


Abstract
Predictions of intrinsic clearance (CLint) from human liver microsomes often underestimate in vivo observations. In this study, a series of five benzodiazepines was used as prototypic CYP3A4 substrates to investigate the prediction of clearance from the less studied alternative in vitro system, cryopreserved human hepatocytes. Formation of the two major metabolites from midazolam, triazolam, diazepam, flunitrazepam, and alprazolam was measured in cryopreserved human hepatocytes from five donors; the kinetics were characterized and CLint values were determined and scaled to predict CLint in vivo. At least one of the two major pathways of metabolic clearance of each benzodiazepine was characterized by autoactivation in hepatocytes; the extent to which this occurred varied depending on substrate and liver (up to 8-fold). Heteroactivation by testosterone of these pathways was also observed (up to 6-fold). The maximum autoactivated clearance was used to predict in vivo CLint (1.6–138 ml/min/kg) which closely agreed with values previously obtained using human liver microsomes. Comparison with in vivo CLint indicates that cryopreserved human hepatocytes systematically underpredict CLint. When three previous studies (documenting CLint for substrates of various enzymes in cryopreserved human hepatocytes using drug depletion-time profiles) were considered as well, the combined data show a consistent underprediction of 5.6-fold. Collectively it is demonstrated that the predicted hepatic intrinsic clearances from cryopreserved hepatocytes show an excellent rank order with in vivo findings but are systematically underpredicting the in vivo value.
The prediction of metabolic clearance of novel drugs plays a critical role in the increasingly highly invested drug discovery and development processes. Although much progress has been made toward understanding the basis for prediction of clearance (Houston, 1994; Iwatsubo et al., 1997; Obach, 1999; Ito and Houston, 2004), a consistently accurate experimental model of metabolic clearance remains a goal. In vitro systems are central to the determination of metabolic clearance, and human hepatocytes are seen as a potentially more accurate, as well as a practical, alternative to the established system of human liver microsomes (Li et al., 1999a,b; Steinberg et al., 1999; Shibata et al., 2002; Soars et al., 2002; Griffin and Houston, 2004). Cryopreserved hepatocytes are readily available commercially and offer convenience suitable for routine use. Recent studies (Lau et al., 2002; Naritomi et al., 2003; McGinnity et al., 2004) have provided valuable information on the utility if this in vitro system for metabolic stability determinations. The advantages of an intact cellular system include the operation of the full spectrum of drug-metabolizing enzyme systems as well as transporter proteins to provide intracellular concentrations comparable to that achieved in the liver in vivo. In addition, the use of hepatocytes allows the potential for different incubation formats (suspension, monolayer, sandwich culture) as well as genetic modification (Mills et al., 2004) which may provide improved in vitro systems in the future.
Underprediction of clearance using human liver microsomes is typical for many drugs (Ito and Houston, 2005). A series of benzodiazepines (alprazolam, diazepam, flunitrazepam, midazolam and triazolam) was selected as prototypic substrates for the major human drug-metabolizing enzyme, CYP3A4, with a range of in vivo clearance over 2 orders of magnitude characterized in a substantial population of human subjects (Rawden et al., 2005). Previously, the intrinsic clearance of each of these drugs in a panel of microsomes from 12 donor livers was found to underpredict clearance in vivo (Rawden et al., 2005). There were two major complications regarding this study with benzodiazepines in human microsomes. First, because the benzodiazepines are highly dependent on CYP3A4 for metabolic clearance, prediction inaccuracy may have resulted from the complexities of the kinetics (Houston and Kenworthy, 1999; Houston and Galetin, 2005). In microsomes, auto- and/or heteroactivation of the CYP3A4-mediated pathways was evident for the benzodiazepines, and it is uncertain whether this phenomenon is unique to this in vitro system. Second, liver variability, in terms of donor individuality (genetic, age, and environmental, etc.), was high and therefore potentially contributed to the disagreement between in vitro and in vivo values.
Previous investigations with human cryopreserved hepatocytes have been limited to metabolic stability studies involving depletion of parent drug (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002). More detailed studies with rat cryopreserved hepatocytes in which classic metabolite formation kinetics were delineated for eight drugs by 15 pathways indicated generally good agreement between freshly isolated cells and in vivo clearance values (Griffin and Houston, 2004). However, certain metabolic pathways, particularly those associated with CYP3A, which show non-Michaelis-Menten kinetics, appeared as outliers. In this study, the kinetic characteristics of 10 pathways of metabolism for five benzodiazepines are investigated in cryopreserved human hepatocytes to compare with analogous microsomal data and to predict in vivo clearance.
Materials and Methods
Chemicals. Midazolam, diazepam, 1′-hydroxymidazolam, 4-hydroxymidazolam, 3-hydroxyflunitrazepam, and desmethylflunitrazepam were donated by Roche Products Ltd. (Welwyn Garden City, UK). Triazolam, 1′-hydroxytriazolam, alprazolam, 1′-hydroxyalprazolam, 3-hydroxydiazepam, flunitrazepam, testosterone, β-glucuronidase, and Krebs-Henseleit buffer were purchased from Sigma-Aldrich Ltd. (Poole, UK). 4-Hydroxytriazolam and 4-hydroxyalprazolam were purchased from BIOMOL International (Exeter, UK). 3-Hydroxydiazepam was donated by Wyeth Laboratories Ltd. (Maidenhead, UK). All solvents were of high-performance liquid chromatographic grade and all reagents were of analytical grade.
