


Abstract
Kinetic parameters (kinact and KI) obtained in microsomes are often used to predict time-dependent inactivation. We previously reported that microsomal inactivation kinetic parameters of diltiazem underpredicted CYP3A inactivation in hepatocytes. In this study, we evaluated the contributions of inactivation and reversible inhibition of CYP3A by diltiazem and its N-desmethyl (MA) and N,N-didesmethyl (MD) metabolites. In human liver microsomes, MA was a more potent time-dependent inactivator of CYP3A than its parent drug, with apparent kinact approximately 4-fold higher than that of diltiazem at a microsomal protein concentration of 0.2 mg/ml. MD did not inactivate CYP3A. Inactivation of CYP3A by diltiazem was dependent on microsomal protein concentration (25, 36, and 41% decrease in CYP3A activity at 0.2, 0.4, and 0.8 mg/ml microsomal protein, respectively, incubated with 10 μM diltiazem over 20 min), whereas inactivation by MA did not seem to be protein concentration-dependent. MA and MD were reversible inhibitors of CYP3A with competitive Ki values of 2.7 and 0.2 μM, respectively. In cryopreserved hepatocytes incubated with diltiazem, time-dependent loss of CYP3A was accompanied by increased formation of MA and MD, with the MA level similar to its KI at higher diltiazem concentrations. In addition, the metabolites appeared to be accumulated inside the cells. In summary, time-dependent CYP3A inactivation by MA seems to be the major contributor responsible for the loss of CYP3A in human liver microsomes and human hepatocytes incubated with diltiazem. These findings suggest that prediction of CYP3A loss based solely on microsomal inactivation parameters of parent drug may be inadequate.
The calcium channel blocker diltiazem is extensively metabolized via multiple pathways including N-demethylation, O-demethylation, and deacetylation (Fig. 1). The demethylation pathways are mediated by cytochrome P450 enzymes primarily in the liver, whereas deacetylation is believed to be carried out by esterases in multiple tissues (Homsy et al., 1995; Sutton et al., 1997). N-Desmethyl diltiazem (MA) is the major metabolite that undergoes further N-demethylation process to form N,N-didesmethyl diltiazem (MD). It has been shown that diltiazem caused clinically significant drug-drug interactions by decreasing the elimination of substrates through inhibition of CYP3A (Jones and Morris, 2002; Jerling et al., 2005). The cause of inhibition has been attributed to both diltiazem and its metabolites. In human liver microsomes (HLMs), MA and MD appeared to competitively inhibit testosterone 6β-hydroxylation with inhibition constants (Ki) of approximately 2 and 0.1 μM (Sutton et al., 1997). However, reversible inhibition may not sufficiently explain the observed drug interactions given that plasma concentrations of diltiazem and its metabolites are lower than their respective Ki values. Time-dependent inactivation (TDI) was also observed in HLMs for both diltiazem (Jones et al., 1999; Dai et al., 2003) and MA (Mathew et al., 2000) and has been suggested to be responsible for the clinically relevant drug interactions. To date, comprehensive study has not been performed to elucidate the contribution of different mechanisms (inhibition versus inactivation) by diltiazem and its oxidative metabolites to diltiazem-induced pharmacokinetic drug interaction.
In a previous report, we evaluated time-dependent CYP3A inactivation by compounds exhibiting this property in cryopreserved human hepatocytes (Zhao et al., 2005). Hepatocytes can serve as an intact system to test the predictability of parameters obtained in HLMs, a much simpler system. A large discrepancy was found to exist between the inactivation predicted by directly applying HLM-generated parameters and that observed in hepatocytes. The discrepancy between the two in vitro systems that differ greatly in complexity suggests that there is some risk of misinterpretation of in vivo outcomes predicted from HLM parameters. Among the inactivators tested, diltiazem appeared to be more potent in hepatocytes than expected from the microsomal kinetic parameters. The discrepancy probably is not due to nonspecific binding, metabolic consumption, or efflux of inhibitor, since these factors result in less potent inhibition in hepatocytes (Zhao et al., 2005).

Metabolic scheme of diltiazem to MA and MD.
The objectives of this study were 2-fold: 1) to reassess the inhibition and inactivation of CYP3A by diltiazem, MA, and MD and identify the major contributor(s) of diltiazem-induced loss of CYP3A, and 2) to evaluate the impact of sequential metabolism on the prediction of drug interaction using microsomal parameters obtained for parent compound.
Materials and Methods
Materials. Cryopreserved human hepatocytes and hepatocyte thawing medium were purchased from In Vitro Technologies (Baltimore, MD). Pooled human liver microsomes from 59 individuals were obtained from a Pfizer Global Research and Development (a division of Pfizer Inc.) in-house supply. HEPES was purchased from Invitrogen (Carlsbad, CA). MA and MD were generous gifts from Tanabe Seiyaku Co., Ltd. (Osaka, Japan). All other reagents were obtained from Sigma (St. Louis, MO) unless otherwise stated.
