


Abstract
Human liver microsomes have typically resulted in marked underprediction of in vivo human intrinsic clearance (CLint); therefore, the utility of cryopreserved hepatocytes as an alternative in vitro system has become an important issue. In this study, 10 compounds (tolbutamide, diclofenac, S-warfarin, S-mephenytoin, dextromethorphan, bufuralol, quinidine, nifedipine, testosterone, and terfenadine) were selected as substrate probes for CYP2C9, 2C19, 2D6, and 3A4, and the kinetics of metabolite formation (n = 14 pathways) were investigated in three individual lots of cryopreserved hepatocytes and in a pool of human liver microsomes. For the majority of the compounds, lower unbound KM or S50 values were observed in hepatocytes compared with microsomes, on average by 50% over a 200-fold range (0.5–140 μM). Expressed on an equivalent liver weight basis, a good correlation between microsomal and hepatocyte Vmax values was observed for most pathways greater than 5 orders of magnitude (0.16–216 nmol/min/g liver). Unbound hepatocyte CLint (CLint,u) values, when scaled to the whole liver (range 0.38–4000 ml/min/kg), were on average 2.5-fold higher than microsomal CLint,u values, with the exception of tolbutamide and diclofenac, for which lower hepatocellular CLint,u values were observed. Hepatocyte predicted CLint values were compared with human in vivo CLint values, and to supplement our data, in vitro data from cryopreserved hepatocytes were collated from four other published sources. These data show that for 37 drugs, there is, on average, a 4.5-fold under-prediction of the in vivo CLint using cryopreserved hepatocytes, representing a significant reduction in prediction bias compared with human microsomes.
Human liver microsomes have traditionally been the most commonly used in vitro system for the prediction of metabolic clearance, in particular, for new chemical entities within drug discovery programs in the pharmaceutical industry. However, the use of this system has typically resulted in an underestimation of clearance, as illustrated by a data set of 55 compounds in which a 9-fold under-prediction of the in vivo intrinsic clearance (CLint) was observed (Ito and Houston, 2005). As a result of this, attention in recent years has been placed on the use of alternative in vitro systems for clearance prediction. Fresh human hepatocytes are envisaged as a potentially more accurate system because of the full complement of both phase I and phase II metabolizing enzymes, along with the presence of transporter proteins, which should result in drug concentrations within the hepatocyte that are equivalent to in vivo concentrations within the liver. However, the limited availability of fresh human tissue and the cost implications involved in the preparation of freshly isolated human hepatocytes have resulted in cryopreserved hepatocytes emerging as the favored alternative, which also have the added advantage of being readily available commercially and more convenient to use (Li et al., 1999).
Two major issues concerning the use of cryopreserved hepatocytes are whether the metabolic activities of specific P450 enzymes are conserved post-thawing after cryopreservation (Li et al., 1999), and whether these hepatocytes retain their functional transporter activity, both of which could confound accurate clearance prediction. Recent studies, however, both in rat (Naritomi et al., 2003; Griffin and Houston, 2004) and in human (Soars et al., 2002; McGinnity et al., 2004) have shown that P450 activities are comparable in both cryopreserved and fresh hepatocytes. Studies have also shown that thawed cryopreserved hepatocytes show no difference in transporter activities compared with fresh hepatocytes in both rat and human (Houle et al., 2003, 2004). These results in combination with metabolic stability studies (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002) illustrate the potential of cryopreserved hepatocytes as an alternative system to microsomes. However, the majority of CLint data from cryopreserved hepatocytes have been obtained using the substrate depletion approach rather than that of metabolite formation, and the availability of KM and Vmax data are limited to only one study (Hallifax et al., 2005).
We have previously characterized metabolite kinetic profiles for a series of five benzodiazepines in cryopreserved hepatocytes, and have compared data to those obtained in microsomes to ultimately assess the utility of hepatocytes for the prediction of human clearance (Hallifax et al., 2005). Overall, a good correlation between microsomal and hepatocyte CLint was observed; therefore, the aims of the present study were to extend this data set to include four additional CYP3A4 substrates, along with six other P450 substrates, to investigate whether this correlation is observed across multiple P450 enzymes. We have determined the metabolite kinetic profiles for 14 pathways involved in the metabolism of quinidine, nifedipine, testosterone, terfenadine, tolbutamide, diclofenac, S-warfarin, S-mephenytoin, bufuralol, and dextromethorphan in three individual lots of cryopreserved hepatocytes and in a pool of human liver microsomes, to enable a more detailed comparison of both the affinity constant and P450 activity in both systems. We have compared the scaled CLint values in both systems after correction for the fraction unbound within the incubation, since the importance of accounting for microsomal and hepatocyte binding has recently been stressed (Austin et al., 2005; Riley et al., 2005; Hallifax and Houston, 2006). Given that the accuracy of metabolic clearance predictions is dependent on the selection of in vivo data against which the predictions are validated, plasma clearance data for each compound have been compiled from a number of studies, to enable robust comparison of the hepatocyte predictions with observed in vivo values.
Materials and Methods
Chemicals. (3S)-3-Hydroxyquinidine, oxidized nifedipine, S-warfarin, 7-hydroxywarfarin, S-mephenytoin, 4-hydroxymephenytoin, bufuralol and 1-hydroxybufuralol were purchased from SAFC Corp. (St. Louis, MO). Azacyclonol was purchased from Acros Organics (Fairlawn, NJ). Terfenadine alcohol was a gift from Pfizer (Sandwich, Kent, UK). All other chemicals and reagents were purchased from Sigma Chemical (Poole, Dorset, UK).
Hepatocyte Thawing Procedure. Cryopreserved human hepatocytes (lots 091, RNG/TPZ, KSE, 104) were purchased from In Vitro Technologies (Baltimore, MD) and were stored in liquid nitrogen until required. Hepatic tissue was obtained from nontransplantable livers; however, there was no information available regarding health status of the donors. Immediately before use, hepatocytes were thawed according to the recommended protocol. In brief, vials of hepatocytes were rapidly thawed in a shaking water bath at 37°C; the contents of the vial were emptied into 48 ml of prewarmed thawing medium (Williams' medium E, pH 7.4). and the suspension was centrifuged at 60g for 5 min at room temperature. The hepatocyte pellet was resuspended in 2.0 ml of prewarmed Krebs-Henseleit buffer (pH 7.4) by gentle inversion, and the cell number and viability were assessed using the trypan blue exclusion test.
