Distribution. PXR tissue distribution was analyzed by qPCR. Human PXR was detected in hPXR mice in liver, duodenum, jejunum, and ileum, but not in lung and heart (Fig. 2A). Similar tissue distribution was reported in humans; however, the expression level of PXR in human liver is much higher when compared with the intestinal tract (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998). PXR expression in the mouse liver was shown to be similar to PXR expression levels in the intestinal tract (Jones et al., 2000). In this hPXR mouse model, human PXR expression levels were similar in liver and gut, a finding analogous to that of mouse PXR expression in WT mice (Fig. 2B).
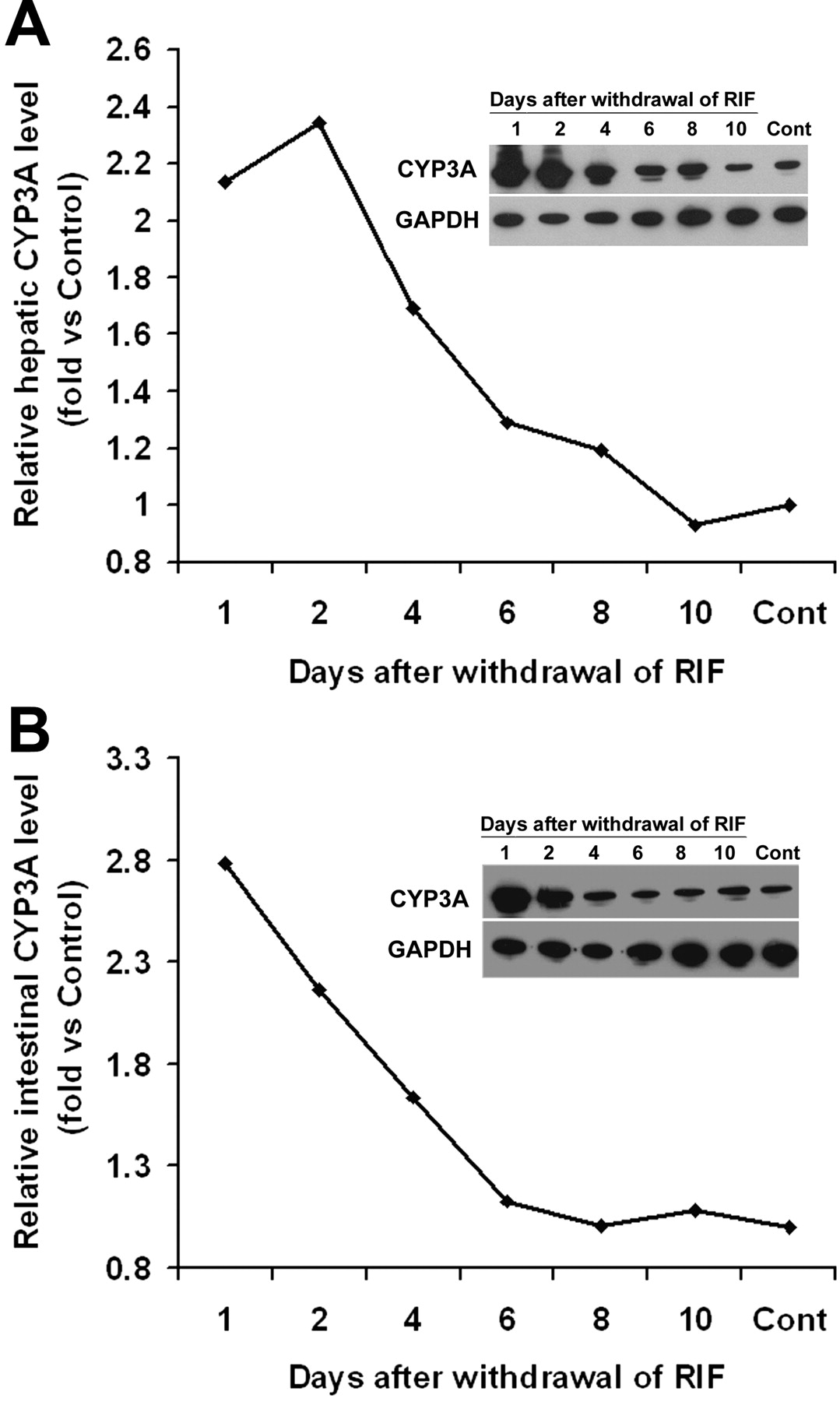
CYP3A Expression and Regulation in hPXR Mice. In the hPXR mouse model, human PXR is selectively expressed in the liver and intestine, the same tissues that express CYP3A. Xenobiotic response of CYP3A in hPXR mice was analyzed by qPCR (Fig. 3A) and Western blot (Fig. 3B). In hPXR mice, CYP3A11 mRNA and CYP3A protein were strongly induced by RIF, a human-specific PXR ligand, but not by PCN, a rodent-specific PXR ligand. In WT mice, CYP3A11 mRNA was strongly induced by PCN, but not by RIF. Neither PCN nor RIF resulted in significant CYP3A11 induction in Pxr-null mice. After the withdrawal of RIF treatment in hPXR mice, hepatic CYP3A protein expression was still remarkably high and took about 1 week to return to the basal level (Fig. 4A). These data indicated that DDIs may still occur in patients who take drugs that are CYP3A substrates soon after withdrawal of drugs that are PXR ligands. The importance of intestinal CYP3A in drug metabolism was addressed in a previous study using a transgenic CYP3A4 mouse model (Granvil et al., 2003). In the hPXR mouse model, induction of intestinal CYP3A protein was also noted, which increased significantly after 3 days of treatment with RIF. Compared with hepatic CYP3A, intestinal CYP3A expression decreased