


Abstract
Because the expression of drug-metabolizing enzymes and drug efflux transporters has been shown in the intestine, the contribution of this tissue to the first-pass effect has become of significant interest. Consequently, a comprehensive understanding of the absorption barriers in key preclinical species would be useful for the precise characterization of drug candidates. In the present investigation, we evaluated the intestinal first-pass effect of midazolam (MDZ) and fexofenadine (FEX), typical substrates for CYP3A and P-glycoprotein (P-gp), respectively, with ketoconazole (KTZ) as a potent dual CYP3A/P-gp inhibitor in cynomolgus monkeys. When MDZ or FEX was administered i.v. at doses of 0.3 or 1 mg/kg, respectively, the plasma concentration-time profiles were not influenced by p.o. coadministration of KTZ (20 mg/kg). On the other hand, when MDZ or FEX was administered p.o. at doses of 1 or 5 mg/kg, respectively, concomitant with a dose p.o. of KTZ (20 mg/kg), significant increases were observed in the area under the plasma concentration-time curves of MDZ or FEX (22-fold in MDZ and 3-fold in FEX). These findings indicate that both CYP3A and P-gp play a key role in the intestinal barrier and that inhibition of intestinal CYP3A/P-gp activities contributes exclusively toward the drug-drug interactions (DDI) with KTZ. Additionally, the Ki values of the antifungal agents, KTZ, itraconazole, and fluconazole, for MDZ 1′-hydroxylation in monkey intestinal and liver microsomes were comparable with those in the respective human samples. These results suggest that monkeys may be an appropriate animal species for evaluating the intestinal first-pass effect of p.o. administered drugs and predicting intestinal DDI related to CYP3A4 and P-gp in humans.
Drug-drug interactions (DDI) can cause serious adverse effects in clinical practice. For example, case reports of DDI resulting in adverse effects have been published for terfenadine and ketoconazole (KTZ) (Honig et al., 1993), felodipine and erythromycin (Bailey et al., 1996), and cerivastatin and gemfibrozil (Staffa et al., 2002). Because most of these in vivo interactions have been interpreted as the result of metabolic inhibition by cytochrome P450 (P450) inhibitors, the prediction of clinically significant DDI in preclinical studies is clearly desirable and has been examined using several in vitro methodologies with hepatocytes and microsomal fractions obtained from human liver. However, the effect of coadministration of P450 inhibitors may be expected to be more pronounced at the level of the intestine compared with the liver based on presumed local differences in concentration during the period of drug absorption. More recently, investigations have focused on the contribution of the intestine to DDI. Some researchers have reported the quantitative prediction of DDI considering the contribution of both intestinal and hepatic interactions (Wang et al., 2004; Galetin et al., 2006; Obach et al., 2006).
It is well known that several enzymes are present in small intestinal epithelial cells (enterocytes) and that they can catalyze substantial metabolism of some p.o. administered drugs, thus exerting a first-pass effect (Kolars et al., 1991; Paine et al., 1996; Kaji and Kume, 2005). In particular, CYP3A4 appears to be the most abundant P450 present in human small bowel (Kolars et al., 1992). It has been suggested that the bioavailability of CYP3A4 substrates such as felodipine, midazolam (MDZ), and cyclosporine might be decreased because of first-pass metabolism in the small intestine (Kato et al., 2003; Galetin et al., 2006). Recent data indicate that the P-glycoprotein (P-gp) in the enterocyte brush border can act as an efflux pump for drugs and may also limit the bioavailability of many of the same drugs that interact with CYP3A4. von Richter et al. (2004) reported that the CYP3A4 protein content was approximately 3 times and the P-gp content approximately 7 times higher in enterocyte homogenates compared with liver homogenates obtained from the same set of 15 patients. Furthermore, characterization of the co-operative role between CYP3A4 and P-gp in limiting oral drug availability has been elucidated in several important clinical studies (Lown et al., 1997; Paine et al., 2002).
The monkey is a species that is widely used in drug-safety evaluation and biotransformation studies by the pharmaceutical industry. Because the monkey is the nearest species to humans in the evolutionary tree, a high level of nucleotide and amino acid sequence identity (more than 90%) has been observed between human and monkey P450s (Komori et al., 1992; Mankowski et al., 1999). In fact, in comparing the pharmacokinetic parameters of approximately 100 xenobiotics obtained from several animals, it has been reported that the monkey provides the most accurate prediction of human pharmacokinetics (Ward and Smith, 2004). The monkey may also serve as a good preclinical surrogate for DDI studies. Therefore, we selected cynomolgus monkeys as an animal model in the current investigation.