Hepatocyte Incubations. Hepatocyte lots were incubated with each compound over a substrate concentration range that was wide enough to detect both the expected KM and Vmax values (see Table 1). For terfenadine experiments, lot KSE was replaced by lot 104. Incubations were performed in duplicate (except for terfenadine, which was performed in triplicate) and were optimized with respect to linearity with incubation time (Table 1) and hepatocyte density (see Table 3).
Incubation and analytical LC-MS/MS conditions for substrates in cryopreserved hepatocytes and human liver microsomes
Binding of compounds in both microsomal and hepatocyte incubations
In brief, cells were resuspended in fresh medium (Krebs-Henseleit buffer, pH 7.4) to twice the required concentration and prewarmed at 37°C. Drug was diluted in Krebs-Henseleit buffer (pH 7.4) (125 μl/well), placed into 24-well polypropylene plates (Hampton Research Corp., Aliso Viejo, CA), and preincubated for 5 min at 37°C in an atmosphere of 5% CO2 and 95% relative humidity. Reactions were initiated by the addition of prewarmed hepatocytes (125 μl of cell suspension/well) followed by gentle swirling of the plates and incubated as described above. Reactions were terminated by immersing the plates in liquid nitrogen. Samples were thawed and incubated with 250 μlof β-glucuronidase with sulfatase activity (200 units/ml in sodium acetate, 60 mM, pH 4.5) for 60 min in a plate shaker at 37°C to hydrolyze any glucuronide/sulfate conjugates. The reactions were terminated by the addition of 0.5 ml of ice-cold acetonitrile containing a 1 μM concentration of the appropriate internal standard. For studies involving tolbutamide and terfenadine, no hydrolysis step was performed. To minimize P450 inhibition or induction potential, the concentration of organic solvent (either dimethylformamide or methanol) was kept below 0.2% (v/v), except for testosterone, nifedipine, and S-mephenytoin for which the solvent concentrations were 1%, 1%, and 0.8%, respectively. However, even at these higher concentrations, no inhibitory effect is expected using the recommended solvent for each P450 enzyme (Easterbrook et al., 2001).
Microsomal Incubations. Pooled human liver microsomes (n = 22 livers) were purchased from BD Gentest (Woburn, MA). All incubations were carried out in duplicate (or triplicate in the case of terfenadine) and were performed under initial rate conditions with respect to incubation time (see Table 1) and microsomal protein concentration (see Table 3). Substrates were preincubated with microsomal protein in phosphate buffer (0.1 M, pH 7.4) for 5 min in a shaking water bath at 37°C. Reactions were initiated by the addition of a cofactor regenerating system (1 mM NADP+, 7.5 mM isocitric acid, 10 mM magnesium chloride, 0.2 unit of isocitric dehydrogenase) to give a final incubation volume of 0.25 ml. Incubations were terminated by the addition of 0.25 ml of ice-cold acetonitrile containing a 1 μM concentration of the appropriate internal standard. For the terfenadine studies, incubations were performed in 96-well polypropylene plates and were terminated by the addition of 0.07 ml of acetonitrile containing 0.2 μM internal standard. The final substrate concentration range studied was the same as that used in the hepatocyte incubations for all compounds (Table 1). Kinetic parameters for tolbutamide and dextromethorphan were taken from previously published data from this laboratory (Brown et al., 2006,Brown et al., 2006).
Determination of Metabolite Concentration. Samples were centrifuged for 10 min at 11,600g (Eppendorf Centrifuge 5413), and an aliquot (10 μl) of the supernatant was analyzed by LC-MS/MS. The metabolites of each substrate were analyzed by LC-MS/MS (Waters 2795 with Micromass Quattro Ultima triple quadrupole mass spectrometer; Waters, Milford, MA) with atmospheric pressure electrospray ionization. The LC column eluate was split and one quarter was delivered into the mass spectrometer where the desolvation gas (nitrogen) flow rate was 600 l/h, the cone gas (nitrogen) flow rate was 100 l/h, and the source temperature was 125°C. Product ions were formed in argon at a pressure of 2 × 10–3 mbar and detection was performed by multiple reaction monitoring of the parent and selected daughter ion. The mass transitions of each substrate, metabolite, and internal standard along with the cone voltage and collision energies are shown in Table 1. For each assay, 12 calibration standards with a blank were prepared in a matrix identical to the incubation extracts and included levels at, below, and above the expected concentrations. The ion chromatograms were integrated and quantified by quadratic regression of standard curves using Micromass QuanLynx 3.5 software (Waters).
Data Analysis. All data were analyzed by nonlinear regression analysis using Grafit 4 (Erithacus Software, Horley, Surrey, UK) using models for either Michaelis-Menten kinetics, biphasic kinetics, substrate inhibition kinetics, or sigmoidal kinetics. The goodness of fit criteria used to select the model with the most appropriate fit comprised visual inspection, consideration of the randomness of residuals, and the standard error of the parameters.
Determination of Microsomal Binding and Hepatic Uptake. The binding of each compound was determined in both microsomal and hepatocyte incubations. The microsomal binding of S-mephenytoin, testosterone, nifedipine, and quinidine was performed using the method of microfiltration as has been described previously (Carlile et al., 1999). Microsomal binding data for the other substrates were obtained from published literature (Carlile et al., 1999; Obach, 1999; Baba et al., 2002; Margolis and Obach, 2003), and the fraction unbound in microsomes (fumic) was calculated using eq. 1, where Ka represents the affinity constant for the drug-protein interaction and [P] represents the microsomal protein concentration. 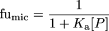
In the absence of human microsomal binding data, fumic values for dextromethorphan were taken from rat data (Witherow and Houston, 1999). For 5 of the 10 compounds (dextromethorphan, S-warfarin, tolbutamide, diclofenac, and quinidine), hepatic uptake experiments had previously been performed using isolated rat hepatocytes to determine the tissue-plasma partition coefficient (Kp) using methods as described in Witherow and Houston (1999). This value was used to calculate the fraction unbound within the hepatocyte incubation (fuinc) using eq. 2 (Witherow and Houston, 1999), where Vcell and Vinc represent the volume of the cell and the volume of the incubation, respectively, and is equal to 5 μl/1000 μl for 1 × 106 cells/ml. The use of eq. 2, however, assumes that active transport processes are not involved in the hepatic uptake of these compounds. 