more rapidly after withdrawal of ligand (Fig. 4B). Other hepatic P450s were also investigated; however, no significant effect was observed on the expression of hepatic CYP1A2, CYP2C, CYP2D, and CYP2E1 P450s in the RIF-activated hPXR mouse line (Fig. 5). In a previous report on another line of PXR-humanized mice (VP-hPXR), CYP2C mRNAs were not induced by RIF (Rosenfeld et al., 2003). The lack of CYP2C induction is of interest since in cultured human hepatocytes, RIF induced CYP2C mRNAs (Raucy et al., 2002b; Ferguson et al., 2005). However, earlier studies in mice did not reveal induction of CYP2C transcripts with the rodent PXR ligand PCN (Guzelian et al., 2006; Jackson et al., 2006). These results suggest the possibility of a species difference in PXR-binding elements controlling CYP2C genes.

PXR tissue distribution in wild-type and PXR-humanized mice. PXR tissue distribution was analyzed by qPCR. A, human PXR tissue distribution in PXR-humanized mice. B, mouse PXR tissue distribution in WT mice. Values were quantified using the comparative CT method, and samples were normalized to β-actin.

Hepatic CYP3A expression and regulation in PXR-humanized mice. Animals were treated with PXR ligands, RIF or PCN, 10 mg/kg/day for 3 days. Animals treated with corn oil were designated as controls (Cont). A, qPCR analysis of CYP3A11 expression. Values were quantified using the comparative CT method, and samples were normalized to β-actin. B, Western blot analysis of mouse CYP3A; GAPDH served as a loading control.

CYP3A expression in liver and intestine of PXR-humanized mice after the withdrawal of RIF treatment. hPXR mice were treated with RIF, 10 mg/kg/day for 3 days. Animals treated with corn oil were designated controls (Cont). CYP3A was detected by Western blot; GAPDH served as a loading control. A, CYP3A expression in liver after the withdrawal of RIF treatment. B, CYP3A expression in small intestine (S. Intestine) after the withdrawal of RIF treatment.