Microsomal Studies.TDI of CYP3A in HLMs. The main body of this study used 1′-hydroxylation of midazolam as the probe reaction for CYP3A in both HLMs and cryopreserved human hepatocytes. TDI by MA in HLMs was performed according to methods described elsewhere (Ito et al., 1998). In brief, the inactivator (MA) was incubated at varying concentrations with HLMs in potassium phosphate buffer (0.1 M, pH 7.4) at 37°C for 5 min before the addition of NADPH. The final concentrations of microsomal protein and NADPH were 0.2, 0.4, or 0.8 mg/ml and 1 mM, respectively. The nominal concentrations of MA in these incubations were 0.25, 0.5, 1, 2, and 4 μM. The experiments were performed in triplicate. At different times after the addition of NADPH (0, 2, 5, and 10 min), 10 μl of the inactivator/microsome mixture (primary incubation) were transferred to 90 μl of potassium phosphate buffer containing 100 μM midazolam and 1 mM NADPH, which had been preincubated at 37°C for 5 min. This resulted in a 10-fold dilution of inactivator in the incubation with midazolam. The mixture was incubated for 1 min and was terminated by the addition of 100 μl of a mixture of acetonitrile/methanol (3:1 v/v) containing alprazolam as internal standard. In parallel, the primary incubation samples at time 0 and at different incubation time points after the addition of NADPH were quenched with acetonitrile/methanol (3:1 v/v) containing alprazolam to evaluate the metabolic consumption of MA over time during the primary incubation.
The effect of microsomal protein concentration on TDI of CYP3A by diltiazem was evaluated by incubating 10 μM diltiazem with 0.2, 0.4, or 0.8 mg/ml microsomal protein. These protein concentrations were the same as those used in the primary incubations with MA. At different times after the addition of NADPH (0, 2, 6, 12, and 20 min), 10 μl of the primary incubation was transferred to 90 μl of potassium phosphate buffer containing 100 μM midazolam and 1 mM NADPH, which had been preincubated at 37°C for 5 min, for the analysis of CYP3A activity. To assess the metabolism of diltiazem during the primary incubation, a separate set of samples was quenched with acetonitrile/methanol (3:1 v/v) containing alprazolam as internal standard for the analyses of diltiazem, MA, and MD.
Since MD is the end product of the N-demethylation processes of diltiazem, dependence of microsomal protein concentration of MD-induced TDI of CYP3A was not evaluated. TDI of CYP3A by MD was measured by incubation of 1, 2, and 4 μM MD with 0.2 mg/ml HLM, the lowest concentration used in the TDI studies with MA and diltiazem. Using the dilution method, as described above, aliquots (10 μl) were taken from the primary incubation at 0 and 12 min.
Reversible inhibition of CYP3A by diltiazem, MA, and MD. A reversible inhibition experiment was conducted under conditions similar to those of the midazolam incubations mentioned above for TDI experiments. Diltiazem, MA, or MD was coincubated with midazolam and HLMs in potassium phosphate buffer (0.1 M, pH 7.4) at 37°C for 5 min before the addition of NADPH. The final concentrations were: 0.02 mg/ml HLM protein, 1 mM NADPH, and varying concentrations of inhibitors (diltiazem: 0.5, 1, 2, and 4 μM; MA: 0.5, 1, 2, and 4 μM; and MD: 0.25, 0.5, 1, and 2 μM) and substrate (midazolam: 1.25, 2.5, 5, and 10 μM). The 0.02 mg/ml HLMs matched the protein concentration used in the secondary incubation of the TDI studies (with 0.2 mg/ml HLMs in primary incubation). This concentration was chosen to minimize nonspecific binding and substrate/inhibitor depletion. The experiments were performed in triplicate. Each reaction lasted for 1 min and was terminated by the addition of 100 μl of a mixture of acetonitrile/methanol (3:1 v/v) containing alprazolam as internal standard.
Kinetics of diltiazem and MA metabolism and the effect of ketoconazole. Formation kinetics of MA and MD was determined in HLMs using diltiazem and MA, respectively, as substrates. The incubation mixture contained 0.2 mg/ml HLM protein, 1 mM NADPH, and varying concentrations of substrates: 6.25, 12.5, 25, 50, 100, and 200 μM diltiazem or 0.8, 1.6, 3.1, 6.3, and 12.5 μM MA. The microsomal concentration of 0.2 mg/ml matched the lowest concentration used in primary incubation of the TDI studies to ensure minimal substrate depletion and nonspecific binding. Substrate and HLM protein were preincubated in potassium phosphate buffer (0.1 M, pH 7.4) at 37°C for 5 min before the addition of NADPH. The effect of CYP3A inhibitor ketoconazole on diltiazem metabolite formation was assessed by coincubating ketoconazole (1 μM) and substrate (10 μM diltiazem or 5 μM MA) for 5 min at 37°C before the addition of NADPH. The experiments were performed in duplicate. The reactions were stopped at 0, 0.5, 1, and 2 min by the addition of an equal volume of a mixture of acetonitrile/methanol (3:1 v/v) containing alprazolam as internal standard. The linear portion of the time course of metabolite formation was used to obtain reaction velocity.