KTZ is an antifungal agent that is one of the most potent CYP3A inhibitors. The inhibitory effects of KTZ on in vitro and in vivo metabolic activities of CYP3A, including MDZ 1′-hydroxylation, have been well studied by many investigators (Olkkola et al., 1994; Wrighton and Ring, 1994; Tsunoda et al., 1999). It has also been reported that KTZ causes clinically relevant interactions with P-gp substrates, including cyclosporine, tacrolimus, verapamil, and fexofenadine (FEX) (Gomez et al., 1995; Floren et al., 1997; Davit et al., 1999; Sandström et al., 1999). Although most of the clinically available P-gp substrates are metabolized by CYP3A because of overlapping substrate specificity, FEX is not metabolized by P450s and is mostly excreted into the urine and feces in the unchanged form (Simpson and Jarvis, 2000). Accordingly, FEX is a suitable drug for the evaluation of DDI by differentiating P-gp from CYP3A4. On the other hand, MDZ has become a widely accepted and validated CYP3A phenotyping probe that is not a P-gp substrate. In the present study, we investigated the effects of KTZ on the pharmacokinetics of MDZ as a CYP3A probe and FEX as a P-gp probe that can be administered i.v. and p.o. to discriminate between intestinal and hepatic activities in cynomolgus monkeys. In addition, the inhibitory kinetics of the antifungal agents in monkey intestinal and liver microsomes was compared with that in respective human samples. Furthermore, we discuss the similarity of DDI related to intestinal metabolism/absorption between monkeys and humans.
Materials and Methods
Chemicals. MDZ, KTZ, and polyethyleneglycol 400 were purchased from Wako Pure Chemical Industries (Osaka, Japan). Itraconazole (ITZ), 1′-hydroxymidazolam, tolbutamide, and β-NADPH were purchased from Sigma-Aldrich (St. Louis, MO). FEX hydrochloride, fluconazole (FLZ), reserpine, and Gelucire 44/14, a self-emulsifying excipient, were obtained from Toronto Research Chemicals (North York, ON, Canada), ICN Biomedicals (Eschwege, Germany), Nacalai Tesque (Kyoto, Japan), and Gattefossé (Lyon, France), respectively. Pooled liver and intestinal microsomes of human (liver microsomes, pooled from 50 donors; intestinal microsomes, pooled from 11 donors) and monkey (liver microsomes, pooled from 5 animals; intestinal microsomes, pooled from 7 animals) were provided by Xenotech (Lenexa, KS). All the other reagents and solvents were of analytical grade and were commercially available.
Animals. Male cynomolgus monkeys, 3.0 to 3.2 kg, were supplied by the Guangxi Primate Center of China (Guangxi, China). Animals were housed in a temperature- and humidity-controlled room with a 12-h light/dark cycle. Animals were fed a standard animal diet (Teklad Global Certified 25% Protein Primate Diet, Harlan Sprague-Dawley, Indianapolis, IN); food was provided ad libitum except for during the overnight periods before dosing. Whenever overnight fasting was used, food was provided after the 8-h blood sample was obtained. All the procedures for the animal experiments were approved by the Animal Ethics Committee of Tanabe Seiyaku.
In Vitro Inhibition Studies. All the incubations were performed in duplicate in 100-μl aliquots of a solution containing 0.08 M potassium phosphate, pH 7.4, 1 mM EDTA, 5 mM MgCl2, 1 mM NADPH, 0.1 mg/ml microsomal protein, and 0.5, 1, 2, and 4 μM MDZ. The incubation mixtures for the determination of Ki values contained the antifungal agents KTZ, ITZ, or FLZ. The final concentrations of KTZ were 0, 0.01, 0.02, 0.05, and 0.1 μM; the final concentrations of ITZ were 0, 0.04, 0.08, 0.16, and 0.32 μM; and the final concentrations of FLZ were 0, 5, 10, 20, and 40 μM. MDZ and the inhibitors were each dissolved in acetonitrile. The final concentration of organic solvent in the incubation mixture was 0.5% (v/v). After preincubation at 37°C for 5 min, the reactions were initiated by the addition of the NADPH solution. Following incubation at 37°C for 3 (monkey samples) or 5 min (human samples), the reactions were terminated by the addition of 100 μl of ice-cold acetonitrile. The samples were spiked with 10 μl of internal standard solution containing 10 μM tolbutamide. After vortexing and centrifugation, the supernatants were transferred to a high-performance liquid chromatography (HPLC) vial for HPLC/mass spectrometry analysis.
In Vivo Studies. The p.o. and i.v. dosing solutions of MDZ were prepared in ultrapure water containing 0.01 M hydrochloric acid. The p.o. and i.v. dosing solutions of FEX were prepared in ultrapure water and 5% (w/v) sugar aqueous vehicle, respectively. The dosing solution of KTZ was dissolved in a polyethyleneglycol 400/10 (v/v) Gelucire 44/14 containing 0.01 M hydrochloric acid (2/8, v/v) vehicle. The same four male cynomolgus moneys were used again after a washout period of at least 2 weeks to investigate the effects of KTZ on the pharmacokinetics of MDZ and FEX. Immediately after p.o. administration of KTZ (5 or 20 mg/kg), MDZ or FEX was administered p.o. or i.v. to the monkeys. The dosages of MDZ and FEX were 1 and 5 mg/kg for p.o. administration and 0.3 and 1 mg/kg for i.v. administration, respectively. To obtain the control values of the pharmacokinetic parameters for MDZ and FEX, the vehicle for KTZ was administered p.o. immediately before p.o. and i.v. administration of MDZ or FEX. For all the studies, blood was collected from the femoral vein at the desired time points and centrifuged to obtain plasma.