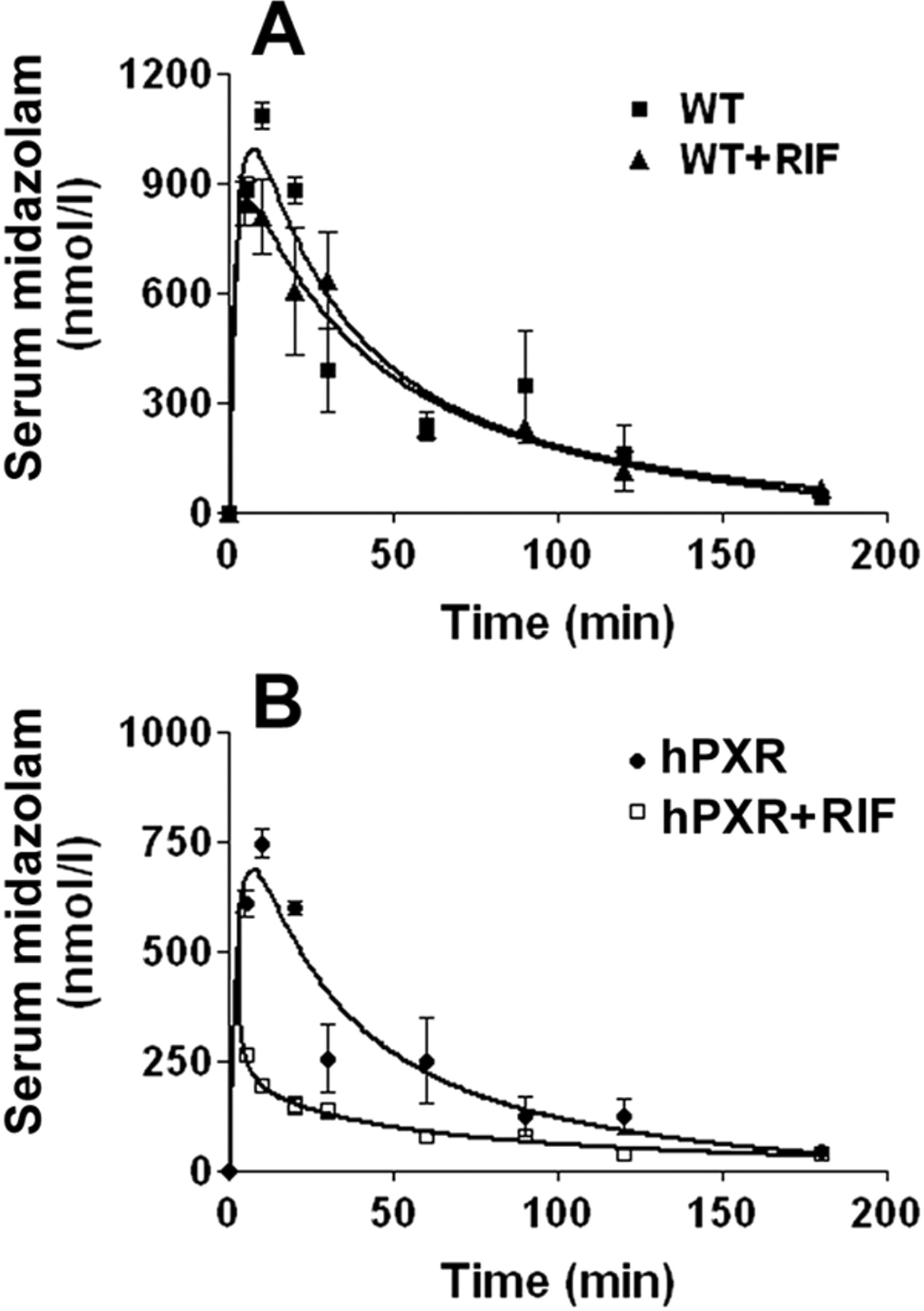
CYP3A Activity and Pharmacokinetic Study of Midazolam in hPXR Mice. In vitro hepatic CYP3A activity was analyzed by using midazolam 1′-hydroxylase as a probe (Thummel et al., 1994). Consistent with CYP3A expression, CYP3A activity increased significantly in hPXR mice treated with RIF. In WT and Pxr-null mice, RIF produced no significant effect on CYP3A activity (Fig. 6, A and B). In hPXR mice treated with RIF, CYP3A activity increased 314% compared with the vehicle-treated control (Fig. 6C). CYP3A activity in hPXR mice after RIF treatment was significantly inhibited (∼90%) by KCZ at 2 μM (Fig. 6D). In vivo, MDZ and RIF interactions were investigated in WT and hPXR mice. MDZ is a short-acting hypnotic-sedative drug metabolized by CYP3A (Gorski et al., 1994). Loss of the pharmacodynamic effects of MDZ was noted in tuberculosis patients undergoing RIF treatment, and human PXR activation and CYP3A induction were proposed as key factors in this DDI (Backman et al., 1996; Niemi et al., 2003). This clinical phenomenon was recapitulated and illuminated by using the hPXR mouse model. In WT mice, RIF has no significant effect on MDZ pharmacokinetics (Fig. 7A; Table 1). However, in hPXR mice pretreated with RIF, the maximal MDZ serum concentration (Cmax) was decreased by 64% and the area under the concentration-time curve (AUC0–180 min) was decreased by 60% (Fig. 7B; Table 1).