Microsomal binding of MA and MD. Microsomal protein binding was determined using equilibrium dialysis as described previously (Obach, 1997; Banker et al., 2003). A multiwell Teflon dialysis apparatus was manufactured at Pfizer Global Research and Development and the dialysis membrane (molecular mass cutoff 12–14 kDa) was purchased from Spectrum Medical Industries (Los Angeles, CA). An aliquot (150 μl, n = 3) of a mixture containing HLMs (0.2, 0.4, and 0.8 mg/ml in 0.1 M potassium phosphate buffer, primary incubation condition for TDI studies) and inactivator (nominal concentrations of 0.25, 0.5, 1, 2, and 4 μM for MA or 0.4 and 4 μM for MD) was placed on one side of the membrane and blank phosphate buffer was added to the other side. The dialysis chamber was maintained at 37°C for 6 h. Samples were quenched with an equal volume of a mixture of acetonitrile/methanol (3:1 v/v) containing alprazolam as internal standard. Sample extraction and analytical methods for the measurement of inactivators and 1-hydroxymidazolam are described below.
Hepatocyte Studies.Loss of CYP3A activity in cryopreserved human hepatocytes incubated with diltiazem. Hepatocyte study was performed using the same pool of donors (three males and two females) and the same conditions as described previously (Zhao et al., 2005). In brief, hepatocytes were thawed in thawing medium (25 ml per 5 million hepatocytes) and centrifuged at 50g at room temperature for 5 min. The cell pellet was reconstituted in 30 ml of incubation medium (Williams' Medium E containing 10 mM HEPES) and centrifuged at 50g at room temperature for 5 min. The resulting cell pellet was reconstituted in 30 ml of fresh incubation medium and cell viability was determined by the trypan blue method (generally between 78 and 82% viability). Cell suspension was placed in a 37°C incubator supplemented with 5% CO2 before use.
Hepatocytes were preincubated in the presence of different concentrations of diltiazem (0.5, 1, 2, 4, 10, and 20 μM) in a 48-well plate for 1 h in a 37°C incubator under 5% CO2. The final volume was 250 μl and the final cell concentration was 0.5 × 106 viable cells/ml. The experiments were performed in triplicate. At the end of preincubation, 200-μl aliquots of the suspension were transferred to 96-well plate microtubes (1.2 ml) and centrifuged at 50g for 5 min at room temperature. One hundred fifty microliters of each supernatant were discarded and the pellets were resuspended after the addition of 150 μl of fresh incubation medium for washing. The suspension was further centrifuged at 50g for 5 min at room temperature and 180 μl of each supernatant were discarded. The postwash pellet was resuspended in 100 μl of fresh incubation medium containing midazolam. At this point, the concentrations were approximately 1 × 106 cells/ml for hepatocytes and 100 μM for midazolam. The suspensions were immediately placed in the incubator (37°C, 5% CO2) and were incubated for 9 min before the addition of 2× volume of a mixture of acetonitrile/methanol (3:1 v/v) containing alprazolam as internal standard. The use of 100 μM midazolam (to ensure saturation condition) and the choice of 9-min incubation time were confirmed by preliminary kinetic studies.
MA and MD Formation in Hepatocytes Incubated with Diltiazem. Free drug concentrations of diltiazem, MA, and MD in hepatocyte suspension (Iu) were believed to be equivalent to concentrations in hepatocyte medium since no protein was added to the medium. Hepatocyte suspensions (incubated at 37°C for 0, 15, 40, and 60 min) were centrifuged at 50g for 5 min to obtain supernatant for the quantitation of Iu. Samples were quenched with an equal volume of acetonitrile/methanol (3:1 v/v) containing alprazolam as internal standard. In parallel, an aliquot of hepatocyte suspension was quenched with an equal volume of acetonitrile/methanol (3:1 v/v) containing alprazolam for the quantitation of total drug concentrations.
LC-MS/MS Methods. The supernatant from samples extracted by organic solvent was analyzed for diltiazem and its metabolites using a Micromass Quattro Ultima mass spectrometer (Waters Corp., Milford, MA) equipped with an Agilent 1100 high-performance liquid chromatography system (Agilent Technologies, Palo Alto, CA) and a LEAP autosampler (LEAP Technologies, Carrboro, NC). Chromatographic separation was performed on a Zorbax C18 column (2 × 50 mm, 5 μm; Agilent Technologies), in line with a Keystone Javelin C18 guard column (2 × 20 mm; Western Analytical Products, Inc., Murrieta, CA), at a flow rate of 0.2 to 0.3 ml/min using a linear gradient elution of A, 50 mM ammonium acetate in water; and B, acetonitrile/methanol (89:11 v/v). Table 1 shows the analytical parameters for each analyte. The analysis of 1-hydroxymidazolam was performed using a Micromass Quattro LC mass spectrometer (Quattro II) equipped with a Waters 2790 high-performance liquid chromatography system (Waters Corp.) as described previously (Zhao et al., 2005).