For compounds for which hepatocyte uptake data were unavailable, the binding within the hepatocyte incubation was calculated by assuming that the extent of binding to microsomes at a concentration of 1 mg/ml was equivalent to binding at 1 × 106 cells/ml (Austin et al., 2005).
Calculation of Intrinsic Clearance. CLint,u was determined from the ratio of Vmax/KM,u for substrates displaying Michaelis-Menten, biphasic, and substrate inhibition kinetics. For substrates displaying sigmoidal kinetics, the equivalent of CLint, the clearance at maximal activation (CLmax) was calculated (Witherow and Houston, 1999). Hepatocyte CLint (μl/min/106 cells) and microsomal CLint (μl/min/mg protein) were scaled to in vivo CLint (ml/min/kg) using a hepatocellularity of 120 × 106 cells g–1 liver, a microsomal recovery value of 40 mg microsomal protein g–1 liver (Hakooz et al., 2006), and a human liver weight of 21.4 g liver kg–1 body weight. After normalizing both data sets to per gram of liver, the unbound CLint values from cryopreserved hepatocytes and human liver microsomes were compared. CLint predictions were assessed from the prediction error (difference between the predicted and observed in vivo value). The bias of CLint prediction was assessed from the geometric mean of the ratio of predicted and observed value [average -fold error (afe)], and the -fold under-prediction calculated using eqs. 3 and 4, respectively. 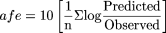
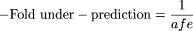
Comparison with in Vivo Clearance Data. The scaled CLint values were evaluated relative to in vivo human clearance data. In vivo CLint values were calculated from the published values of intravenous hepatic clearance (CLH), the fraction unbound in plasma (fup), and the blood to plasma concentration ratio (RB), according to eq. 5, where QH represents hepatic blood flow (20.7 ml min–1 kg–1). Values of hepatic blood clearance (CLb) were calculated by correcting the CLH for the RB value. 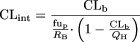
For the majority of compounds, metabolic clearance data were compiled from intravenous studies; however, when such data were unavailable, oral clearance data were used. Therefore, for three compounds (S-mephenytoin, dextromethorphan, and terfenadine), the CLint was calculated by correcting the oral plasma clearance (assuming complete absorption from the gastrointestinal tract) for the fraction unbound in blood (fub) as shown in eq. 6. 
For several CYP3A4 compounds, clearance values after intravenous administration were unavailable; therefore, these studies were removed from the database. Oral clearance values could not be used because the fraction of drug escaping intestinal extraction (FG) could not be assumed to be 1, as a result of the high levels of CYP3A4 in the intestine, resulting in a substantial contribution to first-pass metabolism following oral administration. In the case of two compounds, propofol and naloxone, intravenous plasma clearance values after intravenous dosing were higher than hepatic blood flow; thus, in the absence of oral clearance data, the CLint values were calculated using eq. 6, assuming CLH was just below the value of QH. The in vivo clearance of propofol is very high, and this may reflect a significant renal component in metabolic clearance (Al-Jahdari et al., 2006).
The fup and RB values were compiled from two major sources (Moffat et al., 2004; Thummel et al., 2005). In vivo plasma clearance data were compiled from various literature sources using The University of Washington Metabolism and Transport Drug Interaction Database (http://www.druginteractioninfo.org/DatabaseInfo.aspx). When the RB was unavailable, a value of 1 was assumed for basic and neutral compounds. A value of 0.55 (1 – hematocrit) was assumed for acids because acidic compounds are usually highly bound to plasma proteins, therefore restricting blood cell uptake. For dextromethorphan and nifedipine, the RB value obtained in rats was used, and for bufuralol, a value of 0.80 was assumed since this correlates to that observed with other structurally similar β-blockers with a similar lipophilicity. Predictions of in vivo human CLint were supplemented by using other data previously obtained in this laboratory for five benzodiazepines (Hallifax et al., 2005) and other literature data using cryopreserved hepatocytes (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002).
Results
Metabolite Formation Kinetics. The kinetics of metabolite formation were determined for each of the 10 probe substrates in three individual lots of cryopreserved human hepatocyte suspensions and in a pool of human liver microsomes. For most substrates, the kinetic profiles were described using classic one-site Michaelis-Menten kinetics (hydroxytolbutamide, 3-hydroxyquinidine, 4-hydroxydiclofenac, 4-hydroxymephenytoin, 7-hydroxywarfarin, 3-methoxymorphinan, and azacyclonol). However, two pathways displayed biphasic kinetics (1-hydroxybufuralol and dextrorphan), indicative of both a high-affinity low-capacity site, and a low-affinity high-capacity site. For three CYP3A4 substrates, atypical Michaelis-Menten kinetics were evident with substrate-inhibition kinetics (oxidized nifedipine, terfenadine alcohol, and fexofenadine) and sigmoidal kinetics (6β-hydroxytestosterone). The kinetic profiles of each compound were best described using the same models in both systems and were assigned as described previously under Materials and Methods.
Determination of in Vitro CLint. The in vitro kinetic parameters, Vmax, unbound KM, and S50 (in the case of sigmoidal kinetics) obtained in cryopreserved hepatocytes were compared with those obtained in microsomes (Table 2). To calculate these parameters, the binding of each compound was determined in both microsomes and hepatocytes to give the fumic and fuinc, respectively (Table 3). From these data it can be observed that the majority of the 10 compounds studied displayed relatively little binding at the protein concentrations used within the incubations. Substrates of CYP2C displayed the least binding with fu values in the range of 0.9 and 1, as would be expected because of their acidic nature, whereas the range of fu values for substrates of CYP3A4 and CYP2D6 ranged between 0.58 and 0.85 for microsomal incubations, and between 0.48 and 0.93 for hepatocyte incubations. The only exception was terfenadine, which displayed significant binding in both systems, with fu values of 0.04 and 0.1 in microsomal and hepatocyte incubations, respectively. Regardless of the extent of binding, all data were corrected to give the unbound value. Figure 1 compares the unbound affinity constants (KM,u or S50,u values) for each individual pathway of metabolism (14 pathways) in both human liver microsomes and cryopreserved human hepatocytes. It was observed that for the majority of the pathways, hepatocyte values were typically lower than the corresponding values in microsomes, by an average of 50%. The only exceptions to this were azacyclonol, nifedipine, diclofenac, and the low-affinity bufuralol pathway, where higher KM,u values were observed in hepatocytes.