Acquisition, Storage, and Resuspension of Cryopreserved Human Hepatocytes. Cryopreserved human hepatocytes and hepatocyte isolation kits were purchased from XenoTech LLC (Lenexa, KS); the cells were stored in the vapor phase of liquid nitrogen until use. Immediately before use, vials of hepatocytes were rapidly thawed in a shaking water bath (37°C; 1.75 min) and the live hepatocytes were isolated via centrifugation (50g; 4°C) through a Percoll solution (isolation kit). The pellet of hepatocytes was resuspended by gentle inversion in prewarmed Krebs-Henseleit buffer (pH 7.4; 1–2 ml) and the cell number and viability were determined by the trypan blue exclusion assay (Berry et al., 1991) prior to drug incubations.
Incubations to Determine Metabolite Formation Kinetics. Incubations of cryopreserved human hepatocytes with either midazolam, triazolam (1–100 μM), or diazepam, flunitrazepam, and alprazolam (1–500 μM) were performed under initial rate conditions: linear with respect to cell density (0.1 × 106 live hepatocytes/ml for midazolam and triazolam; 0.5 × 106 live cells/ml for diazepam, flunitrazepam, and alprazolam) and time (5, 45, and 60 min for midazolam, triazolam, and diazepam; flunitrazepam; and alprazolam, respectively).
The cryopreserved human hepatocytes (from five donor livers) were diluted in Krebs-Henseleit buffer (pH 7.4) to twice the required incubation density, placed into 24-well plates (Corning Glassworks, Corning, NY) (125 μl of cell suspension/well), and preincubated for 10 min (37°C) in an atmosphere of 5% CO2 and 95% relative humidity. Primary drug solutions were prepared in methanol or acetonitrile and diluted in Krebs-Henseleit buffer (pH 7.4) to twice the required incubation concentration and warmed (37°C). Reactions were initiated by the addition of preincubated drug in buffer (125 μl/well), followed by gentle swirling of the plates, and incubated under conditions described above. To limit the inhibitory effects of solvent used to no more than 10%, either acetonitrile or methanol was used at a total incubation proportion of 0.5%, depending on the relevant P450 involvement for a particular pathway, according to reported specific inhibitory effects (Hickman et al., 1998; Busby et al., 1999). Reactions were terminated by immersion of the plates in liquid nitrogen. β-Glucuronidase (125 U) in sodium acetate (60 mM, pH 4.5) was added and coincubated (60 min; shaking; 37°C) to cleave any glucuronide metabolites. The reaction was terminated by the addition of acetonitrile (250 μl, containing 0.5 mM clobazam internal standard). Samples were centrifuged (5000g; 5 min), and an aliquot was analyzed by LC-MS/MS. Standard curves were generated for each metabolite using an equivalent concentration of frozen and thawed rat hepatocytes.
Incubations to Determine Metabolite Formation in the Presence of Testosterone. Incubations of cryopreserved human hepatocytes with each substrate (1 μM) in the presence of testosterone at different concentrations (10, 25 and 100 μM) were performed as described above. The final concentration of organic solvent (methanol or acetonitrile) was 1.0% v/v. Samples were further incubated with β-glucuronidase, and analyzed as described above.
Nonspecific Binding of Substrates. An estimate of the nonspecific binding to the incubation vessel (24-well polystyrene plate) was obtained by comparison of the concentration of drug (nominally 1 and 10 μM) in buffer, sampled from a plate, to the equivalent concentration in solvent. The fraction of recovered drug in the buffer was between 0.9 and 1.0, depending on benzodiazepine. A similar range of recovery of unbound drug was obtained from microfiltration after incubation (5 min) with freeze-thawed cell suspension in buffer. It was deemed unnecessary to adjust the assumed drug incubation concentrations for calculation of the results.
Determination of Metabolite Concentration. The two major metabolites of each benzodiazepine substrate (1′-hydroxyalprazolam, 4-hydroxyalprazolam, 1′-hydroxymidazolam, 4-hydrozymidazolam, 1′-hydroxytriazolam, 4-hydroxytriazolam, 3-hydroxydiazepam, nordiazepam, 3-hydroxyflunitrazepam and norflunitrazepam) were quantified by LC-MS/MS. For each assay, nine calibration standards with a blank were prepared in a matrix identical to the incubation extracts and included levels below and above the expected concentrations. The stability of 1′-hydroxyalprazolam and 4-hydroxyalprazolam was checked in incubate extracts of standard prepared in mobile phase using either 0.01 M formic acid (pH 3) or 0.001 M ammonium acetate (pH 6) with acetonitrile (50%). Both metabolites were found to be stable in the ammonium acetate preparations, but not in the formic acid preparations, over a storage period of 96 h. Ammonium acetate was therefore selected for the preparation of the study samples. Each metabolite pair, together with clobazam as internal standard, was separated on a Luna C18(2) 50 × 4.6 mm 3 μm column (Phenomenex, Macclesfield, Cheshire, UK) at 40°C using either a binary or ternary gradient maintained at 1 ml/min by a Waters Alliance 2795 HT LC system (Waters, Milford, MA).
