


Abstract
The ability to use vitro inactivation kinetic parameters in scaling to in vivo drug-drug interactions (DDIs) for mechanism-based inactivators of human cytochrome P450 (P450) enzymes was examined using eight human P450-selective marker activities in pooled human liver microsomes. These data were combined with other parameters (systemic Cmax, estimated hepatic inlet Cmax, fraction unbound, in vivo P450 enzyme degradation rate constants estimated from clinical pharmacokinetic data, and fraction of the affected drug cleared by the inhibited enzyme) to predict increases in exposure to drugs, and the predictions were compared with in vivo DDIs gathered from clinical studies reported in the scientific literature. In general, the use of unbound systemic Cmax as the inactivator concentration in vivo yielded the most accurate predictions of DDI with a mean -fold error of 1.64. Abbreviated in vitro approaches to identifying mechanism-based inactivators were developed. Testing potential inactivators at a single concentration (IC25) in a 30-min preincubation with human liver microsomes in the absence and presence of NADPH followed by assessment of P450 marker activities readily identified those compounds known to be mechanism-based inactivators and represents an approach that can be used with greater throughput. Measurement of decreases in IC50 occurring with a 30-min preincubation with liver microsomes and NADPH was also useful in identifying mechanism-based inactivators, and the IC50 measured after such a preincubation was highly correlated with the kinact/KI ratio measured after a full characterization of inactivation. Overall, these findings support the conclusion that P450 in vitro inactivation data are valuable in predicting clinical DDIs that can occur via this mechanism.
The prediction of drug-drug interactions (DDIs) using in vitro enzyme kinetic data has been an area of increasing advances and sophistication. This has proven to be a valuable endeavor because DDIs remain an important issue in clinical practice and the discovery and development of new drugs. The earlier that the potential for DDIs can be identified in new compounds being studied as potential drugs, the greater the likelihood that this deleterious property can be removed through improved design of the molecule. Also, for those compounds already undergoing clinical trials, in vitro DDI data can be leveraged in the design of adequate and appropriate clinical DDI studies. With our increased understanding of drug-metabolizing enzymes and their roles in the metabolism of specific drugs, a mechanistic approach to assessing DDIs can be taken. The results of clinical DDI studies with one drug can be extrapolated to other drugs that are cleared by the same enzyme.
The alteration of drug-metabolizing enzyme activities can occur by three main mechanisms: reversible inhibition, mechanism-based inactivation, and induction. Confidence in quantitatively extrapolating in vitro results to in vivo varies with these mechanisms. For reversible inhibition mechanisms, recent advances in our ability to predict the magnitude of DDIs from in vitro inhibition data have been made such that for cytochrome P450 (P450) enzymes, increases in exposure can be predicted within 2-fold (Brown et al., 2005; Ito et al., 2005; Obach et al., 2006). Through the use of human hepatocytes in culture, enzyme inducers can be readily identified (Silva and Nicholl-Griffith, 2002). However, the magnitude of predicted DDIs for inducers varies with the source of individual hepatocytes. Recently, in vitro induction data from an immortalized human hepatocyte line, which gives a robust and reliable response, has been shown to yield quantitative predictions of CYP3A induction-based DDIs in vivo (Ripp et al., 2006).
For mechanism-based inactivators, there have been some reports describing the prediction of in vivo DDIs, particularly for CYP3A (Kanamitsu et al., 2000a; Mayhew et al., 2000; Yamano et al., 2001; Ito et al., 2003; Wang et al., 2004; Galetin et al., 2006), as well as one analysis for CYP2D6 (Venkatakrishnan and Obach, 2005). Compared with reversible inhibition, there are some added complexities regarding the prediction of DDIs from inactivation data. The design and conduct of in vitro inactivation studies are more complex than reversible inhibition studies, in that experiments require two steps (preincubation with inactivator followed by dilution and incubation to measure the standard marker activity), and several incubation times are needed to generate inactivation rate constants (kinact). For extrapolation to in vivo, knowledge of the in vivo rate of degradation of the target enzyme in human is needed. Such a value cannot be measured directly, forcing the use of in vitro data in human hepatocytes, animal data (Mayhew et al., 2000), or data modeled from human pharmacokinetic studies of deinduction or recovery following inactivation (Takanaga et al., 2000; Greenblatt et al., 2003; Faber and Fuhr, 2004; Venkatakrishnan and Obach, 2005), and thus greater uncertainty is introduced.
The primary objectives of this work are 2-fold: 1) to devise a simplified in vitro approach whereby mechanism-based inactivators of P450 enzymes can be identified, and 2) to determine whether there is a reliable method that can be used to predict the magnitude of DDIs from in vitro inactivation data across multiple P450 enzymes.
Materials and Methods
Materials. P450 substrates, internal standards, and pooled liver microsomes were the same as those described earlier (Walsky and Obach, 2004). NADPH was from ICN (Aurora, OH). The compounds examined as inactivators were obtained from one of the following sources: Aldrich Chemical Co. (Milwaukee, WI), Sequoia Research Products (Oxford, UK), or Sigma Chemical Co. (St. Louis, MO) with the exception of desethylamiodarone (Synfine, Richmond Hill, ON, Canada) and tienilic acid (Cerilliant, Austin, TX). PPP was synthesized at Pfizer by Dr. James Eggler. Other reagents were obtained from common commercial suppliers.