LC-MS/MS analytical parameters of diltiazem and metabolites
Data Analysis.Microsomal protein binding and correction of metabolic consumption. The percentage of inactivator/inhibitor unbound (percentage free) in HLMs was calculated as the ratio of free concentration to total concentration at equilibrium in the dialysis experiment. Mean percentage of MA remaining at each time point (from three experiments) of the primary incubation was plotted versus time and the area under the percentage remaining-time curve from time 0 to the last time point was calculated using a linear trapezoidal method by Microsoft Excel software (Microsoft Corp., Redmond, WA). A time-averaged factor was then calculated by dividing the area by time (zero to last time of primary incubation) to account for the metabolic consumption. Free MA concentrations in microsomal TDI studies were corrected for both precentage of free and metabolic consumption ([I]u,ave).
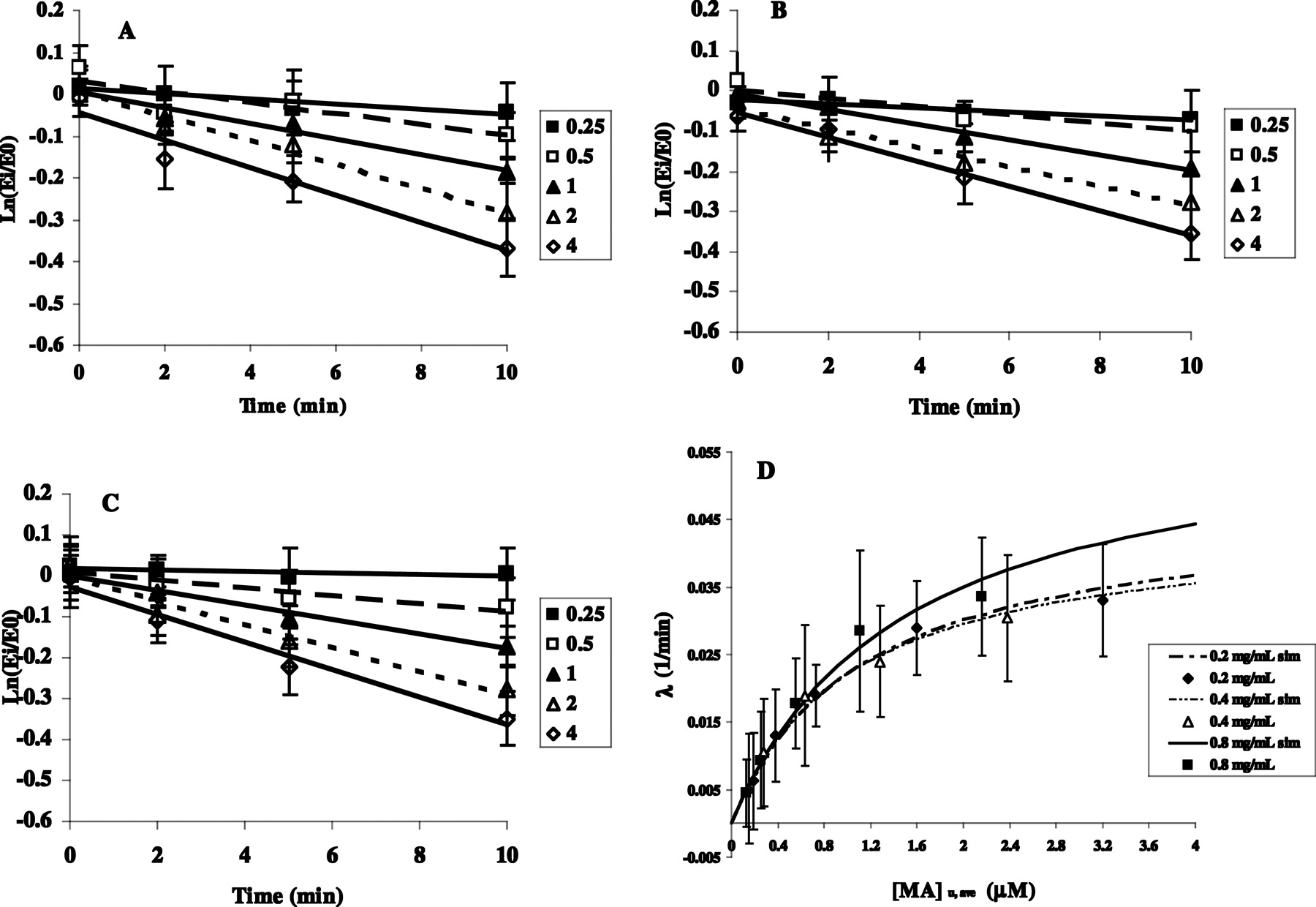
TDI of CYP3A in HLMs. Inactivation parameters in HLMs were obtained by plotting the logarithm of the ratio of the remaining enzyme activity in the presence of inactivator to the activity in the absence of inactivator versus the preincubation time (Fig. 2, A–C). The apparent inactivation rate constant (λ) was determined from the slope of the initial linear phase on the semi logarithmic plots at each nominal inactivator concentration. Maximum inactivation rate constant (kinact) and unbound inactivation constant (KI,u,ave) were obtained by nonlinear regression of the [I]u,ave versus λ data using eq. 1. Data fitting was performed by the numerical module of SAAM II software (Version 1.2.1; SAAM Institute, Seattle, WA). A fractional standard deviation of 10% was assigned to each datum. 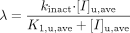 The predicted remaining CYP3A activity in hepatocytes or HLMs was then calculated as a fraction of control without inactivator by eq. 2:
The predicted remaining CYP3A activity in hepatocytes or HLMs was then calculated as a fraction of control without inactivator by eq. 2: 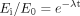 where Ei and E0 are CYP3A activities at specific primary incubation time point and time 0, respectively.