Kinetic parameters obtained for each compound in microsomes and hepatocytes
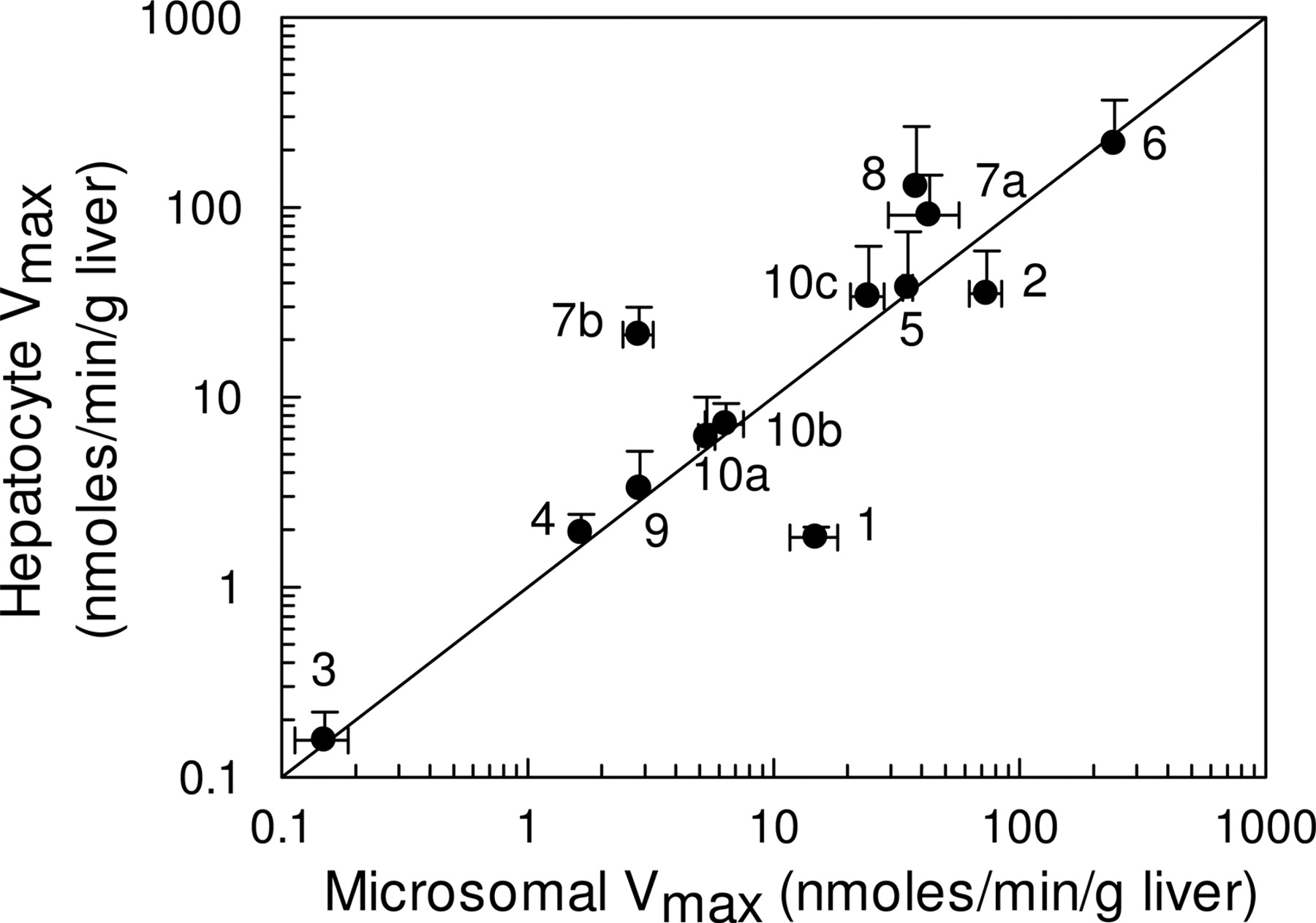
When expressed on an equivalent liver weight basis (using scaling factors as indicated under Materials and Methods), a good correlation was observed between microsomal and hepatocyte Vmax values (Fig. 2), with the exception of diclofenac and tolbutamide, for which lower hepatocellular activities were observed compared with microsomes. For some CYP3A4 substrates, notably nifedipine and terfenadine, higher cellular Vmax values were obtained, with a 10-fold higher value observed for terfenadine in hepatocytes than in microsomes. The CLint,u for each pathway was determined by the ratio of Vmax/KM,u or CLmax in the case of testosterone, and when values were scaled to the whole liver, overall, there was a good correlation between both systems. In fact, for 12 pathways involved in the metabolism of eight compounds (dextromethorphan, S-mephenytoin, quinidine, nifedipine, testosterone, terfenadine, bufuralol, and S-warfarin), hepatocellular CLint,u values were higher than the corresponding microsomal values, on average by 2.5-fold (Fig. 3). Hepatocyte CLint,u values for tolbutamide and diclofenac, however, were on average 30% of the CLint,u observed in microsomes.
Comparison with in Vivo CLint. The CLint values in cryopreserved hepatocytes were compared with human in vivo CLint values. In vivo CLint was calculated from published values of total plasma clearance, the RB, and the fup (as described under Materials and Methods). To ensure the accurate estimation of CLint, plasma clearance data were collated from various in vivo studies in a number of different individuals to ensure that a representative value was obtained. These in vivo data for nine of the probe substrates are shown in Table 4. No in vivo plasma clearance data for testosterone were available. For some compounds such as bufuralol, S-warfarin, and diclofenac, limited intravenous data were available; therefore, plasma clearance values for these substrates should not be regarded as definitive compared with other compounds such as dextromethorphan, for which in vivo data were obtained from a larger population data set. Also, the lack of intravenous studies involving S-mephenytoin meant that the oral bioavailability could not be estimated; therefore, this in vivo clearance value must be regarded as an upper estimate since complete systematic availability cannot be ensured.
In vivo human clearance values for n = 9 compounds in this study and the key parameters for prediction
References for plasma clearance data are available at www.capkr.man.ac.uk.