Single Point Inactivation and IC50 Shift Experiments. Pooled human liver microsomes (0.3–2 mg/ml, depending on which enzyme was assessed) were incubated with inactivators and MgCl2 (3.3 mM) in potassium phosphate buffer (100 mM, pH 7.4) in the absence and presence of NADPH (1.3 mM). In single point inactivation experiments, the concentration of the inactivator used was 10-fold that which gave 25% inhibition when tested under reversible inhibition conditions. (Thus, after dilution of 10× into the subsequent activity assay, the concentration in the minus NADPH control will be at IC25.) In IC50 shift experiments, multiple concentrations of inactivators were used such that the range included the concentration that was 10 times the IC50 measured for reversible inhibition. (Thus, after dilution of 10× into the subsequent activity assay, the concentrations in the minus NADPH controls will span IC50.) The inactivators were delivered in solvent such that the final solvent concentration was less than 1% (v/v). These preincubations (i.e., the “inactivation incubation”) were carried out for 30 min at 37°C. For highly efficient inactivators, a 30-min preincubation time will exceed the time over which the inactivation is first order (i.e., log-linear). However, the selection of a 30-min preincubation period was made to ensure that even weak inactivators could be identified (i.e., to avoid false negatives). Vehicle controls were run to account for any decrease in enzyme activity caused by incubation under these conditions. After the inactivation incubation, a portion of the inactivation mixture (0.02 ml) was added to a mixture containing a standard P450 substrate in 0.18 ml of potassium phosphate buffer (100 mM, pH 7.4) containing MgCl2 (3.3 mM) and NADPH (1.3 mM) for measurement of P450 activities (i.e., the “activity incubation”). The substrates used and their concentrations are listed in Table 1. Substrate concentrations used were proximal to KM values. Incubations were carried out and samples analyzed by high-performance liquid chromatography/mass spectrometry using previously described methods (Walsky and Obach, 2004).
Concentrations of P450 substrates used in single point, IC50 shift, and kinact/KI experiments
Single point inactivation data were analyzed by comparing the percentage inhibition measured when the inactivator was preincubated for 30 min in the presence of NADPH versus that in the absence of NADPH: 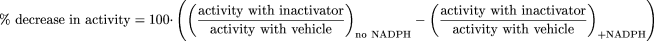
Inactivation Kinetic Experiments to DetermineKI andkinact. Inactivation kinetic experiments were conducted in a manner similar to that described above. In the inactivation preincubation, various concentrations of inactivator were incubated at 37°C with pooled human liver microsomes (0.3–2 mg/ml, depending on which enzyme was assessed), MgCl2 (3.3 mM), and NADPH (1.3 mM) in potassium phosphate buffer (100 mM, pH 7.4). At six time points, aliquots of the inactivation preincubation mixture (0.02 ml) were removed and added to a mixture containing a standard P450 substrate, MgCl2 (3.3 mM), and NADPH (1.3 mM) in potassium phosphate buffer (100 mM, pH 7.4) at 37°C. The substrates used were those described above at concentrations approximately 10 times the KM (Table 1). To determine kobs values, the decrease in natural logarithm of the activity over time was plotted for each inactivator concentration, and kobs values were described as the negative slopes of the lines. Inactivation kinetic parameters were determined using nonlinear regression of the data to the following expression: 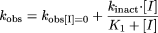 in which [I] represents the concentrations of inactivators in the inactivation preincubations, kobs represents the negative values of the slopes of the natural logarithm of the percentage activity remaining versus inactivation incubation time at various [I], kobs[I] = 0 is the apparent inactivation rate constant measured in the absence of inactivator, kinact is the limit maximum inactivation rate constant as [I]→∞, and KI is the inactivator concentration yielding kobs at the sum of kobs[I] = 0 and 0.5 times kinact.
in which [I] represents the concentrations of inactivators in the inactivation preincubations, kobs represents the negative values of the slopes of the natural logarithm of the percentage activity remaining versus inactivation incubation time at various [I], kobs[I] = 0 is the apparent inactivation rate constant measured in the absence of inactivator, kinact is the limit maximum inactivation rate constant as [I]→∞, and KI is the inactivator concentration yielding kobs at the sum of kobs[I] = 0 and 0.5 times kinact.