where Ei and E0 are CYP3A activities at specific primary incubation time point and time 0, respectively.
Reversible inhibition by MA and MD in HLMs. Competitive inhibition was assumed to be the mechanism of reversible inhibition of CYP3A by MA and MD (Sutton et al., 1997), which was further confirmed by Dixon plots of the inhibition data from this study (data not shown). Inhibition constant (Ki) values and the Michaelis-Menten kinetic parameters for 1-hydroxymidazolam formation (Km and Vmax) were obtained simultaneously by fitting the 1′-hydroxymidazolam formation velocity (v) data according to eq. 3. Because the microsomal concentration (0.02 mg/ml) used in the reversible inhibition experiment was low and the reaction time was kept short, no correction of [I] regarding nonspecific binding and depletion was performed. Data fitting was performed by nonlinear regression analysis using the numerical module of SAAM II software. A fractional standard deviation of 10% was assigned to each datum. 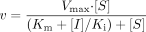

TDI of CYP3A in HLMs by MA. A to C, HLM protein concentration of 0.2, 0.4, and 0.8 mg/ml, respectively; legends show the nominal concentration of MA. Data are mean ± S.D. (n = 3 individual experiments). D, apparent inactivation rate constant (λ) versus [MA]u,ave plot. Lines represent simulations according to eq. 1 and legends show microsomal protein concentrations.
Michaelis-Menten kinetics of diltiazem metabolite formation in HLMs. Metabolite formation velocity was plotted against v/[S] (Eadie-Hofstee plot) for each reaction. In general, each reaction was well described by a simple one-enzyme Michaelis-Menten model (data not shown). Parameters (Km and Vmax) of MA or MD formation were determined by nonlinear regression analysis using the numerical module of SAAM II software eq. 4. A fractional standard deviation of 10% was assigned to each datum. 
Results
TDI of CYP3A by MA and MD in HLMs. MA caused concentration- and time-dependent inactivation of CYP3A (Fig. 2, A–C). To ensure a fair comparison of parameters across varying microsomal protein concentrations, inactivator concentrations were corrected for microsomal binding and metabolic consumption during the primary incubation (Table 2) to obtain [I]u,ave. Thus, the reported KI values for MA are unbound KI (KI,u,ave; Fig. 2D). Inactivation kinetic parameters for MA measured at different microsomal protein concentrations (0.2–0.8 mg/ml) are summarized in Table 3. Microsomal protein concentration seems to have little effect on the TDI parameters. The values for KI,u,ave ranged from 1.0 to 1.5 μM and kinact values from 0.045 to 0.061 min–1. Table 3 also includes parameters obtained from diltiazem at 0.2 mg/ml HLMs for comparison. The maximum rate of inactivation by MA is approximately 4 times faster than that of the parent drug (kinact of 0.045–0.061 min–1 for MA versus 0.012 min–1 for diltiazem). MD did not exhibit measurable time-dependent CYP3A inactivation (Table 4). Microsomal binding and metabolic consumption of MD were minimal.
Time-averaged free concentration ([I]u,ave) of MA in liver microsomes
Time-dependent CYP3A inactivation in HLMs by MA
Lack of time-dependent CYP3A inactivation by MD
Reversible Inhibition of CYP3A in HLMs. Reversible inhibition by diltiazem, MA, and MD was evaluated in HLMs under the conditions for which TDI was evaluated. MA and MD caused concentration-dependent inhibition of CYP3A. The estimated competitive Ki values are summarized in Table 5 and agree with the literature in general (Sutton et al., 1997). MD is approximately 10 times more potent than MA. The percentage free at 0.2 mg/ml HLM protein concentration was near unity for both MA and MD (Tables 2 and 4). Therefore, the competitive Ki values of MA and MD do not require further correction for metabolic consumption and microsomal binding at a 10-fold lower protein concentration (0.02 mg/ml) used in reversible inhibition experiment. The Michaelis-Menten kinetic parameters for 1′-hydroxymidazolam formation (Km and Vmax) were simultaneously fitted with Ki according to eq. 3 for the experiments using MA or MD as inhibitor. The values of parameter estimate (standard error) for Km are 2.4 (0.2) and 2.2 (0.2) μM, and for Vmax, 5.0 (0.1) and 4.9 (0.2) nmol 1′-hydroxymidazolam/min/mg protein from the experiments with MA and MD, respectively. The Km values are similar to that reported by Walsky and Obach (2004), which was approximately 2 μM. As expected from literature reports (Sutton et al., 1997), diltiazem caused minimal inhibition of CYP3A under our experimental conditions (data not shown).