Pharmacokinetics of MDZ in wild-type and PXR-humanized mice pretreated with or without RIF, at 10 mg/kg/day for 3 days
Serum MDZ was detected by LC-MS/MS. Pharmacokinetic parameters were estimated from the serum concentration-time (three mice at each time point) data by a noncompartmental approach using WinNonlin. Data are expressed as means ± S.D.
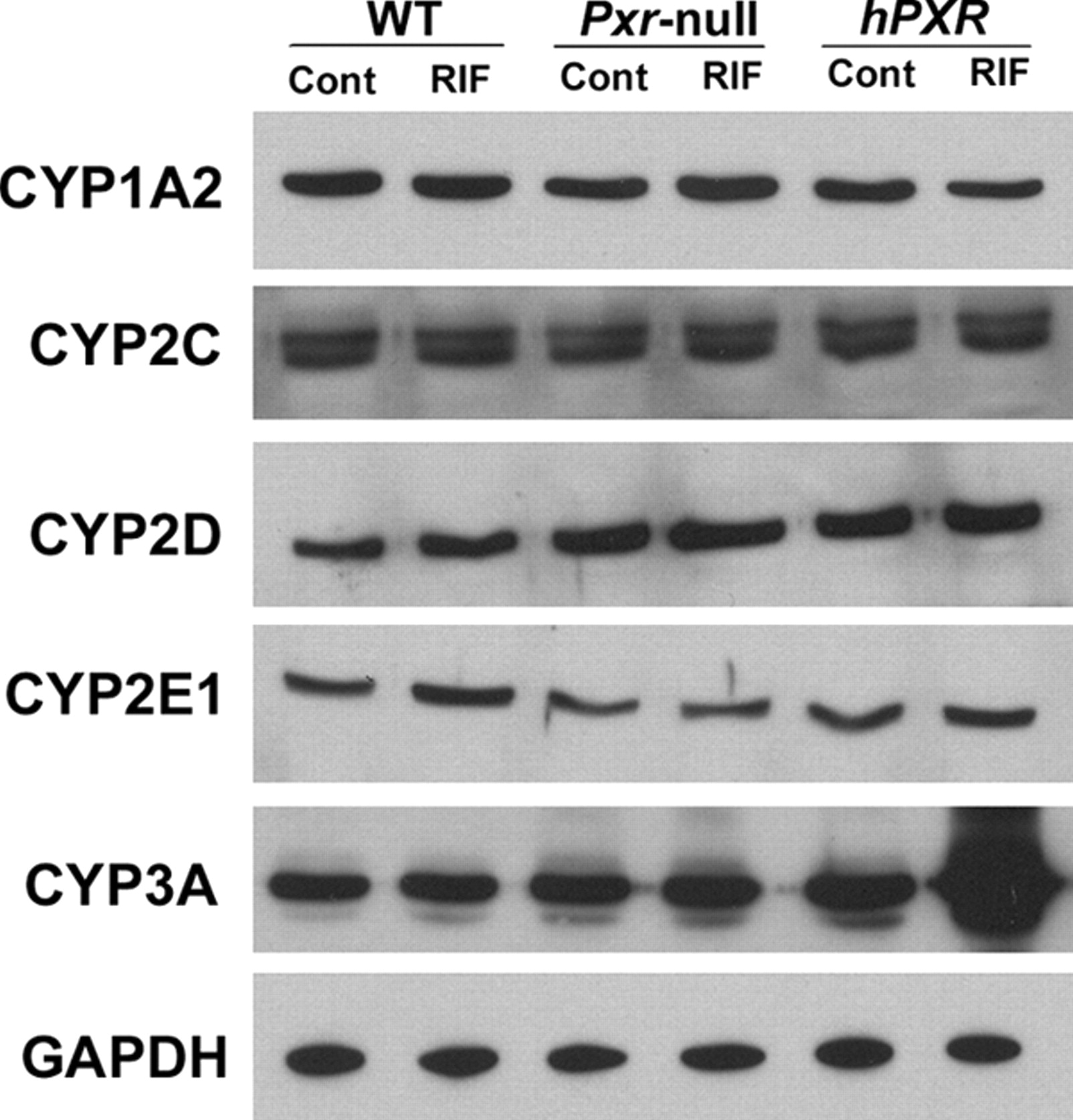
Effect of human PXR ligand treatment on the expression of major P450 isoenzymes in PXR-humanized mice. hPXR mice were treated with RIF, 10 mg/kg/day for 3 days. Animals treated with corn oil were designated as controls (Cont). CYP1A2, 2C, 2D, 2E1, and 3A were detected by Western blotting. GAPDH served as a loading control.