Analytical Procedure.Determination of 1′-hydroxymidazolam concentrations. The concentrations of 1′-hydroxymidazolam in the microsomal reaction mixtures were measured using an Agilent 1100 MSD system (Agilent Technologies, Palo Alto, CA) operating in the positive ion electrospray mode with selected ion monitoring. Chromatography was performed using an Atlantis dC18 column (3-μm particle size, 2.1 × 100 mm, Waters, Milford, MA). The mobile phase consisted of 10 mM acetate ammonium buffer and acetonitrile. The flow rate was 0.3 ml/min, and the initial mobile phase was 80% 10 mM acetate ammonium buffer and 20% acetonitrile. The acetonitrile concentration was increased linearly to 90% over 9 min. Selected ion monitoring was performed at m/z 342 for 1′-hydroxymidazolam and m/z 271 for the internal standard. For the mass spectrometer, the drying gas flow rate was set at 12 l/min, nebulizer pressure at 20 psig, gas temperature at 330°C, and capillary voltage at 3000 V. The fragmenter voltages for 1′-hydroxymidazolam and the internal standard were set at 180 and 90 V, respectively.
Determination of MDZ, FEX, and KTZ concentrations. Two hundred microliters of acetonitrile and 10 μl of internal standard solution containing 1 μg/ml reserpine were added to 100-μl aliquots of the plasma samples. The mixtures were centrifuged, and then an aliquot of the supernatants was analyzed by HPLC/tandem mass spectrometry. Chromatography was performed using an Inertsil ODS-3 column (5-μm particle size, 2.1 × 150 mm, GL Science, Tokyo, Japan) and an HPLC system consisting of Shimazu 10A series (Shimazu, Kyoto, Japan) with a gradient mobile phase of 10 mM formate ammonium and acetonitrile/0.01 M hydrochloric acid (1:1, v/v). The flow rate was 0.3 ml/min. Detection of the analyte and internal standard was performed using a Sciex API 4000 mass spectrometer (Applied Biosystems, Foster, CA) in the positive ion mode using turbo ion spray source at 600°C. The collision gas was set at level 8. Mass transitions (m/z) monitored were 326.0 → 291.9 for MDZ, 502.3 → 466.2 for FEX, 531.0 → 489.3 for KTZ, and 609.4 → 195.0 for internal standard at collision energy of 37, 41, 42, and 50 eV, respectively.
Data Analysis. The inhibition types and the apparent inhibition constant (Ki) values for KTZ, ITZ, and FLZ against 1′-hydroxymidazolam formation were evaluated by graphical analysis with Lineweaver-Burk and Dixon plots. Pharmacokinetic parameters were calculated in individual animals by noncompartmental analysis using WinNonlin Professional (version 4.0.1, Pharsight, Mountain View, CA). Estimation of the terminal elimination rate constant (λ) was performed by log-linear regression of the last three concentration-time points. The terminal elimination half-life (t1/2) was calculated by the relationship t1/2 = 0.693/λ. The area under the plasma concentration-time curve (AUC) was calculated using the trapezoidal rule up to the last measurable concentration, and the AUC was extrapolated to infinity using the λ value (AUCinf). The bioavailability (F) was determined from the dose-corrected AUCinf following p.o. and i.v. administration. The components of the availability of MDZ were further predicted based on the following equation: F = FABS · FG · FH, in which FABS refers to the fraction of the dose absorbed from the gut lumen, FG refers to the fraction of the dose not metabolized by intestinal metabolic enzymes, and FH refers to the fraction of the dose absorbed into the hepatic vein that escapes first-pass effect in the liver. FH was defined as follows: FH = 1 – ERH, in which ERH refers to the hepatic extraction ratio defined as the clearance of i.v. administered MDZ divided by the hepatic blood flow rate in monkeys (45 ml/min/kg) (Davies and Morris, 1993), presuming that the nonhepatic contribution to the clearance of MDZ was negligible and that MDZ exhibited linear pharmacokinetics in the range of plasma concentrations observed in this study. The products FABS · FG and FH were determined and compared with and without the presence of KTZ.