Parameters of reversible inhibition of CYP3A in HLMs by MA and MD
Results from this study are estimates (standard error) obtained using SAAM II software.
Diltiazem-Induced TDI of CYP3A and Sequential Metabolism in HLM: Effect of Microsomal Protein Concentration.Figure 3A shows the time-dependent loss of CYP3A activities in HLMs incubated with 10 μM diltiazem at varying concentrations of microsomal protein (0.2, 0.4, and 0.8 mg/ml). The observed inactivation of CYP3A appears to be dependent on microsomal protein concentrations. Figure 3, B to D, shows the corresponding profiles of parent drug loss (percentage remaining versus time) and metabolite formation (concentration versus time) during the first incubation at varying microsomal protein concentrations. At the end of 20 min, the percentage remaining of diltiazem was protein concentration-dependent (86 ± 5%, 73 ± 12%, and 65 ± 4% at 0.2, 0.4, and 0.8 mg/ml protein, respectively), which corresponds to the formation of sequential metabolites: the higher the protein concentration, the more the metabolites were formed.
CYP3A DependentN-Demethylation of Diltiazem and MA. Michaelis-Menten parameters for MA and MD formation from diltiazem and MA are shown in Table 6. The apparent Km and Vmax are 54 μM and 1881 pmol/min/mg for MA formation, and 19 μM and 385 pmol/min/mg for MD formation from MA, respectively. Sutton et al. (1997) have reported a Km of 23 μM and Vmax ranging from 1.0 to 5.4 nmol MA/min/mg in HLMs from three individuals. The apparent intrinsic clearance (Vmax/Km) for the formation of MA from diltiazem appears to be greater than that for MD formation from MA (Table 6). A classic CYP3A inhibitor, ketoconazole (1 μM), caused more than 90% inhibition of each N-demethylation process.
Michaelis-Menten kinetic parameters of metabolite formation from diltiazem and MA in HLMs
Results are parameter estimates (standard error) obtained by SAAM II software assuming one enzyme reaction.
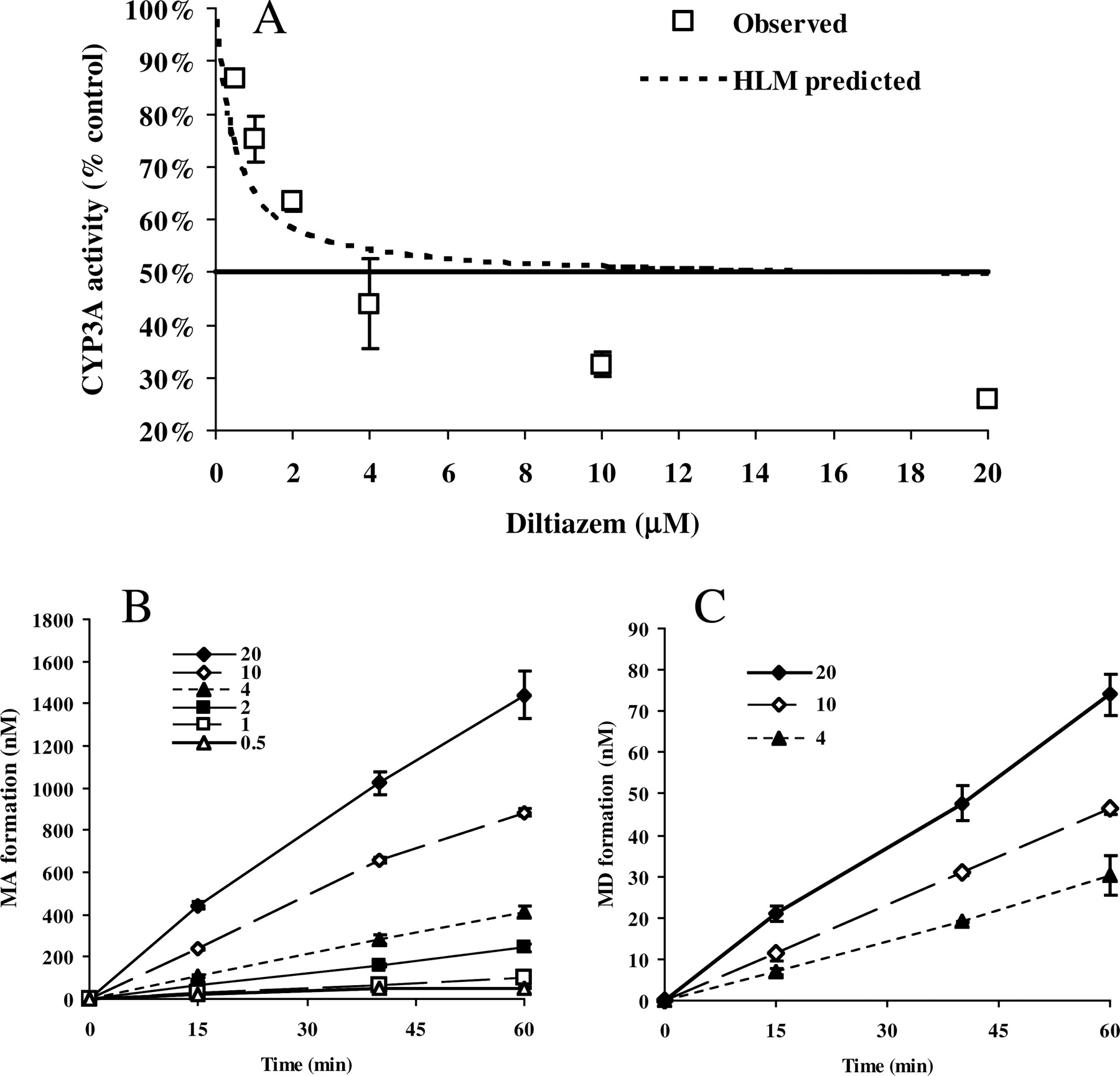
CYP3A Inactivation by Diltiazem in Hepatocytes.Figure 4A shows the loss of CYP3A activity in cryopreserved human hepatocytes incubated with varying concentrations of diltiazem for 60 min. The activity study was performed in hepatocytes washed with fresh medium (at least 20 times dilution; see Materials and Methods). Figure 4, B and C, show the MA and MD formation in hepatocytes incubated with diltiazem. MD formation at diltiazem