Predictions of Drug Interactions. The potential for an inactivator to cause an increase in exposure to a drug as a result of inactivation of hepatic enzymes was assessed using the following equation (Mayhew et al., 2000): 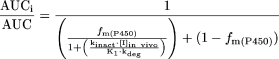 The terms are defined as follows: AUCi/AUC is the predicted ratio of in vivo exposure of a P450-cleared drug with coadministration of the inactivator versus that in control state, fm(P450) is the fraction of total clearance of the drug to which the affected P450 enzyme contributes, kdeg is the first-order rate constant of in vivo degradation of the affected P450 enzyme, kinact is the theoretical maximum inactivation rate constant at infinite inactivator concentration as assessed in vitro, KI is the inactivator concentration yielding a measured inactivation rate at half of kinact, and [I]in vivo is the in vivo concentration of the inactivator. For CYP3A, the impact on extraction by the intestine also needs to be accounted for by including a term for the effect on CYP3A in the intestine (Wang et al., 2004):
The terms are defined as follows: AUCi/AUC is the predicted ratio of in vivo exposure of a P450-cleared drug with coadministration of the inactivator versus that in control state, fm(P450) is the fraction of total clearance of the drug to which the affected P450 enzyme contributes, kdeg is the first-order rate constant of in vivo degradation of the affected P450 enzyme, kinact is the theoretical maximum inactivation rate constant at infinite inactivator concentration as assessed in vitro, KI is the inactivator concentration yielding a measured inactivation rate at half of kinact, and [I]in vivo is the in vivo concentration of the inactivator. For CYP3A, the impact on extraction by the intestine also needs to be accounted for by including a term for the effect on CYP3A in the intestine (Wang et al., 2004): 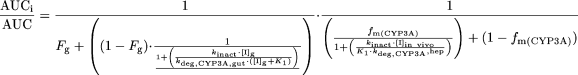 The parameters are as described above, with the addition of the following: Fg is the fraction of the dose of the affected drug that passes through the intestine unchanged after p.o. administration in the control state, [I]g is the concentration of the inactivator in the intestine, fm(CYP3A) is the fraction of the affected drug cleared by hepatic CYP3A, and kdeg,CYP3A,gut and kdeg,CYP3A,hep represent in vivo degradation rate constants for CYP3A in the intestine and liver, respectively.
The parameters are as described above, with the addition of the following: Fg is the fraction of the dose of the affected drug that passes through the intestine unchanged after p.o. administration in the control state, [I]g is the concentration of the inactivator in the intestine, fm(CYP3A) is the fraction of the affected drug cleared by hepatic CYP3A, and kdeg,CYP3A,gut and kdeg,CYP3A,hep represent in vivo degradation rate constants for CYP3A in the intestine and liver, respectively.
The following values were used for the parameters needed for eqs. 3 and 4. Fraction of the affected drug metabolized by P450s (fm(P450)). Values for the fraction of the affected drug metabolized by the P450 enzyme that is inactivated were previously reported (Obach et al., 2006) and had been derived from various sources. These are theophylline fm(CYP1A2) = 0.8; S-warfarin fm(CYP2C9) = 0.91; omeprazole fm(CYP2C19) = 0.87; desipramine fm(CYP2D6) = 0.9; and midazolam and buspirone fm(CYP3A) = 0.93. For bupropion hydroxylation, a value of fm(CYP2B6) = 0.95 was used based on estimates from in vitro data (Hesse et al., 2000), and for caffeine a value of fm(CYP1A2) = 0.95 was made using a combination of quantitative human metabolism data reported from control subjects receiving radiolabeled drug (Rodopoulos et al., 1995) and in vitro data describing P450 enzymes involved in metabolic pathways (Ha et al., 1996).
Concentration of the inactivator in vivo ([I]). For the term [I], described as the concentration of inactivator available to the enzyme, the systemic steady-state Cmax, the systemic steady-state unbound Cmax (defined as fu× Cmax), and the estimated unbound steady-state Cmax at the inlet to the liver (i.e., Cmax,u,inlet) (Kanamitsu et al., 2000b) as: 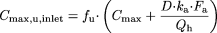 in which D is the dose of the inactivator, ka is the oral absorption rate constant of the inactivator, Fa is the fraction of the inactivator absorbed following p.o. administration, Cmax is the systemic steady-state maximum concentration of the inactivator, fu is the unbound fraction of inactivator in plasma, and Qh is hepatic blood flow (1450 ml/min).
in which D is the dose of the inactivator, ka is the oral absorption rate constant of the inactivator, Fa is the fraction of the inactivator absorbed following p.o. administration, Cmax is the systemic steady-state maximum concentration of the inactivator, fu is the unbound fraction of inactivator in plasma, and Qh is hepatic blood flow (1450 ml/min).
For CYP3A, the concentration of the inactivator in the enterocyte during absorption ([I]g) was also considered and defined as (Rostami-Hodjegan and Tucker, 2004): 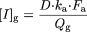 Parameters are the same as described above, with Qg representing enterocytic blood flow (248 ml/min).
Parameters are the same as described above, with Qg representing enterocytic blood flow (248 ml/min).
In Vivo P450 degradation rate constants (kdeg). Under normal conditions, the rate of de novo biosynthesis of P450 enzymes should equal the rate of degradation. Experimentally measured values for such a parameter in humans are not obtainable; therefore, these values must be estimated. In this analysis, where available, kdeg values for each P450 enzyme were estimated by modeling the time course of deinduction or recovery following inactivation of oral clearance of substrates specific for various P450s. This was accomplished using data from well designed studies in the clinical pharmacokinetic literature for CYP1A2 (Faber and Fuhr, 2004), CYP2D6 (Liston et al., 2002; Venkatakrishnan and Obach, 2005), and intestinal CYP3A (Greenblatt et al., 2003) based on the time course of deinduction following smoking cessation, recovery following paroxetine inactivation, and recovery following inactivation by grapefruit juice, respectively. The resulting kdeg estimates (per minute) were 0.000296, 0.000226, and 0.000481 for CYP1A2, CYP2D6, and intestinal CYP3A, respectively. For hepatic CYP3A, an initial estimate of 0.000321/min was used based on the kinetics of deinduction of the oral clearance of verapamil (Fromm et al., 1996), general clinical pharmacologic understanding of the kinetics of induction and deinduction of CYP3A (Thummel et al., 2000; Lin, 2006), and additionally supported by in vitro estimates of CYP3A turnover in primary human hepatocytes using pulse-chase methods (Pichard et al., 1992). For the other enzymes (CYP2B6, CYP2C9, and CYP2C19), a mean value of the above-described estimates for hepatic P450s (0.00026/min) was used because clinical pharmacokinetic data to support similar calculations were not available.