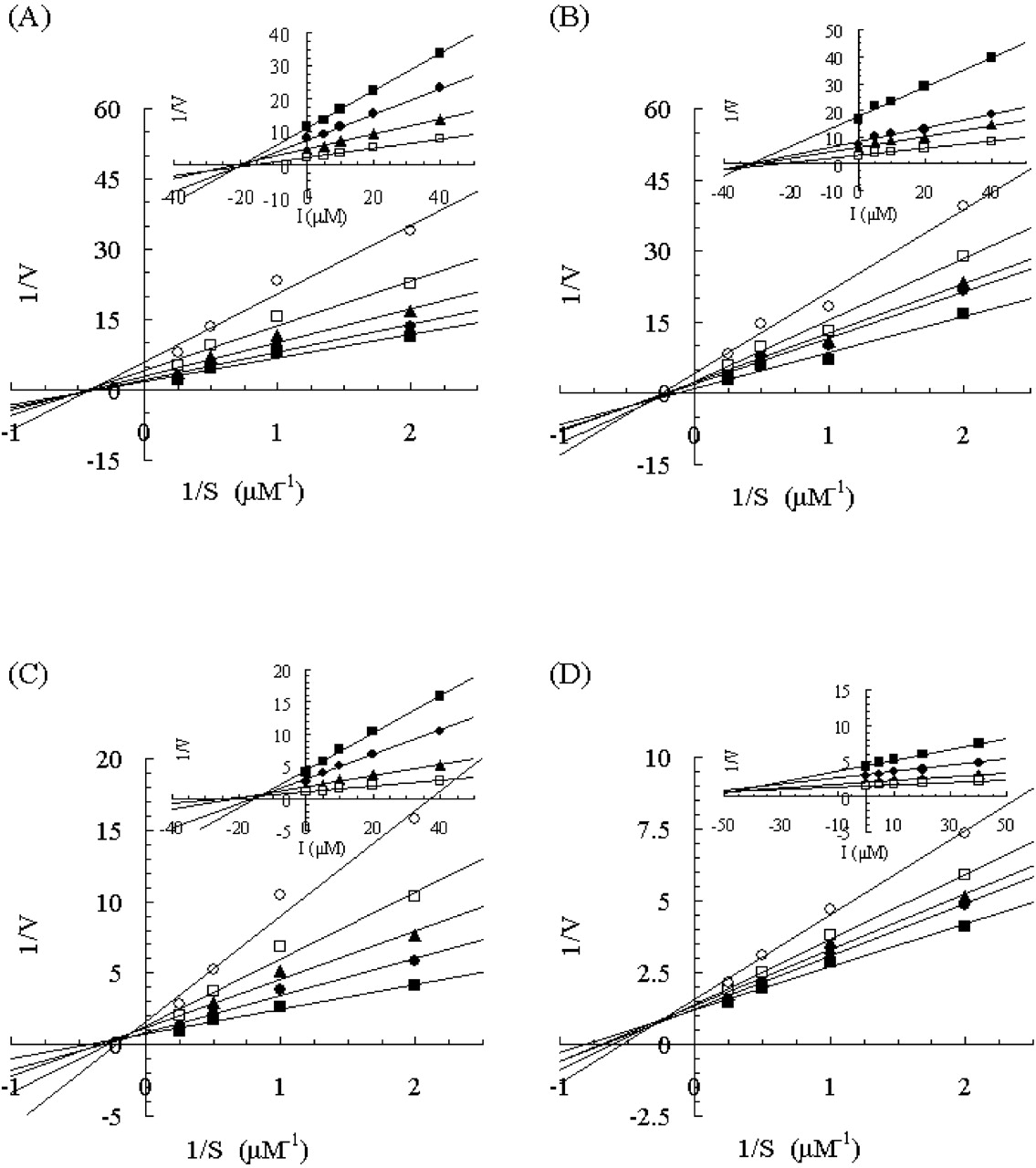
Lineweaver-Burk plots of the effect of KTZ on 1′-hydroxymidazolam formation in human and monkey liver and intestinal microsomes. Insets represent the corresponding Dixon plots. Results from incubations with human liver microsomes (A), human intestinal microsomes (B), monkey liver microsomes (C), and monkey intestinal microsomes (D) are shown. Each point represents the mean of duplicate incubations. MDZ (0.5, 1, 2, or 4 μM) was incubated without KTZ or with 0.01, 0.02, 0.05, or 0.1 μM KTZ. Lines represent the linear regression of transformed data. The rate of 1′-hydroxymidazolam formation is expressed in picomoles per minute per milligram.
Statistical Analysis. All the statistical tests were performed using the Prism software package (version 3.02, GraphPad, San Diego, CA). Statistical differences in the pharmacokinetic parameters were calculated by a two-tailed paired Student's t test with Bonferroni correction. In all the cases, a probability level of p < 0.025 was considered significant.
Results
Effects of Antifungal Agents on 1′-Hydroxymidazolam Formation. The incubation conditions for 1′-hydroxymidazolam formation were evaluated using both liver and intestinal microsomes obtained from both humans and monkeys. The accumulation of 1′-hydroxymidazolam was proportional to the reaction time (human samples, up to 5 min; monkey samples, up to 3 min) and to the microsomal protein concentrations (both human and monkey samples, 0.05–0.1 mg/ml) in the reaction mixtures (data not shown). The inhibitory mechanism of the antifungal agents KTZ, ITZ, and FLZ on 1′-hydroxymidazolam formation was determined in all samples. Lineweaver-Burk and Dixon plots for the inhibition of 1′-hydroxymidazolam formation by KTZ, ITZ, and FLZ are depicted in Figs. 1, 2, and 3, respectively. The average Ki values and inhibition types are summarized in Table 1. Inspection of the reciprocal plots indicated that KTZ was a noncompetitive inhibitor in monkey liver and intestinal microsomes, as well as in human liver and intestinal microsomes. Among the three azole antifungal agents, KTZ was the most potent inhibitor with Ki values of 0.025 and 0.015 μM in monkey liver and intestinal microsomes, respectively, which were similar to those in the respective human samples (liver microsomes, 0.032 μM; intestinal microsomes, 0.044 μM). ITZ was found to be a competitive inhibitor in human liver and intestinal microsomes with Ki values of 0.13 and 0.099 μM, respectively, whereas it noncompetitively inhibited 1′-hydroxymidazolam formation in monkey liver and intestinal microsomes with Ki values of 0.096 and 0.062 μM, respectively. As compared with these, FLZ exhibited a weak inhibition with higher Ki values >10 μM in all the microsomal samples, even though the inhibition type was different between humans and monkeys (noncompetitive and mixed-type, respectively).