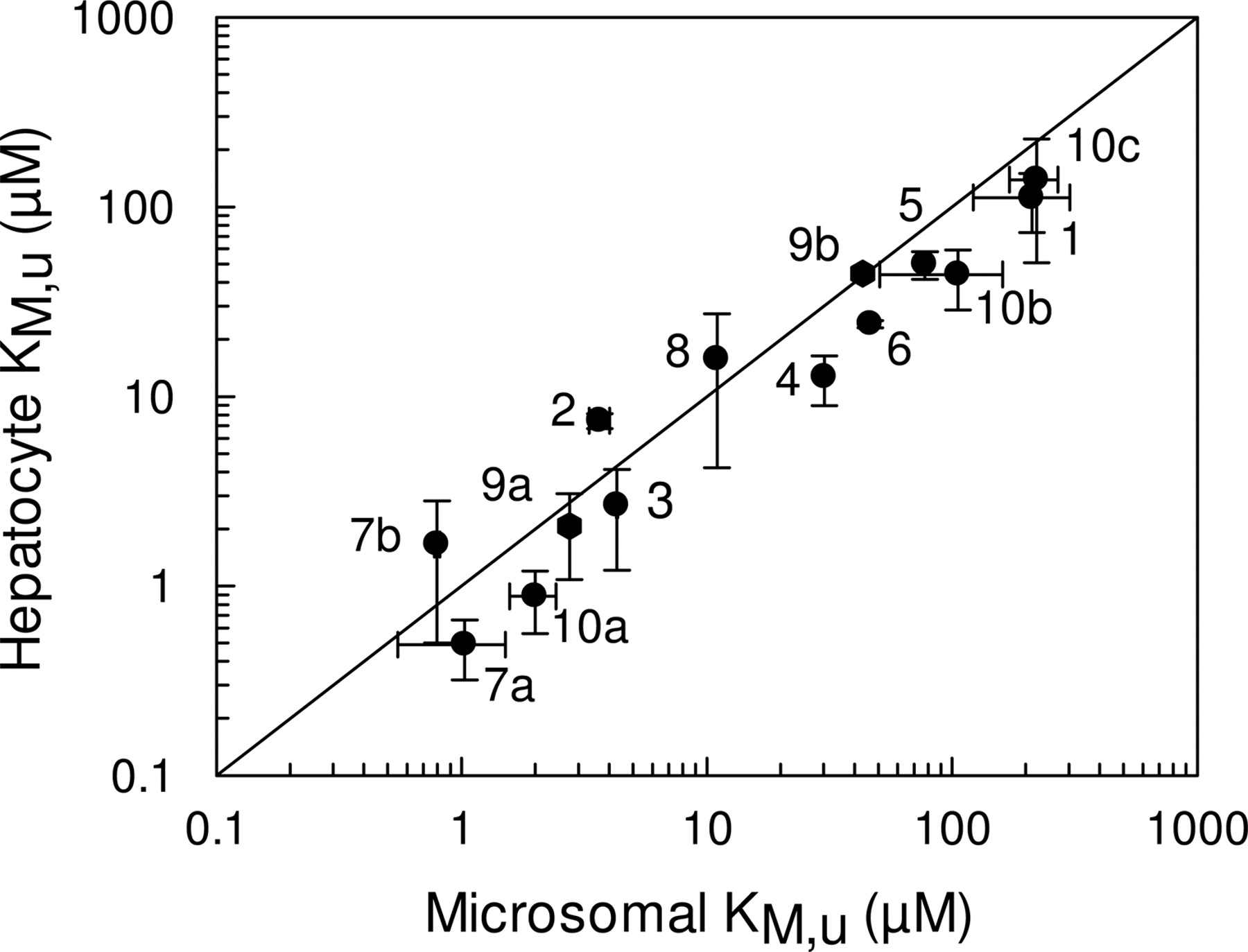
Comparison of KM,u (or S50,u) values for 14 pathways in human liver microsomes and human cryopreserved hepatocytes. The metabolites represented are 1, hydroxytolbutamide; 2, 4-hydroxydiclofenac; 3, 7-hydroxy-(S)-warfarin; 4, 4-hydroxymephenytoin; 5, 3-hydroxyquinidine; 6, 6β-hydroxytestosterone; 7a, terfenadine alcohol; 7b, azacyclonol; 8, oxidized nifedipine; 9a, 1-hydroxybufuralol (high-affinity pathway); 9b, 1-hydroxybufuralol (low-affinity pathway); 10a, dextrorphan (high-affinity pathway); 10b, dextrorphan (low-affinity pathway; and 10c, methoxymorphinan. The solid line represents the line of unity. Data are represented as mean ± S.D. of three determinations.
To supplement our data, in vitro parameters using cryopreserved hepatocytes were also included from four other published literature sources (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002; Hallifax et al., 2005), resulting in an additional 27 compounds. In some instances, compounds had been studied by more than one laboratory; thus, for seven substrates, the mean in vitro CLint was determined. In vivo CLint values were calculated from in vivo data as described previously under Materials and Methods, and are displayed in Table 5.
In vivo human clearance values for n = 28 compounds with the key parameters for prediction
References for plasma CL data are available at www.capkr.man.ac.uk
Table 6 compares the observed and predicted CLint values using cryopreserved hepatocytes for compounds in the present study and the literature (n = 37). Compounds were grouped into four categories according to the accuracy of CLint predictions (≤2-fold, 2-fold ≥ 5-fold, 5-fold ≥ 10-fold, and ≥10-fold). The major P450s responsible for metabolism of the 37 compounds along with the contribution of glucuronidating phase II metabolizing enzymes are also presented to assess whether prediction accuracy is systematic for a certain group of P450s. Comparison of CLint values for the nine compounds in this study involved the predicted CLint,u. The extent of binding within the incubation for the five benzodiazepines involved in the study of Hallifax et al. (2005) had previously been determined, and fu values in both microsomal and hepatocyte incubations ranged between 0.69 and 0.94 for the protein concentrations used within the incubations (Rawden et al., 2005). Therefore, the CLint,u values for these five compounds were also compared with the in vivo CLint. Corrections for binding with the other literature data were also possible. In the study by Shibata et al. (2002), incubations were performed in 100% human serum; therefore, the CLint in vitro was corrected for the fraction unbound in plasma, because it was assumed that the impact of binding to plasma proteins would be greater than that of binding to hepatocytes in reducing the concentration available within the incubation for metabolism. In the study by Soars et al. (2002), microsomal binding values at a concentration of 2 mg/ml were reported, and because hepatocyte incubations were performed at a concentration of 2 × 106 cells/ml, the extent of binding was assumed to be the same (Austin et al., 2005). Hepatocyte binding values for the compounds studied by Lau et al. (2002) were reported by Riley et al. (2005). Analysis of these data show that for the 37 compounds, there is a systematic under-prediction of in vivo human clearance using cryopreserved hepatocytes, with an average 4.5-fold under-prediction observed.