In addition, empirical predictions of the magnitude of DDIs were explored using IC50 values measured after a 30-min preincubation of inactivator, human liver microsomes, and NADPH. Equations used for these predictions were those described previously (Obach et al., 2006): 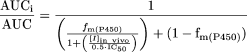 and for CYP3A:
and for CYP3A: 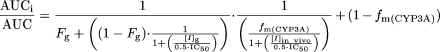 in which IC50 is the value measured in an activity assay after the inactivator had been preincubated for 30 min with microsomes and NADPH.
in which IC50 is the value measured in an activity assay after the inactivator had been preincubated for 30 min with microsomes and NADPH.
Accuracies of prediction methods were assessed using the geometric mean -fold error: 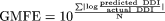 and the root mean squared error:
and the root mean squared error:  in which N is the total number of predictions.
in which N is the total number of predictions.
Results
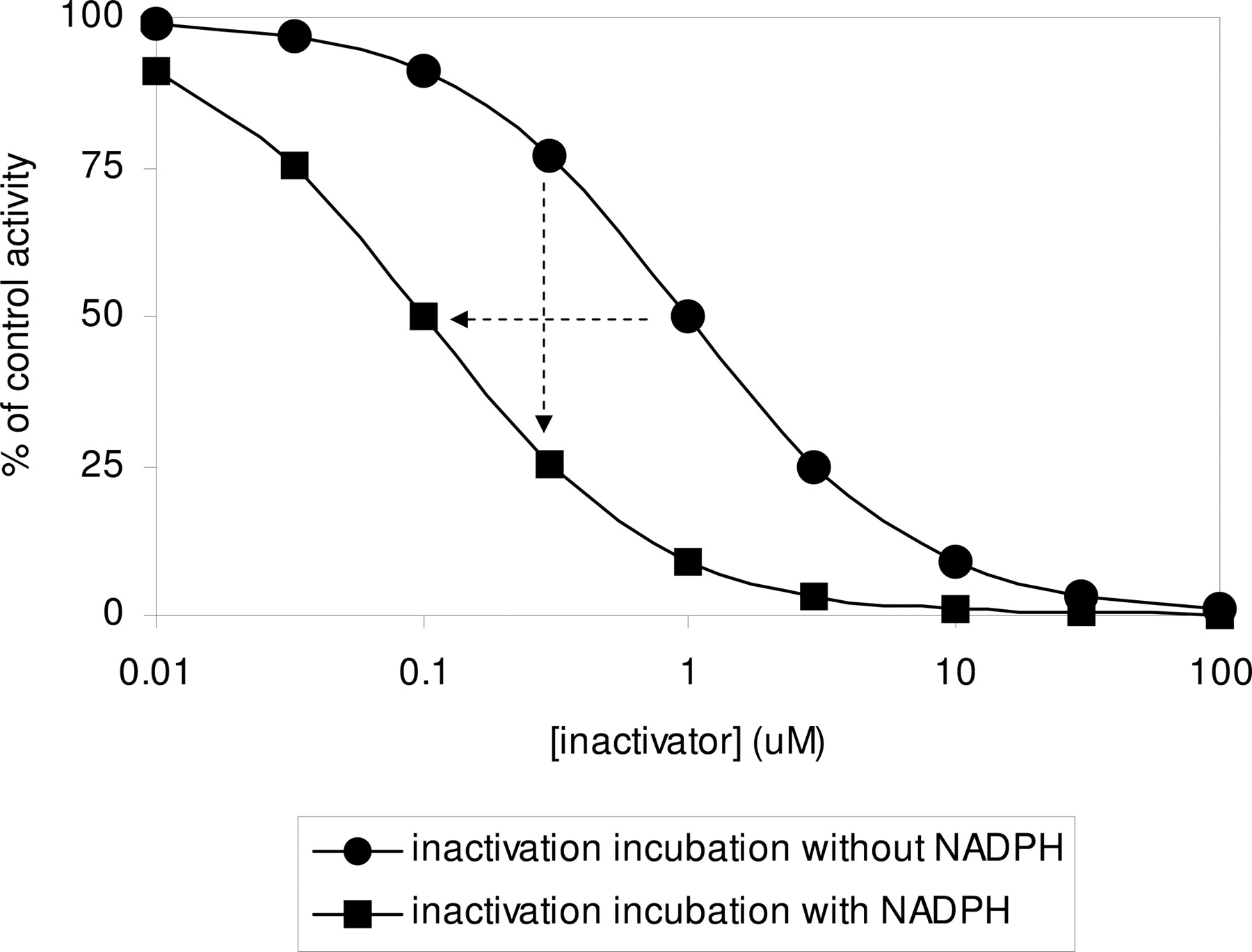
Characterization of Single Point Inactivation. Compounds known to be inactivators for various human P450 enzymes were incubated in an inactivation preincubation at a single concentration representing 10-fold of the concentration known to cause 25% inhibition in the activity incubation. These same compounds were also tested for the other P450 enzymes for which it had not been shown to cause inactivation. This concentration was selected because, in theory, it should be at the most sensitive point for detecting a mechanism-based inactivator (refer to Fig. 1). The data showing the percentage decrease in activity caused by a 30-min incubation of inactivators in the presence of NADPH versus that in the absence of NADPH are shown in Table 2. With the exception of ticlopidine/CYP2C19 and rotonavir/CYP3A, for those combinations where inactivation was expected the percentage of decrease in activity was at least 30%. These include furafylline and zileuton (CYP1A2) (Racha et al., 1998; Lu et al., 2003), ticlopidine, PPP, and N,N′,N″-triethylenethiophosphoramide (thioTEPA) (CYP2B6) (Chun et al., 2000; Richter et al., 2004, 2005), desethylamiodarone (CYP2C8) (Polasek et al., 2004), tienilic acid (CYP2C9) (Melet et al., 2003), methylenedioxymethamphetamine (MDMA) and paroxetine (CYP2D6) (Bertelsen et al., 2003; Heydari et al., 2004), and diltiazem, erythromycin, and verapamil (CYP3A) (Mayhew et al., 2000; McConn et al., 2004; Wang et al., 2004; Ernest et al., 2005). With a few exceptions (e.g., desethylamiodarone, thioTEPA), these compounds did not show appreciable inactivation for other P450 enzymes. As a negative control, montelukast, which potently inhibits several P450 enzymes (Walsky et al., 2005), did not show inactivation for any of the enzymes. Ticlopidine showed only an 11% decrease in CYP2C19 activity when incubated in the presence of NADPH. This could be partially because the incubation time for assessing CYP2C19 S-mephenytoin hydroxylase activity (40 min) is longer than the 30 min inactivation incubation time, which would be expected to blunt any observable difference. Thus, a decrease in activity of 15% was identified as a cutoff value for identifying inactivators for P450 enzymes, except for CYP2C19 for which a cutoff value of 10% was identified.