For 1′-hydroxyalprazolam, 4-hydroxyalprazolam, 3-hydroxydiazepam, and nordiazepam, an initial mobile phase of 90% 0.001 M ammonium acetate/10% acetonitrile was ramped immediately to 62% 0.001 M ammonium acetate/38% acetonitrile at 1 min and immediately to 74% acetonitrile/26% 0.001 M ammonium acetate at 3 min. The initial ratio was immediately reestablished at 4 min and maintained to 5 min. The retention times were approximately 3.4 (4-hydroxyalprazolam), 3.6 (1′-hydroxyalprazolam), 4.3 (3-hydroxydiazepam, nordiazepam), and 4.4 (clobazam) min.
For 3-hydroxyflunitrazepam, norflunitrazepam, 1′-hydroxymidazolam, 4-hydroxymidazolam, 1′-hydroxytriazolam, and 4-hydroxytriazolam, an initial mobile phase of 90% 0.001 M ammonium acetate/10% acetonitrile was ramped immediately to 44.5% 0.001 M ammonium acetate/42% acetonitrile/13.5% 0.01 M formic acid at 1 min and immediately to 78% acetonitrile/13.5% 0.01 M formic acid/8.5% 0.001 M ammonium acetate at 3 min. The initial ratio was immediately reestablished at 4 min and maintained to 5 min. The retention times were approximately 2.8 (4-hydroxymidazolam), 3.2 (1′-hydroxytriazolam, 4-hydroxytriazolam), 3.3 (1′-hydroxymidazolam), 3.4 (3-hydroxyflunitrazepam), 3.5 (norflunitrazepam), and 4.2 (clobazam) min.
The compounds were detected and quantified by atmospheric pressure electrospray ionization MS/MS using a Micromass Quattro Ultima triple quadrupole mass spectrometer. The LC column eluate was split and one fourth was delivered into the mass spectrometer where the desolvation gas (nitrogen) flow rate was 600 l/h, the cone gas (nitrogen) flow rate was 100 l/h, and the source temperature was 125°C.
Using positive ion mode, protonated molecular ions were formed using a capillary energy of 3.5 kV and cone energies of 39 V (3-hydroxydiazepam), 71 V (3-hydroxyflunitrazepam), 74 V (nordiazepam), 76 V (norflunitrazepam), 78 V (clobazam), and 80 V (1′-hydroxyalprazolam, 4-hydroxyalprazolam, 1′-hydroxymidazolam, 4-hydroxymidazolam, 1′-hydroxytriazolam, and 4-hydroxytriazolam). Product ions formed in argon at a pressure of 2 × 10–3 mbar and at collision energies of 12 eV (3-hydroxydiazepam, m/z 301.05→255.3), 15 eV (3-hydroxyflunitrazepam, m/z 330.05→284.35), 20 eV (4-hydroxyalprazolam, m/z 325.05→280.3; clobazam, m/z 301.05→259.35), 22 eV (norflunitrazepam, m/z 300.0→254.35), 25 eV (nordiazepam, m/z 271.05→208.35; 4-hydroxymidazolam, m/z 342.0→234.3), 30 eV (1′-hydroxyalprazolam, m/z 325.05→297.35; 1′-hydroxymidazolam, m/z 342.0→203.3; 1′-hydroxytriazolam, m/z 359.05→176.3), and 35 eV (4-hydroxytriazolam, m/z 359.05→273.3) were monitored as ion chromatograms, which were integrated and quantified by quadratic regression analysis of standard curves using Micromass QuanLynx 3.5 software.
The accuracy of the method was assumed to be adequate since the concentrations were calculated from calibration standards prepared in the same way as the extracts (spike calibration). Values were accepted if the internal standard ratio was greater than a value equal to the calibration regression intercept plus approximately 10 times the estimated standard deviation of the intercept. Repeatability precision was considered adequate if duplicate sample values were within 10% of each other.
In Vitro Kinetics. Metabolite formation rates were analyzed by nonlinear regression analysis using GraFit (v. 3.0, Erithicus Software, Horley, UK) using models for either Michaelis-Menten kinetics or sigmoidal kinetics (Hill equation, eq. 1). The goodness of fit criteria used to select the model with the most appropriate fit comprised visual inspection, consideration of the randomness of residuals and the standard error of the parameters. 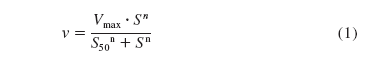 where v is the velocity of the reaction, Vmax is the maximum velocity, S50 is the substrate concentration at half-maximum velocity, S is substrate concentration, and n is the Hill coefficient denoting sigmoidicity.
where v is the velocity of the reaction, Vmax is the maximum velocity, S50 is the substrate concentration at half-maximum velocity, S is substrate concentration, and n is the Hill coefficient denoting sigmoidicity.