Kinetic parameters for the inhibition of 1′-hydroxymidazolam formation by KTZ, ITZ, and FLZ
Effect of KTZ on the Pharmacokinetics of MDZ in Monkeys. The contribution of CYP3A to the first-pass effect in monkey intestine was evaluated by coadministering MDZ, a probe substrate for CYP3A, along with KTZ, as a CYP3A inhibitor. The plasma concentration-time profiles of MDZ with or without coadministration of KTZ p.o. are presented in Fig. 4. The effects of KTZ on the pharmacokinetic parameters of MDZ are summarized in Table 2. We confirmed that the vehicle for KTZ did not affect the plasma concentration-time profiles of MDZ administered p.o. (data not shown). As shown in Table 2, the pharmacokinetic parameters of MDZ after i.v. administration were not significantly affected by p.o. treatment with 5 or 20 mg/kg KTZ. On the other hand, when MDZ was administered p.o. concomitant with a p.o. dose of KTZ, the plasma concentrations of MDZ increased markedly. The values of Cmax, AUCinf, and F for MDZ were increased 6.3- to 8.7-fold, 6.0- to 21.7-fold, and 6.7- to 20.9-fold, respectively, depending on the dose of KTZ. Statistically significant increases were observed in the t1/2, AUCinf, and F for MDZ following p.o. coadministration with 20 mg/kg KTZ compared with the control values following p.o. coadministration of the KTZ vehicle. The Cmax for MDZ exhibited a tendency to increase, depending on the dose of KTZ; however, this change was not statistically significant (5 mg/kg KTZ, p = 0.0615; 20 mg/kg KTZ, p = 0.0820). The plasma concentrations of KTZ were simultaneously determined, and the pharmacokinetic parameters for KTZ are summarized in Table 4. No obvious differences were observed in the parameters for KTZ with concomitant p.o. or i.v. administration of MDZ. The mean values of Cmax and AUCinf increased supraproportionally with the dose of KTZ.
Pharmacokinetic parameters of MDZ following intravenous (0.3 mg/kg) or oral (1 mg/kg) administration of MDZ with a concomitant oral dose of vehicle or KTZ (5 or 20 mg/kg)
Each value represents the mean ± S.D. of four monkeys.
Pharmacokinetic parameters of KTZ following oral (5, 20 mg/kg) administration of KTZ concomitant with MDZ (0.3 or 1 mg/kg) or FEX (1 or 5 mg/kg)
Each value represents the mean ± S.D. of four monkeys.

Lineweaver-Burk plots of the effect of ITZ on 1′-hydroxymidazolam formation in human and monkey liver and intestinal microsomes. Insets represent the corresponding Dixon plots. Results from incubations with human liver microsomes (A), human intestinal microsomes (B), monkey liver microsomes (C), and monkey intestinal microsomes (D) are shown. Each point represents the mean of duplicate incubations. MDZ (0.5, 1, 2, or 4 μM) was incubated without ITZ or with 0.04, 0.08, 0.16, or 0.32 μM ITZ. Lines represent the linear regression of transformed data. The rate of 1′-hydroxymidazolam formation velocity is expressed in picomoles per minute per milligram.
Effect of KTZ on the Pharmacokinetics of FEX in Monkeys. The barrier function of P-gp in monkey intestine was investigated using coadministration of FEX as a probe substrate for P-gp and KTZ as a P-gp inhibitor. The plasma concentration-time profiles and pharmacokinetic parameters of FEX, with or without p.o. coadministration of KTZ are shown in Fig. 5 and Table 3, respectively. As in the case of MDZ, we confirmed that the vehicle for KTZ did not affect the plasma concentration-time profiles of p.o. administered FEX (data not shown). Following p.o. administration of FEX with a concomitant p.o. dose of KTZ (5, 20 mg/kg), the plasma concentrations of FEX increased considerably. However, no obvious change was observed after concomitant i.v. administration of FEX with KTZ even at a dose of 20 mg/kg. The values of Cmax, AUCinf, and F for FEX were increased 1.3- to 1.9-fold, 2.2- to 2.6-fold, and 2.2- to 2.6-fold, respectively, by p.o. coadministration with KTZ. Statistically significant increases were observed in the values of AUCinf and F for FEX only after p.o. coadministration with 5 mg/kg KTZ compared with the vehicle control. On the other hand, the t1/2 values did not change even with 20 mg/kg KTZ. Again, there were no obvious differences in the pharmacokinetic parameters of KTZ with concomitant p.o. or i.v. administration of FEX (Table 4).