CYP3A activity analysis in liver microsomes of WT, Pxr-null, and PXR-humanized mice treated with RIF, 10 mg/kg/day for 3 days. Midazolam 1′-hydroxylation was used as the probe for CYP3A activity and analyzed by LC-MS/MS. CYP3A activity in the control group (Cont) was set as 100% for each animal line. Data are expressed as means ± S.D., n = 3. A, CYP3A activity in WT mice. B, CYP3A activity in Pxr-null mice. C, CYP3A activity in hPXR mice. *, P < 0.01 compared with control. D, inhibition on CYP3A activity by KCZ in hPXR mice with RIF pretreatment. *, P < 0.01 compared with no KCZ group.
Discussion
In the current study, a humanized PXR mouse model was generated by BAC transgenesis in Pxr-null mice and contained the complete human PXR gene, including the 5′- and 3′-flanking sequences. In the hPXR mouse model, PXR was selectively expressed in the liver and intestine, the same tissue expression pattern as CYP3A. Treatment with PXR ligands showed a clear species difference between WT and hPXR mice in response to xenobiotics, which suggests that this BAC transgenic hPXR mouse model is useful for the investigation of human PXR function in vivo.

Effect of RIF on midazolam pharmacokinetics in WT and PXR-humanized mice. hPXR mice and WT mice were pretreated with RIF, 10 mg/kg/day for 3 days. A, time course of serum MDZ in WT mice with or without RIF pretreatment. B, time course of serum MDZ in hPXR mice with or without RIF pretreatment.
PXR, a member of the nuclear receptor superfamily, plays an important role in regulating CYP3A and other genes involved in drug metabolism and elimination (Kliewer, 2005). The most common clinical implication of human PXR activation is the occurrence of DDIs. This is because CYP3A P450s, predominantly CYP3A4, metabolize over 50% of clinically used drugs. This phenomenon is ably illustrated by RIF, with which hundreds of drugs have been reported to result in DDIs during combined therapy (Niemi et al., 2003). RIF, a first-line antitubercular drug, has been used for more than 40 years, and DDIs with RIF were noted soon after its clinical introduction, such as the interaction between RIF and oral contraceptives (Rocher et al., 1971). However, for drugs in development, it is difficult to predict, using rodents, the likely occurrence of clinical drug interactions with RIF. This is due to species differences between rodents and humans in the response to PXR ligands (Jones et al., 2000; LeCluyse, 2001; Tirona et al., 2004). Therefore, the use of a hPXR mouse model would provide a solution to this problem through the expression of the human receptor in mouse liver and intestine.
High-throughput in vitro PXR activation and binding assays have been used to identify PXR ligands and CYP3A inducers, and it was concluded that the PXR reporter gene assay is a reliable and complementary method to evaluate CYP3A induction (Luo et al., 2002; Zhu et al., 2004). However, the general problem remains, that of extrapolating from in vitro findings to the clinical situation in vivo. It has been speculated that a PXR-humanized mouse model might be informative regarding human DDIs (Xie and Evans, 2002). A previously reported hPXR mouse model, Alb-VP-hPXR, was generated expressing a constitutively active human PXR in the liver of Pxr-null mice (Xie et al., 2000). The limit of this hPXR mouse model was hepatic-specific PXR expression, which did not reflect the full characteristics of human PXR, especially for its role in gut. The hPXR mouse model generated by BAC transgenesis in the current study showed PXR expression and CYP3A induction both in liver and gut. Investigation of the metabolism and pharmacokinetics of MDZ illustrated the potential effectiveness of using the hPXR mice for the study of the induction of CYP3A and thus its associated DDIs. The interaction between RIF and MDZ could not have been predicted from studies in WT mice because murine PXR is not activated by RIF (see Fig. 3). We take the interaction between RIF and MDZ in hPXR mice to be a paradigm for clinical interactions between PXR ligand drugs and CYP3A substrates. Not all drug metabolism mediated by CYP3A P450s can be attributed to the predominant form CYP3A4. Approximately 20% of human livers express CYP3A5, and the occurrence of the WT CYP3A5*1 allele displays considerable interethnic heterogeneity (Daly, 2006). Moreover, CYP3A5 is thought to account for 17% to 50% of hepatic CYP3A protein, and this may also be the same for intestinal CYP3A5. It would also appear from pharmacogenetic studies that CYP3A5 may contribute little, if anything, to the metabolism of classical CYP3A4 substrates, such as midazolam, cyclosporine, nifedipine, and docetaxel (Daly, 2006). However, for the immunosuppressant drug tacrolimus, multiple reports have established that CYP3A5 is a major contributor to its metabolism (Daly, 2006). The tissue distribution, induction, and clinical relevance of the minor P450 forms CYP3A7 and CYP3A43 remain more poorly understood. Nevertheless, clinical interactions between drugs that are PXR ligands and drugs that are CYP3A4, CYP3A5, CYP3A7, and CYP3A43 substrates would be expected to be predicted from the hPXR mouse model.