Intrinsic Clearance. CLint was determined from the ratio Vmax/KM (for pathways following Michaelis-Menten kinetics); its equivalent (for non-Michaelis-Menten kinetics), CLmax (maximum activated clearance), was determined using eq. 2 (Houston and Kenworthy, 1999) or directly from the maximum v/S.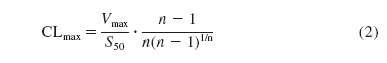
CLint and CLmax values were scaled to in vivo units using scaling factors of 120 × 106 hepatocytes g–1 liver (Houston and Carlile, 1997) and 21 g liver kg–1 body weight.
Comparisons with Microsomal and in Vivo Clearances. The kinetic parameter values and intrinsic clearances from cryopreserved hepatocytes were compared with the equivalent data from a similar study using human liver microsomes from our laboratory (Rawden et al., 2005) after normalizing both data sets to per gram of liver using scaling factors.
Scaled intrinsic clearances from cryopreserved hepatocytes were evaluated relative to a database of in vivo human studies reported previously (Ito and Houston, 2005). Predictions of intrinsic clearance for the benzodiazepines were supplemented by other data from the literature using cryopreserved human hepatocytes. Four sources (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002; Naritomi et al., 2003) were identified. Data from the former three sources were used due to the consistency in bias (average fold underprediction 5–6) and variability (relative mean square error 363–559) in contrast to the Naritomi study (Naritomi et al., 2003; average fold underprediction 14 and relative mean square error 3563).
Results
Incubation Time and Cell Concentration Optimization. The rate of metabolism of diazepam, flunitrazepam, and alprazolam was linear with cell density up to 1 × 106 live hepatocytes/ml, and with time up to 120 min of incubation. Triazolam and midazolam metabolism was linear to 0.25 × 106 live hepatocytes/ml and for 60 and 10 min, respectively.
Metabolite Formation Kinetics. The kinetics of the formation of the two metabolites for each of the five benzodiazepines were characterized. In the case of midazolam, triazolam, and alprazolam, these pathways comprised 1′- and 4-hydroxylation, whereas for diazepam and flunitrazepam they comprised 3-hydroxylation and demethylation. 1′-Hydroxymidazolam, 3-hydroxyflunitrazepam, 3-hydroxydiazepam, and 4-hydroxyalprazolam were major metabolites (>90% of total metabolic clearance for their respective drugs). For triazolam, there was approximately an equal contribution from the two pathways of clearance. These findings are totally consistent with several human hepatic microsomal studies (see Rawden et al., 2005). Average values from the five donor livers for the kinetic parameters for each of the pathways are summarized in Table 1.
Kinetic parameter values from cryopreserved human hepatocytes for midazolam, triazolam, alprazolam, flunitrazepam, and diazepam
Values are means from five donors ± S.D.
To compare the activity of individual livers and show the concentration-dependence of the kinetics, the data are presented as clearance plots where rate of metabolite formation/substrate concentration is plotted as a function of substrate concentration (Fig. 1). For certain pathways (1′-hydroxylation and 4-hydroxylation of triazolam and 1′-hydroxylation of alprazolam), clearance was consistently concentration-dependent at subsaturating concentrations, reaching a maximum at a finite concentration, signifying autoactivation in all livers (Fig. 1, A and B).
There were large differences in clearance between livers for all pathways (Fig. 1). There were also differences between livers in the extent of autoactivation and the concentrations at which maximum activation was reached; this phenomenon was absent in some livers for certain pathways. The presence of autoactivation for two or more substrates was not always consistent for particular livers. For the 3-hydroxylation and demethylation of flunitrazepam and diazepam, activation was evident for several but not all livers (Fig. 1, C and D). A similar inconsistency between donor livers was seen for the 4-hydroxylation of alprazolam (Fig. 1B). A linear clearance at low substrate concentrations, as associated with Michaelis-Menten kinetics, was observed in all livers for both hydroxylations of midazolam with no evidence of activation (Fig. 1E).
Whereas pathways that follow Michaelis-Menten kinetics yielded an intrinsic clearance which can be directly extrapolated to predict in vivo, for pathways involving activation kinetics, clearance was dependent on concentration. At maximum activation, the clearance may be considerably greater than that at lower drug concentrations. To illustrate the magnitude of this effect, the data are presented in terms of the fold increase of clearance from the minimum observed, at low substrate concentrations, to the maximum activated clearance (CLmax) (Table 1). The largest autoactivation effects occur for triazolam hydroxylations (5.9- and 8.6-fold) and alprazolam 1′-hydroxylation (7.5-fold).

Clearance (v/S) as a function of substrate concentration in cryopreserved hepatocytes from five different human livers for 1-hydroxylation (left) and 4-hydroxylation (right) of triazolam (A), alprazolam (B), midazolam (E), and 3-hydroxylation (left) and demethylation (right) of flunitrazepam (C) and diazepam (D). Each symbol represents a different liver.