Pharmacokinetic parameters of FEX following intravenous (1 mg/kg) or oral (5 mg/kg) administration of FEX with a concomitant oral dose of vehicle or KTZ (5, 20 mg/kg)
Each value represents the mean ± S.D. of four monkeys.
Discussion
In the present study, we evaluated the intestinal first-pass effect involving CYP3A and P-gp in cynomolgus monkeys using typical substrates, MDZ and FEX, respectively, and determined the degree of DDI with KTZ, a potent dual CYP3A/P-gp inhibitor. Because it is well known that KTZ causes a relevant DDI when coadministered with MDZ or FEX in humans, it would be useful to investigate whether this DDI could be reproduced in monkeys. In particular, there have been many reports with regard to human intestinal and hepatic CYP3A activity using MDZ as an in vivo probe (Olkkola et al., 1994; Thummel et al., 1996; Gorski et al., 1998; Tsunoda et al., 1999). In addition, KTZ and clarithromycin caused marked inhibition of CYP3A activity that was greater in the intestine compared with the liver (Gorski et al., 1998; Tsunoda et al., 1999). These results showed that the intestine could be a major site for CYP3A-mediated first-pass metabolism, the extraction ratio of which was greater than that of the liver (approximately 0.6 versus 0.3). Our results indicate that the monkey intestine is also a predominant site for first-pass metabolism because the intestinal availability (FG) was much lower than the hepatic availability (FH) (2.3 versus 69.6%; Table 2), assuming that the p.o. dose of MDZ was completely absorbed into gut wall regardless of species (human, FABS = 1) (Gorski et al., 1998; Kanazu et al., 2005; Sakuda et al., 2006). As compared with the intestinal availability between monkeys and humans (approximately 40%), the contribution of the intestine to the first-pass metabolism of MDZ is suggested to be much greater in monkeys than in humans. The results of the current investigation are consistent with recent published data (Sakuda et al., 2006).

Lineweaver-Burk plots of the effect of FLZ on 1′-hydroxymidazolam formation in human and monkey liver and intestinal microsomes. Insets represent the corresponding Dixon plots. Results from incubations with human liver microsomes (A), human intestinal microsomes (B), monkey liver microsomes (C), and monkey intestinal microsomes (D) are shown. Each point represents the mean of duplicate incubations. MDZ (0.5, 1, 2, or 4 μM) was incubated without FLZ or with 5, 10, 20, or 40 μM FLZ. Lines represent the linear regression of transformed data. The rate of 1′-hydroxymidazolam formation velocity is expressed in picomoles per minute per milligram.

Effect of p.o. coadministration of KTZ (•, 0 mg/kg; ▪, 5 mg/kg; ▴, 20 mg/kg) on the plasma concentration time profiles of MDZ after i.v. (A) and p.o. (B) administration to monkeys at the doses of 0.3 and 1 mg/kg, respectively. Insets represent the linear scale of the plasma concentration time profiles of MDZ. Each point represents the mean ± S.D. of data obtained from four monkeys, except where denoted. The point with an asterisk indicates the mean ± S.D. of three monkeys.
In monkeys, no changes were observed for any of the plasma pharmacokinetic parameters for i.v. MDZ during KTZ treatment (Fig. 4A; Table 2), indicating that KTZ did not inhibit hepatic CYP3A activity at a p.o. dose of 20 mg/kg. On the other hand, the Cmax and AUCinf of MDZ were dramatically increased after p.o. dosing with KTZ, resulting in an increase of intestinal availability from 2.3% to 48.5% (20.6-fold) and a 20.9-fold increase in the oral bioavailability (Fig. 4B; Table 2). These results suggest that the inhibition of intestinal CYP3A activity contributes exclusively to the DDI with KTZ in monkeys. However, despite the fact that there were no changes in total body clearance (CLtot) and distribution volume at steady state (Vdss) after i.v. MDZ dosing with 20 mg/kg KTZ, the t1/2 of MDZ plasma concentration after p.o. dosing was prolonged significantly, approximately 2-fold, during KTZ treatment at a dose of 20 mg/kg but not at 5 mg/kg. Although this mechanism was not clearly established in this investigation, it might be related to prolongation of the apparent absorption period of MDZ, which was estimated by the delay in achieving the mean Tmax during treatment with 20 mg/kg KTZ (from 1.8 h to 3.5 h; Table 2).
The pharmacokinetic changes of MDZ by treatment with KTZ have been reported previously (Kanazu et al., 2004), in which 5 mg/kg p.o. MDZ was administered to female cynomolgus monkeys in the presence or absence of 5 mg/kg KTZ. Compared with our results (F, 6.7-fold; Table 2), there is an obvious difference in the effect of KTZ on the pharmacokinetics of MDZ, in which the oral bioavailability of MDZ was increased only 2.2-fold. There are several differences in the experimental conditions between our investigation and those performed previously (including the gender of the monkeys and the dosage of MDZ), and a definitive reason for this discrepancy is unknown. Also, because the dose-normalized AUC of MDZ without KTZ was 3.5-fold greater in the previous report compared with our current investigation, it would appear that administration of a 5-fold higher dose of MDZ might have caused the saturation of the first-pass metabolism in the intestine and/or the liver. The Michaelis-Menten constants for 1′-hydroxymidazolam formation were less than 3 μMin monkey intestinal and liver microsomes, which were comparable with human samples (data not shown). Considering the local concentration during the period of drug absorption, it may be possible for MDZ to partially escape from first-pass metabolism at the intestinal site. MDZ is employed as a typical and sensitive CYP3A probe at doses ranging from 2 to 15 mg/person in clinical DDI studies. Therefore, the suitable dosage of MDZ should be as low as possible in DDI studies using monkeys.