Accuracy of CLint predictions for n = 37 drugs from human cryopreserved hepatocytes
Where in vitro data is taken from more than one published source, the mean in vitro CLint value is calculated.

Comparison of 14 Vmax values obtained in human liver microsomes and human cryopreserved hepatocytes. The metabolites represented are 1, hydroxytolbutamide; 2, 4-hydroxydiclofenac; 3, 7-hydroxy-(S)-warfarin; 4, 4-hydroxymephenytoin; 5, 3-hydroxyquinidine; 6, 6β-hydroxytestosterone; 7a, terfenadine alcohol; 7b, azacyclonol; 8, oxidized nifedipine; 9a, 1-hydroxybufuralol (high-affinity pathway); 9b, 1-hydroxybufuralol (low-affinity pathway); 10a, dextrorphan (high-affinity pathway); 10b, dextrorphan (low-affinity pathway); and 10c, methoxymorphinan. Vmax data are expressed on an equivalent weight basis (grams of liver). The solid line represents the line of unity. Data are represented as mean ± S.D. of three determinations.

Comparison of 14 CLint,u values obtained in human liver microsomes and human cryopreserved hepatocytes. The metabolites represented are 1, hydroxytolbutamide; 2, 4-hydroxydiclofenac; 3, 7-hydroxy-(S)-warfarin; 4, 4-hydroxymephenytoin; 5, 3-hydroxyquinidine; 6, 6β-hydroxytestosterone; 7a, terfenadine alcohol; 7b, azacyclonol; 8, oxidized nifedipine; 9a, 1-hydroxybufuralol (high-affinity pathway); 9b, 1-hydroxybufuralol (low-affinity pathway); 10a, dextrorphan (high-affinity pathway); 10b, dextrorphan (low-affinity pathway); and 10c, methoxymorphinan. The solid line represents the line of unity. The dashed line represents the 2.5-fold average higher value (calculated using eqs. 3 and 4) observed in hepatocytes compared with microsomes when normalized to kilograms of liver weight. Data are represented as a mean of three determinations.
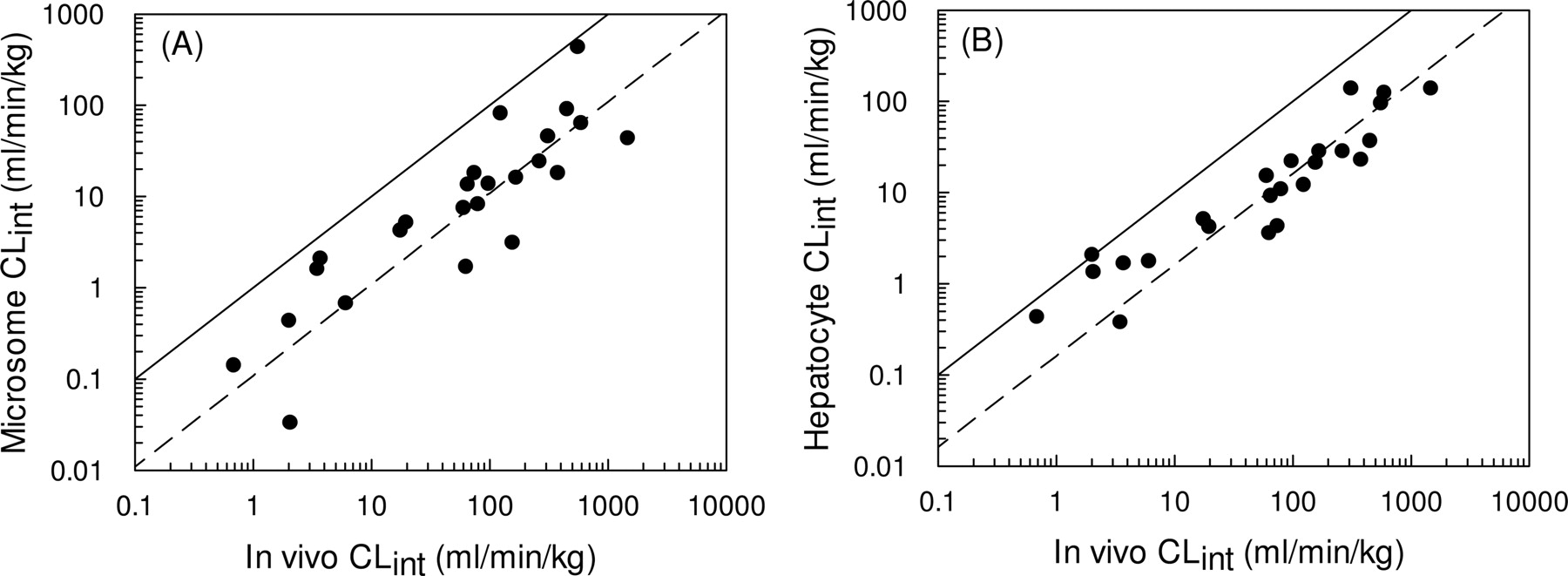
Twenty-five compounds were identified for which in vivo predictions of CLint could be made from both microsomes (see Fig. 5A) and hepatocytes (see Fig. 5B). From this data set, a 9-fold and a 6-fold under-prediction were observed based on data from microsomes and hepatocytes, respectively.
Discussion
Recent investigations have focused on the utility of cryopreserved hepatocytes as an alternative in vitro system for the prediction of human clearance (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002; Naritomi et al., 2003; Hallifax et al., 2005). However, these studies, with one exception (Hallifax et al., 2005), have determined in vitro CLint via substrate depletion rather than metabolite formation assays, resulting in very limited KM and Vmax data. Frequently, the CLint determined is based upon the total rather than the unbound drug concentration, with the assumption that the fraction unbound in blood and within the incubation are equal, and therefore, binding corrections are unnecessary (Lau et al., 2002). This is clearly not the case for acidic compounds in which extensive binding to albumin is usually observed, compared with negligible binding to microsomal protein or hepatocytes (Ito and Houston, 2005). Therefore, the aim of the present study was to provide a comprehensive analysis of CLint predictions obtained using the metabolite formation approach for a range of P450 substrates, and also to assess the effects of nonspecific binding within the incubation matrix on CLint predictions.