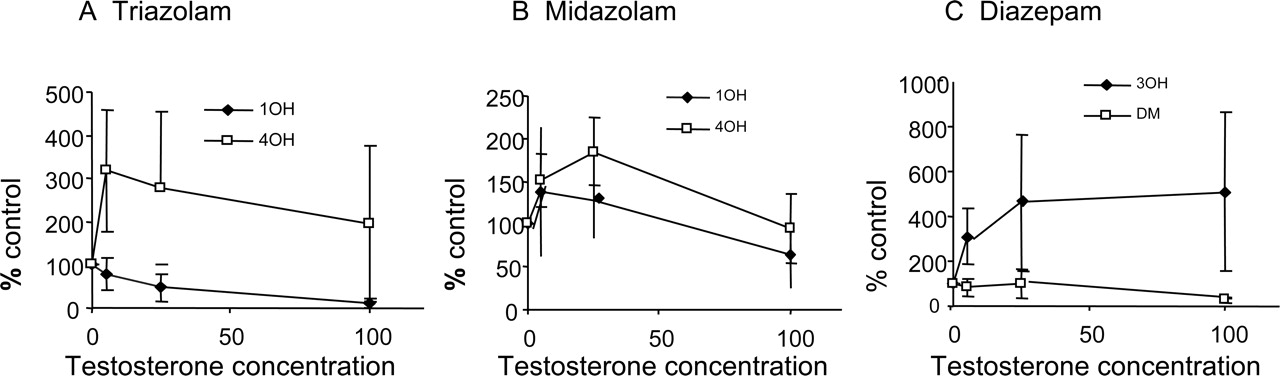
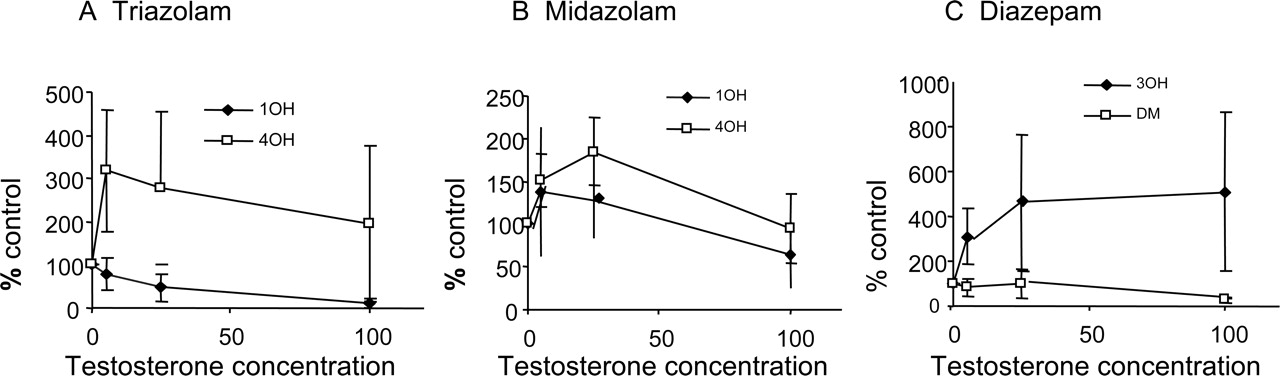
Heteroactivation of human hepatocyte clearance (percentage control) by testosterone (μM) for triazolam (A), midazolam (B), and diazepam (C). Mean data ± S.D. are shown.
The Effect of Coincubation with Testosterone. Heteroactivation was investigated by comparing the rates of formation of each metabolite for each substrate, at low concentration, in the presence of testosterone (5–100 μM). Increased activity in the presence of testosterone was observed for at least one pathway for each substrate, and two general types of responses were seen (Fig. 2). For example, for 4-hydroxylation of triazolam (Fig. 2A), the mean maximum effect (approximate mean 300% of control) occurred at the lowest testosterone concentration; the effect was reduced with increasing testosterone concentration. In contrast, for the triazolam 1′-hydroxylation pathway, there was inhibition in hepatocytes. The profiles for alprazolam were qualitatively and quantitatively similar to those for triazolam. For midazolam, a substrate showing no evidence of autoactivation, heteroactivation of approximately 200% at low testosterone concentrations was observed (Fig. 2B).
Diazepam and flunitrazepam showed a different trend with testosterone activation, in which the effect was maintained over the heteroactivation concentration range used. In each case, 3-hydroxylation was activated (up to approximately 600% of control), but inhibition of the other (demethylation) pathway occurred (Fig. 2C).