Illustration of the single point inactivation and IC50 shift experiments. The two theoretical curves represent IC50 determinations after previous 30-min incubation of enzyme and P450 inactivator in the absence and presence of NADPH. In the IC50 shift, the horizontal dashed arrow shows a 10-fold decrease in IC50 (in this case from 1.0 to 0.1 μM) when the inactivation incubation is done in the presence of NADPH. In the single point approach, the vertical dashed arrow represents the change in the percentage of control activity when the enzyme is incubated with inactivator at 10 times the IC25 for 30 min in the presence of NADPH before assessment of activity. In this case, IC25 in the absence of NADPH is approximately 0.3 μM. When the enzyme is incubated with inactivator at 0.3 μM in the presence of NADPH, followed by assessment of P450 activity, there is a 50% decrease in activity (75% to 25%).
Percentage change in inhibition of human P450 enzymes with a 30-min incubation of inactivator, human liver microsomes, and NADPH
IC50 Shift Results. For those agents that showed a 15% (or 10% for CYP2C19) decrease in activity when incubated for 30 min at 10-fold the IC25, a determination of the decrease in the IC50 was measured. Inactivators were incubated at several concentrations spanning the value of 10 times the reversible inhibition IC50 for 30 min in the presence or absence of NADPH, followed by a 10-fold dilution into an activity assay incubation. A list of the IC50 values generated with and without NADPH in the inactivation incubation is in Table 3, and an example is shown in Fig. 2A. The range of IC50 shifts spanned from a >800-fold decrease (inactivation of testosterone 6β-hydroxylase by erythromycin) to 1.6-fold (inactivation of midazolam 1′-hydroxylase by ritonavir).

Example of mechanism-based inactivation data. In this example, the inactivation of CYP2B6 by thioTEPA is shown. A, an IC50 shift plot. The IC50 values in this example are 0.099 and 3.8 μM for preincubation with and without NADPH, respectively. B, inactivation plots. The values for kobs are determined as the negative slopes of the natural logarithm of the percentage of control versus time. C, nonlinear regression to determine KI and kinact for the inactivation of CYP2B6 by thioTEPA. Values determined in this example were 5.3 μM and 0.17/min, respectively. The value for kobs[I] = 0 was 0.0039/min.
IC50 values for mechanism-based inactivators of human P450 enzymes following a 30-min incubation with human liver microsomes in the absence and presence of NADPH
Inactivation Kinetics. Values for kinact and KI were measured for those compounds showing IC50 shifts, and these are listed in Table 4, with an example shown in Fig. 2. (Refer to the Supplemental Information for plots for all the inactivators.) A practical limit for measuring kinact values appears to be about 0.005/min and depends to some extent on the enzyme being studied. Based on kinact values alone, the compounds with the greatest capacity to inactivate various P450 enzymes were ritonavir (CYP3A), MDMA (CYP2D6), ticlopidine (CYP2B6), and tienilic acid (CYP2C9). When the potency is also included to generate kinact/KI ratios, the compounds with the greatest capability to inactivate P450 enzymes are ritonavir (CYP3A), ticlopidine (CYP2B6), tienilic acid (CYP2C9), paroxetine (CYP2D6), and furafylline (CYP1A2). Comparing values for kinact/KI and the IC50, the shifted IC50, and the -fold shift in IC50 revealed that the greatest correlation existed with the shifted IC50 (Fig. 3). A correlation also existed between the shifted IC50 and KI, which may indicate that the shifted IC50 contains elements of the inactivator potency. This indicates that the -fold change in IC50 alone is not the most important predictor of the efficiency of an inactivator but rather what the potency is after conducting a 30-min incubation with the inactivator before measurement of activity. Some compounds exhibited substantial -fold changes in IC50, yet the overall potency was not high (e.g., MDMA and CYP1A2, thioTEPA and CYP2C8, etc.).