Unlike monkeys, it has been reported that i.v. MDZ exhibited a 5-fold increase in the AUC in humans when coadministered p.o. with three doses of 200 mg of KTZ every 12 h (Tsunoda et al., 1999). KTZ plasma concentrations in humans ranged from 0.1 to 4.9 μg/ml (0.2–9.2 μM), clearly exceeding the in vitro Ki values (Table 1) at most time points, even after correcting for the binding to plasma protein (ca. 97%) (Martinez-Jorda et al., 1990). As shown in Table 4, the plasma concentrations of KTZ in monkeys after 20 mg/kg p.o. dosing was around one-tenth of that in humans, although the monkeys were administered p.o. approximately 6-fold higher dose compared with that in humans (3.3 mg/kg, assuming that human body weight is 60 kg). It appears that the apparent species differences in the sensitivity to KTZ after i.v. dosing of MDZ are caused by the level of systemic exposure to KTZ between monkeys and humans.
It is well known that there are substantial differences in substrate specificity and inhibitor sensitivity of CYP3A across species (Ghosal et al., 1996; Eagling et al., 1998; Perloff et al., 2003). To compare the inhibitory effects of the azole antifungal agents KTZ, ITZ, and FLZ on MDZ metabolism in monkeys and humans, we conducted in vitro studies using liver and intestinal microsomes. The rank order of the inhibitory potency in monkey liver and intestinal microsomes, KTZ > ITZ > FLZ, was identical to that in human microsomes (Table 1). Moreover, the respective Ki values of the azole antifungal agents were very similar in both monkey and human samples. A number of investigators have reported the inhibitory potency of antifungal agents toward CYP3A using human liver and intestinal microsomes. Although significant differences in the Ki values have been observed in the range of 0.0037 to 0.18 μM for KTZ and 0.28 to 2.3 μM for ITZ even when using MDZ as a probe (Wrighton and Ring, 1994; von Moltke et al., 1996; Gibbs et al., 1999; Wang et al., 1999), our results were fairly consistent with the previous data. KTZ was found to be a noncompetitive inhibitor in both monkey liver and intestinal microsomes, as well as in human samples. As for ITZ and FLZ, different types of inhibition were observed between human and monkey microsomes; nevertheless, the respective Ki values of these inhibitors were nearly identical in both human and monkey microsomes. The types of inhibition with ITZ and FLZ observed in human liver microsomes are in agreement with those reported previously (Wrighton and Ring, 1994; von Moltke et al., 1996; Gibbs et al., 1999; Wang et al., 1999; Isoherranen et al., 2004). In humans, CYP3A4 is a predominant isoform that accounts for more than 30% of the total P450 content in the liver (Shimada et al., 1994) and 70% in the intestine (McKinnon et al., 1995). In cynomolgus monkeys, CYP3A8, which exhibits more than 90% amino acid sequence identity to CYP3A4, is the only reported CYP3A isoform and has been shown to be the predominant form of the CYP3A subfamily expressed in the liver (Komori et al., 1992). It has been also reported that P450 MI-2 and MI-3, belonging to the CYP4F and CYP3A subfamilies, respectively, are expressed in cynomolgus monkey intestine (Hashizume et al., 2001). However, information regarding the enzymatic characteristics of the P450 isoforms present in monkey intestine and liver is scarce, and further detailed experiments are necessary to accurately extrapolate DDI related to P450 from monkeys to humans.

Effect of p.o. coadministration of KTZ (•, 0 mg/kg; ▪, 5 mg/kg; ▴, 20 mg/kg) on the plasma concentration time profiles of FEX after i.v. (A) and p.o. (B) administration to monkeys at doses of 1 and 5 mg/kg, respectively. Each point represents the mean ± S.D. of data obtained from four monkeys.