An earlier hPXR model (Xie et al., 2000) has been used to study the role of the P-glycoprotein export pump in the blood-brain barrier. P-glycoprotein provides a principal mechanism by which drugs are excluded from the brain. Although the plasma levels of methadone in control and RIF-induced hPXR mice were similar, the antinociceptive effects of methadone were about 70% reduced in the RIF-induced hPXR mice (Bauer et al., 2006). In addition to characterizing an important interaction between RIF and methadone, at the level of PXR, this report also demonstrates that PXR expression in the blood-brain barrier in hPXR mice can also induce expression of P-glycoprotein through regulation by PXR of the MDR1 (ABCB1) gene (Bauer et al., 2006). This represents another potential application of hPXR mice in drug development, the identification of PXR ligands that may exclude drugs from the brain via the induction of brain capillary P-glycoprotein. It will be possible, therefore, to evaluate this model for its combined utility in the prediction of pharmacokinetic drug-drug interactions at the level of both metabolism and transport.
Although a vast number of CYP3A substrates are known, less is understood about the ability of drugs to act as PXR ligands and thus induce hepatic and gut CYP3A activity and P-glycoprotein in the blood-brain barrier. It has been speculated that the development of new PXR ligands might be avoided through appropriate in silico screening procedures (Ekins and Erickson, 2002). Because the ligand-binding domain of PXR is large and flexible, this permits a diverse range of ligands to interact with PXR. It is therefore reasonable to consider every new drug candidate in preclinical development as a potential PXR ligand, with the undesirable effects on both drug metabolism and distribution. A pharmacophore for human PXR ligands has been developed and tested, and was able to distinguish the most potent PXR activators from weaker ones (Ekins and Erickson, 2002). The final test of a novel human PXR ligand would be to examine it in the hPXR mouse model described here, to evaluate its potential for causing drug-drug interactions in the clinic.
In conclusion, a PXR-humanized mouse model was generated successfully by BAC transgenesis, and it may become a useful tool for the investigation and prediction of pharmacokinetic DDIs.
Acknowledgments
J.R.I. is grateful to U.S. Smokeless Tobacco Company for a grant for collaborative research. We thank John R. Buckley for technical assistance.
Footnotes
-
This work was supported by the National Cancer Institute Intramural Research Program, and in part by National Institutes of Health (National Institute of Allergy and Infectious Diseases) Grant U19 AI067773-02.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.012831.
-
ABBREVIATIONS: DDI, drug-drug interaction; PXR, pregnane X receptor; P450, cytochrome P450; BAC, bacterial artificial chromosome; hPXR, PXR-humanized transgenic mice; RIF, rifampicin; PCN, pregnenolone 16α-carbonitrile; MDZ, midazolam; PCR, polymerase chain reaction; qPCR, quantitative real-time PCR; Ct, threshold cycle; LC-MS/MS, liquid chromatography-coupled tandem mass spectrometry; Cmax, maximal serum concentration; AUC, area under the concentration-time curve; 1′-OH-MDZ, 1′-hydroxymidazolam; UTR, untranslated region; WT, wild-type; Fwd, forward; Rev, reverse; bp, base pair(s); mAB, monoclonal antibody; Ab, antibody; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; KCZ, ketoconazole.
- Received September 8, 2006.
- Accepted November 7, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}