Comparison of the relationships between kinact/KI and IC50 (A), shifted IC50 (B), and -fold shift in IC50 (C), as well as between shifted IC50 and KI. The term “shifted IC50” refers to the value measured after the inactivator has been preincubated with liver microsomes and NADPH for 30 min.
Inactivation kinetic values for mechanism-based inactivators of human P450 enzymes
Prediction of DDIs.Utilizing shifted IC50 values. The generation of IC50 values after a 30-min preincubation of the inactivator, liver microsomes, and NADPH may represent a scaled-down empirical surrogate of a full determination of inactivation kinetic parameters, considering the excellent correlation to the kinact/KI ratio, an established measure of inactivation efficiency (Ernest et al., 2005). In a previous report, it had been shown that the magnitude of DDIs could be reliably predicted for reversible inhibitors when the estimated unbound portal vein Cmax was used, along with reversible inhibition constants (Obach et al., 2005). However, it was observed that this approach was not generally accurate for compounds known to be mechanism-based inactivators. Thus, IC50 values generated after an inactivation preincubation (i.e., “shifted” IC50 values) were used in eqs. 7 and 8, which had been previously shown to yield accurate predictions of DDIs for reversible inhibitors when combined with estimates of unbound portal vein Cmax values. Results are listed in Table 5, and a plot of predicted DDIs versus actual values from clinical studies is shown in Fig. 4A. A comparison of the shifted and nonshifted IC50 values yields mixed results. For some compounds, the shifted IC50 values yielded more accurate predictions (e.g., paroxetine, ritonavir), whereas for others the prediction was less accurate (e.g., tienilic acid, erythromycin). Preincubation led to the identification of diltiazem, paroxetine, and verapamil as perpetrators of DDIs, whereas no preincubation would have misidentified these drugs as noninactivators. In almost every case, the actual DDI magnitude was between the values predicted from shifted and nonshifted IC50 values.
Predictions of drug interactions for mechanism-based inactivators using IC50 values gathered after a 30-min incubation with or without NADPH and the prediction method applicable for reversible inhibition
Utilizing inactivation kinetic parameters. In Table 6, predictions of the magnitude of DDIs are listed using three different values for in vivo concentration of the inactivator ([I]in vivo): total systemic Cmax, unbound systemic Cmax, and estimated unbound Cmax at the inlet to the liver. In almost all the cases, predictions yielded greater values than the actual DDI values. Total systemic Cmax yielded over-predictions; in some cases, these were very inaccurate (e.g., erythromycin, tienilic acid, and verapamil), and in others, predictions of DDIs greater than 2-fold would be made but the actual value was less than 2-fold (e.g., ticlopidine/theophylline and zileuton). Similar observations were made when the estimated unbound portal vein Cmax was used for [I]. Unbound systemic Cmax values generally provided the most accurate predictions. Geometric mean -fold errors for predictions using total systemic Cmax, unbound systemic Cmax, and estimated unbound portal vein Cmax were 2.50, 1.64, and 2.63, respectively.
Predictions of drug interactions for mechanism-based inactivators using in vitro inactivation parameters

Comparison of predicted versus actual DDIs for mechanism-based inactivators. In A, the predictions were made using shifted IC50 values and the equation for reversible inhibition (eqs. 7 and 8). In B, predictions were made using inactivation kinetic parameters and eqs. 3 and 4, using unbound systemic concentrations of the inactivator in the prediction. Note that the predicted value for erythromycin (57-fold) is off-scale in A.
Discussion
Among scientists studying drug metabolism it has been a long-standing goal to develop methods whereby in vitro data can be used for making reliable predictions of various pharmacokinetic phenomena such as clearance and DDIs. An increased understanding of the enzymes and transporters involved in drug metabolism and disposition has led to an increasing ability to make such predictions. Drugs that are known to cause increases in exposure to other drugs commonly cause these by reversible inhibition or irreversible inactivation of metabolizing enzymes. In previous reports, an ability to predict DDIs for those compounds that reversibly inhibit human P450 enzymes was shown (Obach et al., 2005, 2006). However, in these reports, under-predictions of DDIs for several drugs using reversible inhibition data were made, as a result of these compounds actually being irreversible inactivators of drug-metabolizing enzymes. For individual drugs, others have shown that DDIs caused by irreversible inactivation of P450 enzymes can be predicted (Kanamitsu et al., 2000a; Mayhew et al., 2000; Yamano et al., 2001; Ito et al., 2003; Wang et al., 2004; Venkatakrishnan and Obach, 2005; Galetin et al., 2006). In these reports, the investigators have used various values for the input parameters such as enzyme degradation rate constants derived from animal or hepatocyte data. However, to date, the prediction of DDIs caused by P450 inactivators for a comprehensive set of drugs has not been reported. In the present work, attempts to make DDI predictions for inactivators using a set of drugs that spans multiple P450 enzymes were made, albeit the number of drugs for which this can be done (particularly for non-CYP3A enzymes) is still limited compared with the set used to predict DDIs for reversible inhibitors (Obach et al., 2005).