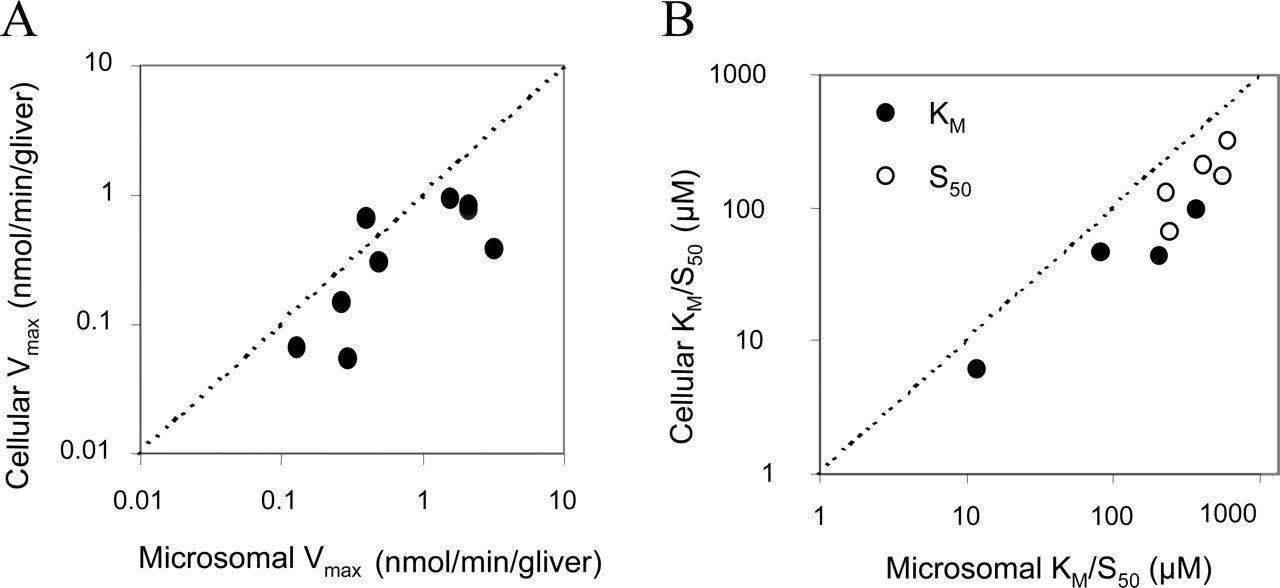
Comparison of Vmax (A) and KM (or S50) (B) from cryopreserved human hepatocytes with human liver microsomes, for two pathways of metabolism for five benzodiazepines. Microsomal data are from Rawden et al. (2005).
Intrinsic Clearance. Kinetic parameters (Vmax, KM, or S50 and n) listed in Table 1 have been compared with previously reported values in microsomes using analogous methodology. When expressed on an equivalent liver weight basis, both Vmax values were linearly correlated and hepatocellular Vmax values were consistently lower than microsomal Vmax values (Fig. 3A). There was also linear agreement between KM (or S50) in the two systems; again, hepatocyte values were consistently lower (Fig. 3B). CLint for each pathway was assessed by either the ratio of the Michaelis-Menten parameters (Vmax/KM) or the maximum activated clearance (CLmax). There was good agreement between CLint for microsomes and hepatocytes (scaled to a whole liver) over the range of about 1 to 300 ml/min/kg (Fig. 4). However, when compared with the intrinsic clearances calculated from human in vivo clearances (Rawden et al., 2005), there was a general underprediction (4-fold on average).
It is of interest to compare these findings obtained from our detailed metabolite kinetic studies with previously published studies (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002) using the substrate depletion approach (Fig. 5). Although the level of experimental detail was less than in the current study (only one substrate concentration and no metabolite formation monitored), the overall trend in this supplementary data set is similar to the current data set. However, in comparison to the current studies, the variability (assessed by the root mean square prediction error) is 3- to 5-fold greater. Using the total data set of 45 drugs, there is an average 5.6-fold underprediction of in vivo CLint from human cryopreserved hepatocytes.
Discussion
Recent advances in cryopreservation have enabled human hepatocytes to be successfully frozen, stored, and thawed with retention of both phase I and II enzyme activities (Li et al., 1999; Steinberg et al., 1999). The subsequent commercial availability of cryopreserved human hepatocytes has enabled routine use of a cellular rather than subcellular in vitro system for hepatic clearance prediction. However, this in vitro system has not been fully explored for its utility in characterizing drug clearance, particularly for substrates for CYP3A4, which are known to display complex microsomal kinetics. This study reports the assessment of cryopreserved human hepatocytes to quantitatively predict metabolic clearance of a series of five benzodiazepines.

Comparison of CLint from cryopreserved human hepatocytes with human liver microsomes, for 10 pathways of metabolism for five benzodiazepines. Microsomal data are from Rawden et al. (2005).