concentrations of 0.5, 1, and 2 μM is not reported because nothing was observed at the given detection limits. At 10 and 20 μM diltiazem, MA concentration increased to 0.88 and 1.44 μM after 60 min, which approached its microsomal KI,u,ave and Ki values (Tables 3 and 5). At 20 μM diltiazem, the MD level reached 74 nM after 60 min, which is approximately 30% of its competitive Ki value of 0.2 μM (Table 5). Washing with medium will result in at least 20 times dilution, making MA and MD levels in the medium of activity experiments significantly below their competitive Ki values (2.7 and 0.2 μM for MA and MD, respectively; Table 5).
Table 7 summarizes the free fraction of inhibitor in hepatocyte incubation (fc), which is calculated by dividing free concentration in incubation medium (Iu) by total concentration. The fc values for diltiazem are near unity, whereas low recovery was apparent for MA and MD (fc from 0.50 to 0.66), suggesting the possible trapping of both metabolites inside the cells upon formation. Therefore, intracellular concentrations of diltiazem metabolites are expected to be higher than the observed total concentrations in Fig. 4, B and C.
Free fraction (fc) of diltiazem and its sequential metabolites in hepatocyte incubations over time
Free fraction was calculated as the ratio of supernatant concentration to total concentration, mean (SD), n = 3 individual incubations.
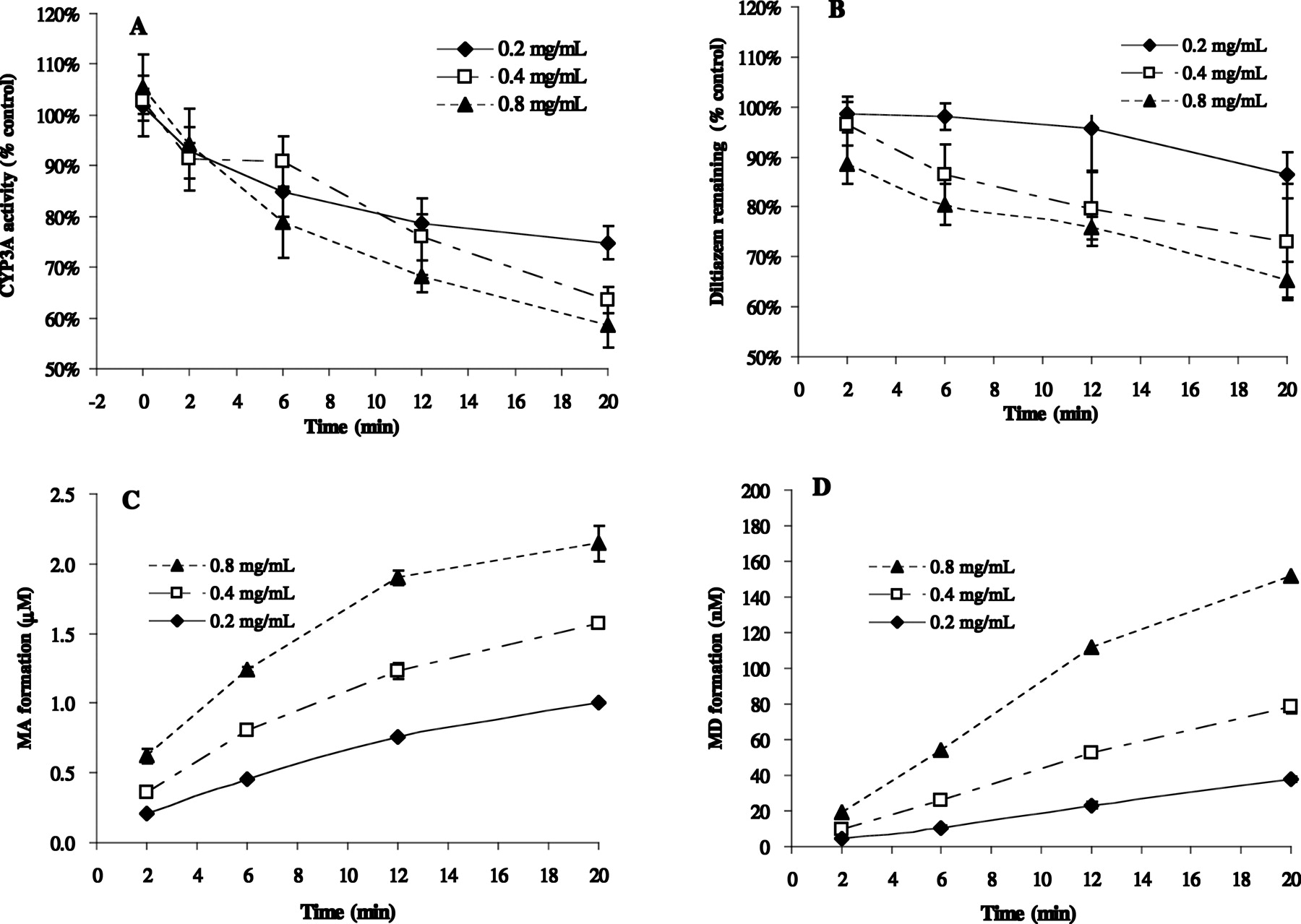
Loss of CYP3A activity (A), metabolic consumption of diltiazem (B), and metabolite formation (C and D) upon incubation of HLMs with 10 μM diltiazem. Legends show microsomal protein concentration. Data are mean ± S.D. (n = 4–5 individual experiments for A and B, n = 3–5 individual experiments for C and D).
Discussion
Identification of the Mechanism Responsible for Diltiazem-Induced Loss of CYP3A. The results of the present study implicate TDI by MA as the major contributor responsible for diltiazem-induced loss of CYP3A. This conclusion is supported by the following findings: 1) the primary metabolite of diltiazem, MA, is a more potent time-dependent CYP3A inactivator than its parent drug; 2) the dependence of enzyme inactivation on microsomal protein concentrations for diltiazem, but not for MA, suggests a significant contribution of this sequentially formed primary metabolite to in vivo drug interaction formerly attributed to the parent compound; and 3) among the different mechanisms induced by diltiazem and its sequential metabolites MA and MD, time-dependent CYP3A inactivation by MA seems to be the major contributor responsible for diltiazem-induced loss of CYP3A in HLMs and human hepatocytes.