In the present report, a hierarchy of in vitro inactivation approaches was presented. It was shown that a simple experiment that uses a single concentration of inactivator that is preincubated with human liver microsomes in the presence and absence of NADPH for 30 min before measurement of a P450 marker activity can distinguish irreversible inactivators from noninactivators (Table 2). Such an experimental design can be used in early stages of drug research in which hundreds of compounds are considered to select those compounds devoid of an ability to inactivate P450 enzymes. A more complex experimental design, which is still simpler than a full determination of inactivation kinetics, is the determination of IC50 after a 30-min preincubation in the presence and absence of NADPH. Inactivators show a lower IC50 when preincubated with NADPH, and the resulting “shifted” IC50 could be useful for predicting the magnitude of a DDI when combined with the estimated unbound portal Cmax of the inactivator and inserted into equations used for predicting DDIs for reversible inhibitors (Obach et al., 2006). Although this approach is empirical, and the shifted IC50 values measured would depend on elements of the experimental design (e.g., preincubation time, dilution factor, etc.), it does appear to be a reasonable approach to making initial predictions of the magnitude of DDIs when conducted in the manner described in this report. Furthermore, use of this approach can be rationalized by the excellent correlation observed between the “shifted” IC50 and the kinact/KI value, the latter being an established measure of inactivator efficiency.
The use of kinact and KI in predicting DDIs for inactivators has been the most frequently reported approach. In the present study, application of this approach was attempted across multiple known inactivators and seven human P450 enzymes. The complexity underlying predicting DDIs for inactivators must be appreciated; there are multiple parameters that need to be considered, each with their own sources of uncertainty. Also, there are aspects of in vitro experimental design that can yield variability and inaccuracy. These have been discussed in a recent article by Ghanbari et al. (2006). The four parameters that are most important to making accurate predictions of DDIs caused by inactivators are 1) the in vivo rate of enzyme degradation, kdeg, 2) the relevant in vivo concentration of the inactivator, [I], available to the target enzyme, 3) the in vitro inactivation kinetic parameters (KI and kinact), and 4) the fraction of the clearance of the affected drug that is mediated by the inactivated enzyme. The first three input parameters are discussed below, whereas the importance of the fourth has been previously discussed (Ito et al., 2005; Obach et al., 2006). Furthermore, it must be kept in mind that the clinical drug interaction data used to test the accuracy of predictions from in vitro inactivation data are derived from reports that used a variety of study designs, each of which have influence on the magnitude of the interaction (e.g., timing of the dosing, etc.).
In previous reports, the value used for kdeg for P450 enzymes frequently was a value derived from rat (kdeg ≈ 0.0008/min) (Mayhew et al., 2000). In the present work, values for kdeg were used that were derived from modeling of the time course of reversal of DDIs caused by induction or inactivation of P450 enzymes in human study subjects. The values obtained were substantially lower than the rat value, were within values reported in Ghanbari et al. (2006), and when used generally led to over-prediction of the magnitude of DDIs (i.e., lower kdeg leads to greater DDI). Actual measurements of P450 enzyme kdeg values in humans in vivo are not obtainable with technology presently available, leaving this parameter as one important source of potential error in DDI predictions for inactivators.
The use of estimated unbound hepatic inlet concentrations of inhibitors occurring during the absorption phase, as surrogates for free concentrations in the liver, proved to be a reliable value for in vivo inhibitor concentration in prediction of DDI magnitude for reversible inhibition (Obach et al., 2006). Such a value has face validity as the target enzymes are in the liver (and for CYP3A in the gut as well); thus, it is expected that this tissue, and hence the target P450 enzyme, would be exposed to considerably greater concentrations of inhibitor than those reflected by systemic concentrations. However, when used for the prediction of DDIs caused by inactivators, estimated free portal concentrations generally led to over-predictions of the magnitude of DDIs (Table 6). Rather, use of the free systemic Cmax of the inactivator yielded the most accurate predictions of DDIs when combined with inactivation data (Table 6). Although this was observed for the compounds examined in this report, continued testing of the use of free systemic Cmax for predicting DDIs for P450 inactivators is warranted. An explanation for the discrepancy between the value for [I] that is most useful for DDI prediction for reversible inhibitors versus irreversible inactivators is not readily apparent and merits further exploration.