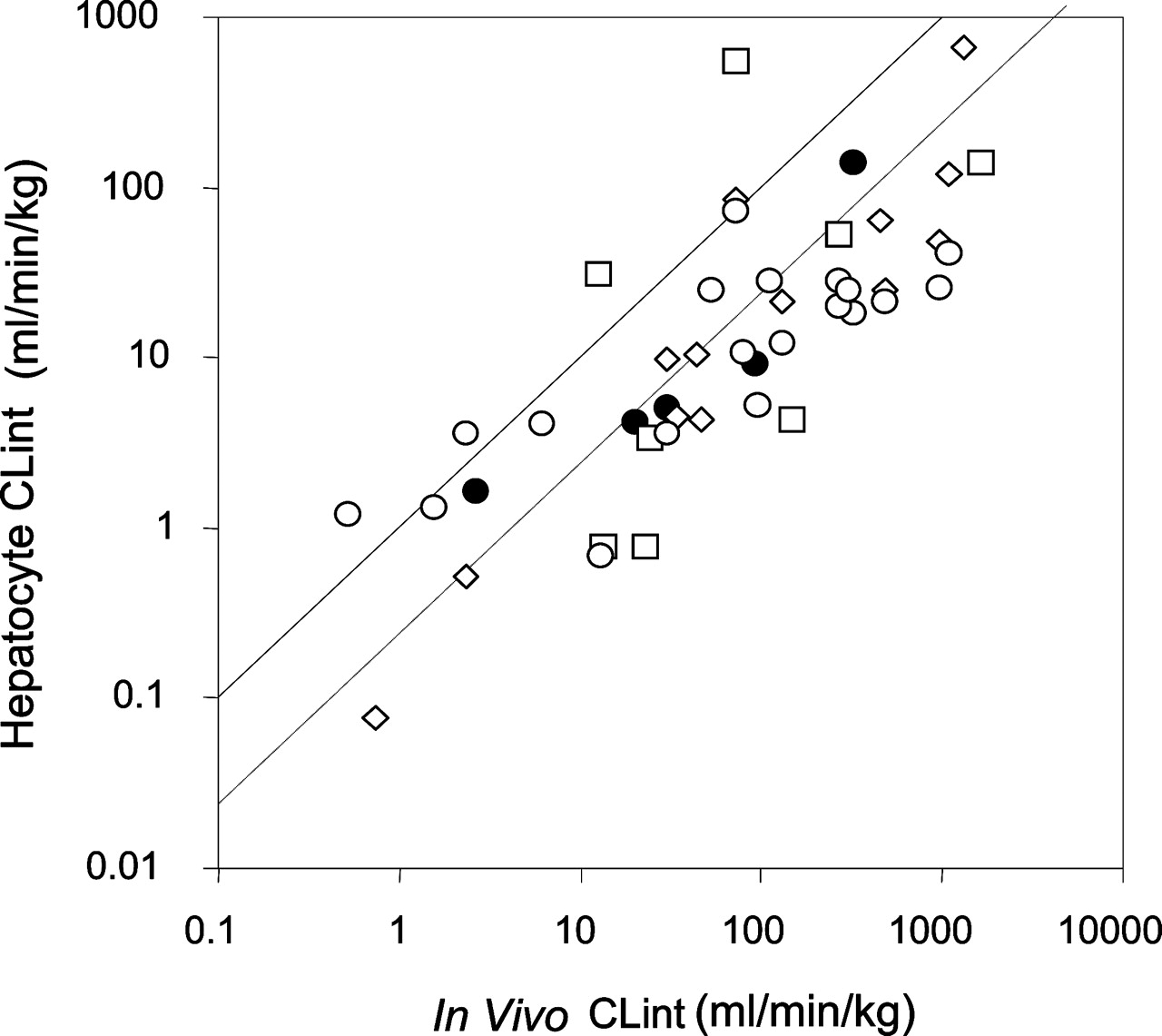
Comparison of CLint from cryopreserved human hepatocytes for five benzodiazepines (this study) and various drugs (literature) with human in vivo studies (total n = 45). The solid line is the line of identity and the dashed line is the regression line when the slope of unity is maintained. The change in intercept indicates the bias. •, this study; □, Soars et al. (2002); ⋄, Shibata et al. (2002); ○, Lau et al. (2002).
Underprediction of in vivo clearance using human liver microsomes is typical for many drugs (Ito and Houston, 2005), as exemplified by the benzodiazepines (Rawden et al., 2005). However, for these drugs, there are two major concerns: first, the benzodiazepines are highly dependent on CYP3A4 for metabolic clearance, and kinetic complexities might have contributed to prediction inaccuracy (Houston and Kenworthy, 1999; Houston and Galetin, 2005); and, second, liver variability, in terms of donor individuality (genetic, age, and environmental factors, etc.), was high and may have contributed to the disagreement between in vitro and in vivo results (Rawden et al., 2005).
In this study, in vitro clearance was measured by metabolite formation kinetics over a range of substrate concentrations for the two major pathways for five benzodiazepines. A high incidence of concentration-dependent clearance was observed at subsaturating substrate concentrations, providing for the first time clear evidence of autoactivation of benzodiazepine metabolism in hepatocytes. Previously, autoactivation of the major pathways was found to be common in microsomes for these drugs (see Rawden et al., 2005); this study has demonstrated that this kinetic phenomenon occurs in human cryopreserved hepatocytes and complements an earlier study in fresh rat hepatocytes with dextromethorphan (Witherow and Houston, 1999).
Autoactivation was demonstrated for four of the five benzodiazepine substrates in all livers. There were large differences in the magnitude of effect (2- to 10-fold increase in clearance) in hepatocytes between the different livers. Some livers are refractory to autoactivation but not consistently for all pathways. Similarly, there were large differences in autoactivation between the pathways. Considering the differences in the concentration dependence of activation, the apparent absence of activation in some cases may have been due to the limit of analytical sensitivity.