The KM,u (or S50,u) values for the 14 pathways of metabolism in the present study were typically lower in hepatocytes than those observed in microsomes, by an average of 50% over a 200-fold range (Fig. 1). This observation is consistent with previous data obtained with five benzodiazepines, where KM values were lower for all nine pathways of metabolism (Hallifax et al., 2005). The lower KM values in hepatocytes would suggest that an increased unbound drug concentration is available to the enzyme compared with that in a microsomal incubation, which may be indicative of some membrane uptake transporter involvement. However, there is little evidence to suggest that any of these 10 compounds are in fact substrates for hepatic transporters, so that currently there is no explanation for this phenomenon. The data obtained in the present study and in the study of Hallifax et al. (2005) enable comparisons for 23 pathways of metabolism and represent the only studies to date for which CLint has been determined in cryopreserved hepatocytes via the use of metabolite formation.
P450 activities, when scaled to per gram of liver, show good agreement between systems greater than 5 orders of magnitude (Fig. 2). In only two cases, values in hepatocytes were higher than those in microsomes (CYP3A4 substrates; nifedipine and terfenadine), but lower CYP2C activities were observed for both tolbutamide and diclofenac. For the CYP2C9 substrates, this could suggest the possibility of differential P450 loss through the cryopreservation process, since there is some evidence in rat to suggest that the CYP2C isoforms may be compromised during cryopreservation (Hewitt and Utesch, 2004). However, the fact that S-warfarin activity (also a CYP2C9 probe) is comparable in both microsomes and hepatocytes would appear to rule out this possibility and suggest that these discrepancies are associated with specific issues related to tolbutamide and diclofenac, such as the contribution of acyl-glucuronidation to the elimination of diclofenac (Kumar et al., 2002). Phase I oxidative metabolism is often followed by phase II conjugation of metabolites, but it is clear that for a significant number (at least 15) of the 37 compounds in this database, direct conjugation catalyzed by UGT enzymes is also a major pathway of metabolism (Table 6). Although the data presented in Table 6 suggest that poor CLint predictions (≥10-fold in vivo CLint) are observed for compounds that are directly conjugated, this is clearly not a systematic trend since good predictions (≤2-fold in vivo CLint) are also observed for the UGT substrates, codeine, naloxone, and propofol. There does not appear to be any trend between the accuracy of CLint predictions and metabolism by any particular UGT enzyme (Table 6). It is also interesting to note that for three compounds that only undergo phase I oxidative metabolism with no sequential conjugation processes (caffeine, theophylline, and terfenadine), microsomes do not predict as well as hepatocytes. However, tolbutamide is an exception to this trend. Conversely, for those compounds that do undergo sequential metabolism, e.g., dextromethorphan, hepatocytes predict more accurately than microsomes for approximately 50% of the compounds (data not shown). It is also evident that there is no bias in prediction accuracy toward compounds metabolized by a particular P450 or, indeed, compounds of a certain chemical class (acid/base/neutral).
Overall, higher CLint,u values (range 0.38–4000 ml/min/kg liver) were observed in hepatocytes, and were on average 2-fold higher than those observed in microsomes for 8 of the 10 compounds (Fig. 3). The higher CLint values in hepatocytes appear to result from the combined effect of comparable P450 activities in both systems, and an apparent higher P450 affinity using hepatocyte preparations. The CLint values of diclofenac and tolbutamide were lower in hepatocytes, resulting from a higher cellular KM and a lower cellular Vmax value, respectively. Previous studies with five benzodiazepines showed that comparable CLint values were obtained in both systems (Hallifax et al., 2005), in contrast to the higher CLint values in hepatocytes observed in this study. Lower Vmax values in hepatocytes were the main difference from the present study; however, sources of both microsomes and hepatocytes were different. This result highlights the difficulties in comparing in vitro methods because of variability between the livers investigated in each particular study (Rawden et al., 2005). It is unlikely that the lower hepatocyte activities observed for the five benzodiazepines occur as a result of their atypical kinetics, since this was not evident with other 3A4 substrates in the present study, such as testosterone and nifedipine, whose kinetic profiles were described by sigmoidal and substrate inhibition kinetics, respectively. Although it has recently been emphasized that the experimentally determined CLint is not necessarily the same as the unbound CLint (Austin et al., 2005), correction for nonspecific binding had little effect on CLint predictions for those compounds investigated in the present study, with the exception of terfenadine, for which the observed CLint was 10-fold lower than the unbound CLint. Consideration of the data presented by Riley et al. (2005) would suggest that this is a common situation.

Comparison between observed in vivo CLint and CLint predicted using cryopreserved hepatocytes for 37 substrates. Open symbols represent predictions from literature data (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002). ▴ represents benzodiazepine data taken from Hallifax et al. (2005) and • represents the nine compounds in the present study. The solid line represents the line of unity, and the dashed line represents a 4.5-fold under-prediction (calculated using eqs. 3 and 4).
The CLint values predicted from cryopreserved hepatocytes in the present study were supplemented with other in vitro data from the published literature (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002; Hallifax et al., 2005) to enable in vivo clearance predictions for a total of 37 studies (Fig. 4). Although these 37 studies include CLint data obtained from both metabolite formation (n = 14) and substrate depletion (n = 23) approaches, the overall trend of a 4.5-fold under-prediction was the same in all data sets. This systematic under-prediction using both methods supports the claim that for the metabolite formation studies, the major metabolites were indeed analyzed. Also, the fact that a similar general trend was observed across published data from three different laboratories in which studies were performed with different livers is encouraging. However, due to the complexities of inter-liver differences alluded to earlier, plus inter-laboratory issues confounding the comparison, we believe detailed comparison must be cautious.