The experimental design used in inactivation experiments can have an impact on the measured parameters (Yang et al., 2005; Ghanbari et al., 2006). In the present studies, an approach was used wherein the inactivator was incubated with enzyme and cofactor for periods of time before dilution of the mixture into incubations in which the marker activity was measured. A major advantage of such an approach includes limiting reversible competitive inhibition because of dilution of the inactivator in the incubation where marker activity is measured and the use of saturating index substrate concentrations in the activity assay. Comparison of activities to incubations that were preincubated with NADPH in the absence of inactivator is important for P450 enzymes because reactive oxygen species that can inactivate the enzyme can be generated in the presence of NADPH and absence of inactivator or substrate. This phenomenon can be accounted for by including the kobs at [I] = 0 in the nonlinear fitting of the kobs versus [I] relationship (e.g., Fig. 2C), as was done in this report. Use of linearized data to estimate inactivation kinetic parameters can under-value this important factor. A disadvantage of using the dilution approach resides in the potential for inactivators to nonspecifically bind to microsomal protein in the inactivation incubation because greater protein concentrations (i.e., >0.1 mg/ml) are needed. This factor was not accounted for in the present study; however, correction for any nonspecific binding would only cause the KI to decrease and lead to even greater over-predictions of DDIs. With the exception of CYP2C19, the protein concentrations used in inactivation incubations were still relatively low (≤0.3 mg/ml) (Table 1) compared with other reports.
Contour plots tracking combinations of the ratio of kinact and kdeg and the ratio of [I] and KI that would yield identical magnitudes of DDIs are shown in Fig. 5. Inactivators with high kinact can cause DDIs even at low concentrations relative to KI (top left side of the plot), whereas weaker inactivators (low kinact) that are highly potent (low KI) can also cause DDIs (right side of the plot) and approach the behavior of reversible inhibitors rather than inactivators. By plotting the coordinates for each inactivator, a clustering can be observed for these compounds. In the region bounded by lower and upper limits of reliable in vitro measurements of kinact, inactivators that cause marked DDIs tend to reside toward the upper right, whereas those that do not cause DDIs reside in the lower left. Predictions of DDIs could be made for those drugs for which inactivation kinetic data have been measured in this report but for which there are no in vivo DDI data by judging whether they reside on the plot near the cluster of known perpetrators of DDIs or known nonperpetrators of DDIs. Thus, it would be expected that MDMA should cause DDIs of concern for CYP2D6-cleared drugs (point 6b on the plot) and thioTEPA should cause DDIs of concern for CYP2B6-cleared drugs (point 3a), whereas ticlopidine, thioTEPA, and MDMA should not cause interactions with drugs cleared by CYP3A, CYP2C19, or CYP1A2, respectively (points 5d, 3c, and 6a). Continued testing of the application of this contour plot is required to determine whether it is broadly applicable to aid inactivation DDI risk assessment for new molecular entities based on where they fall on the contour map relative to known clinically established DDI perpetrators and nonperpetrators.

The relationship between DDI magnitude, [I]/KI, and kdeg/kinact for P450 enzymes. The magnitude of the DDI will be dependent not only on the concentration of the inactivator in vivo relative to its inhibitory potency ([I]/Ki), as is the case for reversible inhibitors, but also on the relationship between the inactivation rate and the rate of degradation of enzyme (kinact/kdeg). The colored contours represent the magnitude increases in exposure of an affected drug (AUCi/AUC) ranging from 1.1-fold increases (violet line) to 50-fold increases (red line), assuming that oral clearance is entirely mediated via hepatic metabolism by the affected enzyme (i.e., fm(P450) = 1). The individual inactivators are indicated by number: 1, furafylline with CYP1A2; 2, zileuton with CYP1A2; 3a, thioTEPA with CYP2B6; 3b, thioTEPA with CYP2C8; 3c, thioTEPA with CYP2C19; 3d, thioTEPA with CYP3A; 4, tienilic acid with CYP2C9; 5a, ticlopidine with CYP1A2; 5b, ticlopidine with CYP2B6; 5c, ticlopidine with CYP2C19; 5d, ticlopidine with CYP3A; 6a, MDMA with CYP1A2; 6b, MDMA with CYP2D6; 7a, paroxetine with CYP2D6; 7b, paroxetine with CYP3A; 8, diltiazem with CYP3A; 9, erythromycin with CYP3A; 10, ritonavir with CYP3A; and 11, verapamil with CYP3A. The color of the number indicates the magnitude of DDI caused by the inactivator: red text, DDI >5×; blue text, 2×<DDI <5×; violet text, DDI <2×; black text, no clinical DDI data available. The horizontal dashed lines represent practical upper and lower limits for detecting inactivation in vitro (2/min > kinact > 0.005/min; kdeg ≈ 0.00032/min).
In summary, the data presented in this report show that in vitro inactivation kinetic data for human P450 enzymes can be useful in predicting in vivo DDIs, when combined with the systemic unbound concentration of inactivator, the fraction of the affected drug metabolized by the inactivated P450, and estimates of in vivo degradation rates of enzyme. Uncertainty in the multiple parameters needed for these predictions must be appreciated. Additionally, some abbreviated experimental approaches that can identify mechanism-based inactivators were presented. These can be used in early drug discovery research when the number of new compounds requiring investigation for this undesired property can exceed the capacity for conducting complete characterizations of inactivation kinetics.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.012633.
-
ABBREVIATIONS: DDI, drug-drug interaction; P450, cytochrome P450; PPP, 2-phenyl-2-(1-piperidinyl)propane; thioTEPA, N,N′,N″-triethylenethiophosphoramide; MDMA, methylenedioxymethamphetamine.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Received August 30, 2006.
- Accepted November 2, 2006.

- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}