Heteroactivation of metabolism of the same five benzodiazepines by testosterone was previously reported for human microsomes (Rawden et al., 2001). It has been speculated that autoactivation kinetics is an in vitro phenomenon and that, in vivo, the enzymes may be naturally activated by endogenous (or exogenous) substrates or activators (Witherow and Houston, 1999). The high degree of activation seen in hepatocytes, in this study, is evidence that the concentration of heteroactivators of CYP3A4 in hepatocytes following isolation is low. A comparison between hepatocytes and microsomes of heteroactivation by testosterone indicates that, in general, the hepatocyte effect was both qualitatively and quantitatively similar to that found in microsomes (Rawden et al., 2001). It remains a moot point whether hepatocytes are heteroactivated in vivo but subsequently lose substantial activation due to loss of activators during the processes of liver isolation and cell isolation. Although the circulating testosterone plasma concentration in vivo is <1 μM (Griffin and Ojeda, 2000), there have been three studies demonstrating heteroactivation in of CYP3A4 in vivo (Tang et al., 1999; Egnell et al., 2003; Chen et al., 2004) via concomitant drug administration.
Activation kinetics have important implications for the prediction of clearance. There is no constant clearance (at subsaturating substrate concentrations), as is characteristic of Michaelis-Menten kinetics, which can be scaled to provide an in vivo prediction. The equivalent “intrinsic clearance” is CLmax (Houston and Kenworthy, 2004), the clearance at the substrate concentration for maximum autoactivation (Table 1). In the hepatocytes, there was a maximum activation corresponding to an 8-fold increase from the clearance at the lowest substrate concentration. This means that prediction of in vivo clearance, if based on a clearance estimate determined at low drug concentration (as might be the case using the widely practiced assay of drug depletion), might be substantially underestimated. The testosterone-activated increase in clearance (at low substrate concentrations) was of a similar magnitude to that of autoactivation. Heteroactivation has been demonstrated for other CYP3A4 substrates in human cryopreserved hepatocytes (Mäenpää et al., 1998; Ngui et al., 2000, 2001) and hepatic microsomes (Kenworthy et al., 2001; Schrag et al., 2001; Nakamura et al., 2002; Patki et al., 2003).
Both Vmax and KM (or S50) for these drugs were found to be consistently lower for hepatocytes than for microsomes. Therefore, the intrinsic clearance values calculated are in numerical agreement (on an equivalent basis relative to body weight) for the two systems (Fig. 4) since they reflect the combined effect of lower metabolic activity and an apparent higher affinity in hepatocytes. This agreement was consistent over the wide range of CLint values (1–300 ml/min/kg) observed for the metabolic reactions for this group of benzodiazepines and indicate that, overall, cryopreserved hepatocytes possess P450 activity similar to that of hepatic microsomes.
In this study, underprediction may have resulted from loss of CYP3A4 activity in cryopreserved cells, which would be supported by the lower Vmax values observed in this study relative to microsomes. However, good agreement in activity between fresh and cryopreserved hepatocytes has been reported for both rat (Naritomi et al., 2003; Griffin and Houston, 2004) and human (McGinnity et al., 2004). If the lower KM (or S50) observed for hepatocytes in this study resulted from accumulation of unbound drug in the cells due to active uptake, this would provide at least a partial explanation of the microsomal underprediction. Diazepam, one of the benzodiazepines studied, has been shown to accumulate substantially (up to 50-fold) in rat hepatocytes (Jones and Houston, 2004) and, based on physiochemical properties, it would be anticipated that the other four benzodiazepines would behave similarly. However, other possible contributing explanations cannot be ignored. For example, it is possible that underprediction (from both systems) of these substrates is due to the effect of potent heteroactivation in vivo. Also, it cannot be excluded that the donor livers may be unrepresentative of those individuals involved in the in vivo study.
In summary, the kinetics of metabolite formation for five benzodiazepines in cryopreserved human hepatocytes was similar to that observed in human liver microsomes, both in terms of turnover rates and auto- and heteroactivation. These latter findings imply a lack of inherent heteroactivation in hepatocytes and confirm previous observations with fresh hepatocytes from rat livers (Witherow and Houston, 1999). There was considerable variability in the effects of activation between the livers; indeed, there was variability in activity in general. Overall, it can be concluded that for the P450-mediated clearance of these benzodiazepines, the use of cryopreserved hepatocytes provides in vivo predictions similar to that observed with hepatic microsomes. Based on a data set of 45 drugs, there is on average a 5.6-fold underprediction of in vivo intrinsic clearance when the current data are supplemented with previous studies using drug depletion-time profiles.
Acknowledgments
We thank Michael Griffin for compiling the data presented in Fig. 5.
Footnotes
-
Financial support for this project was provided by the following Centre for Applied Pharmacokinetic Research (CAPkR) Consortium members: Bristol Myers-Squibb, GlaxoSmithKline, Novartis, Pfizer, Roche, and Servier. Part of this study was presented at the 15th International Symposium on Microsomes and Drug Oxidations in Mainz, Germany, July 4–9, 2004.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.005389.
-
ABBREVIATIONS: LC, liquid chromatography; MS/MS, tandem mass spectrometry; CLint, intrinsic clearance; CLmax, maximum activated clearance.
- Received April 28, 2005.
- Accepted September 16, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}