The 4.5-fold under-prediction illustrated in Fig. 4 represents a significant improvement in the accuracy of clearance predictions compared with the 9-fold under-prediction observed in the microsomal data set discussed previously (Ito and Houston, 2005). Both data sets are relatively extensive, containing 55 compounds in the microsomal database and 37 compounds in the hepatocyte database. However, not all the same compounds were studied in both systems, and this improvement in prediction accuracy could be biased as a result of the particular compounds involved. Subsequent analysis found that 25 compounds were common to both in vitro systems, and in fact, a 9-fold under-prediction of in vivo CLint was observed for the microsomes (Fig. 5A), compared with a 6-fold under-prediction observed for the hepatocytes (Fig. 5B). Therefore, it is reasonable to conclude that the use of cryopreserved hepatocytes does represent a significant improvement compared with microsomes for the prediction of in vivo CLint.
Although the results of this study would appear to favor the use of cryopreserved hepatocytes over microsomes for the prediction of clearance because of the reduced bias, it is clear that hepatocytes are still not fully quantitative for prediction, and there are a number of possible explanations for this. For example, like most in vitro systems these hepatocyte studies involved static incubations and will possibly contain excretory products normally cleared from the system. Also, the majority of the in vivo plasma clearance data used to validate the CLint predictions are typically compiled from studies in young healthy volunteers receiving no concurrent medications. Detailed donor information on the livers used in the preparation of hepatocytes is generally incomplete; therefore, the donor livers may be unrepresentative of those individuals from whom the in vivo plasma clearance data are obtained. Donor livers used in the preparation of hepatocytes are commonly obtained from waste material from patients undergoing a partial hepatectomy or from multi-organ donors, and the source of the liver will have implications for the preparation of viable and metabolically functional hepatocytes because of differences in the procurement and storage of the tissue. It is unknown to what extent differences in the procurement of the liver, in addition to the processing of liver tissue to prepare hepatocytes, will affect the metabolic and transport functions of hepatocyte preparations, and this is clearly an area that requires further investigation.
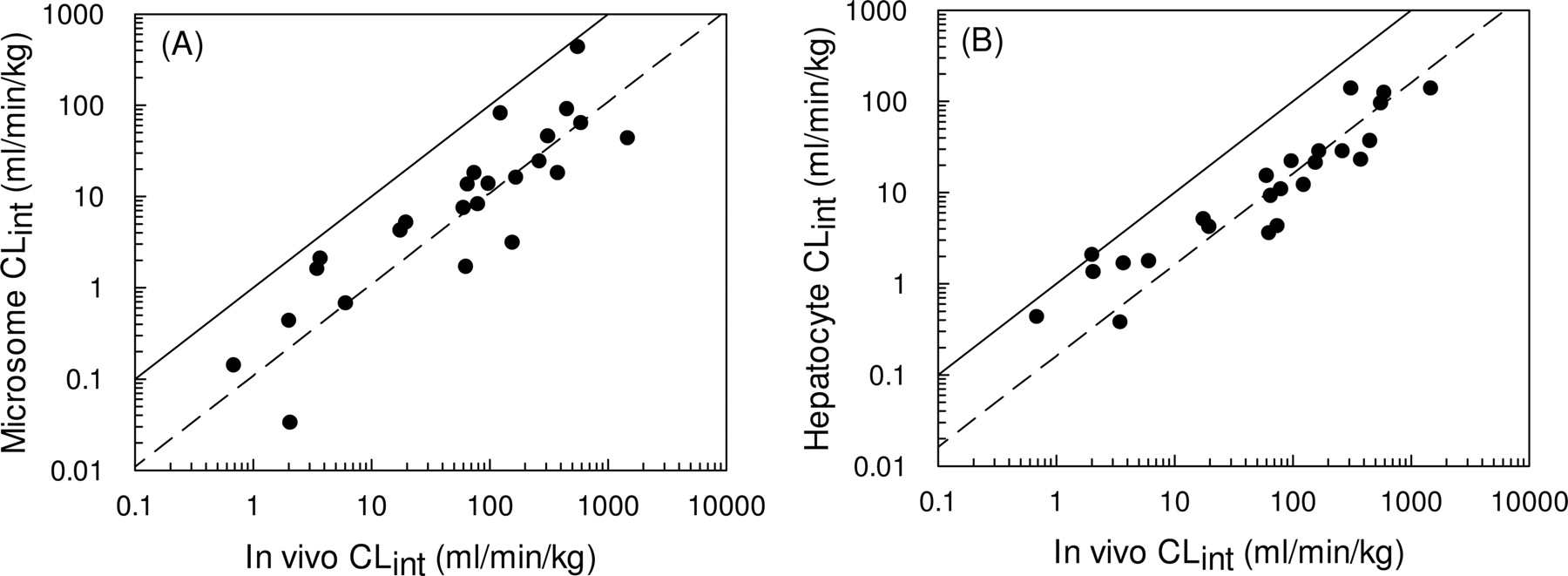
Comparison between observed CLint and CLint predicted using microsomes (from Ito and Houston, 2005) (A) and hepatocytes for the same 25 compounds (B). The solid line represents the line of unity, and the dashed line represents a 9-fold and a 6-fold under-prediction in microsomes and hepatocytes, respectively (calculated using eqs. 3 and 4).
Acknowledgments
We thank Sue Murby for valuable assistance with the LC-MS/MS.
Footnotes
-
This work was funded by a consortium of pharmaceutical companies (GlaxoSmithKline, Novartis, Pfizer, and Servier) within the Centre for Applied Pharmacokinetic Research at the University of Manchester. M.G. was financially supported by a GlaxoSmithKline studentship. Part of this study was presented at the 9th European ISSX Meeting, June 4–7, 2006, Manchester, UK (Brown et al., 2006,Brown et al., 2006).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.011569.
-
ABBREVIATIONS: CLint, total intrinsic clearance; CLint,u, unbound intrinsic clearance; CLmax, clearance at maximal activation; CLH, hepatic clearance; P450, cytochrome P450; UGT, UDP-glucuronosyltransferase; fub, fraction unbound in blood; fup, fraction unbound in plasma; RB, blood-to-plasma ratio; QH, hepatic blood flow; Ka, the affinity constant for the drug-protein interaction; fumic, fraction unbound in microsomal incubation; fuinc, fraction unbound in hepatocyte incubation; Kp, tissue-plasma partition coefficient; Vcell, volume of the hepatocyte; Vinc, volume of the incubation.
- Received June 21, 2006.
- Accepted November 17, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}