FEX has been considered to be a suitable in vivo probe to investigate the role of P-gp-mediated transport and has been subjected to a number of DDI studies (Hamman et al., 2001; Drescher et al., 2002; Dresser et al., 2003; Wang et al., 2002; Kharasch et al., 2005; Yasui-Furukori et al., 2005). These clinical reports have suggested that intestinal P-gp was responsible for these interactions. By contrast, Tannergren et al. (2003a,b) have shown that concomitant administration of verapamil or KTZ did not increase the effective jejunal permeability of FEX using a human in vivo jejunal perfusion technique, suggesting that transporters related to the first-pass liver extraction might be inhibited instead of the intestinal transporters. As such, there are conflicting views regarding the target organ responsible for the DDI of FEX. Because FEX was not significantly metabolized in monkey liver and intestinal microsomes, as with human samples (data not shown), the urinary and/or biliary excretion must also govern the overall disposition of the drug in monkeys. When FEX was coadministered p.o. with KTZ (5 mg/kg), the Cmax and AUC increased significantly by approximately 2-fold without prolongation of the t1/2 (Fig. 5B; Table 3), although the number of animals used in this study might be insufficient to discern a slight difference in the pharmacokinetics of FEX after coadministration with KTZ. On the other hand, none of the parameters, including CLtot and Vdss, were affected when FEX was administered i.v. with KTZ, even at a p.o. dose of 20 mg/kg (Fig. 5A; Table 3). These results suggest that the DDI between FEX and KTZ is based on alteration of the intestinal transport, probably through inhibition of P-gp. The mechanisms for the DDI related to FEX appear to be complex, as multiple transport proteins such as organic anion transporting polypeptides (OATP) may also be involved in the intestinal absorption of FEX (Cvetkovic et al., 1999; Dresser et al., 2002; Nozawa et al., 2004). In fact, it has been reported that grapefruit juice decreased the oral bioavailability of FEX through OATP inhibition in the intestine (Dresser et al., 2002, 2005). Furthermore, KTZ has also been shown to inhibit OATP, as well as P-gp (Cvetkovic et al., 1999; Yasuda et al., 2002; Petri et al., 2004). These results raise the possibility that combined inhibition of both P-gp and OATP may contribute toward the intestinal DDI between FEX and KTZ. In fact, the concomitant administration of 20 mg/kg KTZ tended to affect the absorption rate of FEX with a prolonged Tmax from 1.8 ± 0.5 to 4.0 ± 2.8 h (p = 0.19; Table 3).
Interestingly, the extent of DDI in monkeys is quantitatively consistent with the previous report in humans, in which coadministration of 120 mg of FEX twice daily with 400 mg of KTZ once daily resulted in substantial increases in steady-state FEX plasma levels (Cmax, 2.4-fold; AUC, 2.6-fold) (Davit et al., 1999). This is also comparable with those shown with other P-gp inhibitors verapamil (Cmax, 2.9-fold; AUC, 2.5-fold) and ITZ (Cmax, 1.9-fold; AUC, 2.7-fold) (Yasui-Furukori et al., 2005; Shimizu et al., 2006). In addition, in all these DDI studies with FEX in humans, the t1/2 values were not altered, consistent with our monkey studies. P-gp knockout mice are widely used as an animal model for in vivo investigations regarding P-gp function. However, the increase of intestinal absorption observed in P-gp knockout mice (6.5-fold in the case of FEX) was significantly greater than that in monkeys and humans (Tahara et al., 2005). This may be explained by incomplete inhibition of P-gp activity by KTZ, opposing effects of additional inhibition of OATP, and/or species differences in P-gp expression/activity in the intestine. A recent study using P-gp ATPase assays indicated that the binding affinities of 21 P-gp substrates to rhesus monkey P-gp were much closer to those of human P-gp than beagle dog P-gp (r2 = 0.83 versus r2 = 0.36) (Xia et al., 2006). Furthermore, it has been reported that the renal DDI between famotidine and probenecid via an organic anion transporter (namely, OAT3) in humans could be reproduced in cynomolgus monkeys but not in rats (Tahara et al., 2006). Consequently, monkeys may be also used to predict DDI involving drug transporters, such as P-gp.
In conclusion, we have investigated the barrier functions of CYP3A and P-gp in monkey intestine and the reproducibility of DDI in human intestine using MDZ as a CYP3A probe and FEX as a P-gp probe. We found that the oral bioavailability of FEX, along with that of MDZ, was considerably lower in monkeys and that both intestinal availabilities were significantly enhanced by a concomitant p.o. dose of KTZ, a prototypic dual CYP3A/P-gp inhibitor. Because these results were comparable with those in humans, in vivo studies in monkeys might provide a sensitive model for the prediction of intestinal DDI in humans. To confirm the availability of monkeys in DDI studies, our ongoing studies are focused on the effects of mechanism-based inhibitors, such as erythromycin and verapamil, which likely cause a clinically relevant DDI in the intestine rather than the liver.
Acknowledgments
We thank Dr. Toru Ishizuka, Director of Exploratory Toxicology and DMPK Research Laboratories, for his interest and encouragement. We also thank Hiroshi Ito, Megumi Hiraga, Takashi Sasaki, and Yutaka Fujisawa for technical assistance.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.011288.
-
ABBREVIATIONS: DDI, drug-drug interaction(s); KTZ, ketoconazole; P450, cytochrome P450; MDZ, midazolam; P-gp, P-glycoprotein; FEX, fexofenadine; ITZ, itraconazole; FLZ, fluconazole; HPLC, high-performance liquid chromatography; AUC, area under the plasma concentrationtime curve; CLtot, total body clearance; Vdss, distribution volume at steady state; OATP, organic anion transporting polypeptide.
- Received June 3, 2006.
- Accepted November 29, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}