Inhibitory metabolites have been shown to play an additional role of inhibiting enzymes other than the parent compound. Isoherranen et al. (2004) have demonstrated that itraconazole metabolites are equally or more potent CYP3A4 inhibitors compared with the parent drug in HLMs. Consequently, these inhibitory species may contribute to the inhibition of CYP3A4 observed in vivo after administration of itraconazole. Similarly, this report demonstrates that sequential metabolites of a time-dependent inactivator can significantly contribute to the observed drug interaction.
Inactivation of CYP3A by diltiazem and MA results from the formation of a metabolite intermediate complex between ferrous prosthetic heme iron and a highly oxidized nitroso metabolite (Jones et al., 1999; Mayhew et al., 2000). Because MD is not a TDI of CYP3A (Table 4), we hypothesize that MA causes mechanism-based inactivation by forming reactive intermediate that modifies the enzyme upon formation. In addition, MA and MD have been reported to be competitive inhibitors of CYP3A (Sutton et al., 1997). It is generally believed that drug interaction arising from coadministration of diltiazem with CYP3A substrates in vivo is mainly due to enzyme inactivation rather than reversible inhibition by diltiazem and its metabolites. This conclusion is supported by the facts that the observed free plasma concentrations of MA are much lower than its Ki (Mayhew et al., 2000), and plasma levels of MD appear to be negligible (Molden et al., 2003). In addition, a prolonged half-life of diltiazem was observed in hypertension patients taking multiple doses (Montamat and Abernethy, 1987), consistent with a time-dependent auto-inactivation of diltiazem metabolism. In fact, CYP3A is the major isoform responsible for N-demethylation reactions of diltiazem (Sutton et al., 1997). The inhibiting effect of ketoconazole in this study further confirmed the role of CYP3A in N-demethylation pathways for diltiazem (Table 6). In this report, we comprehensively assessed the contribution of different mechanisms to the loss of CYP3A activity by conducting microsomal studies to obtain inhibition/inactivation parameters of the sequential N-desmethyl metabolites of diltiazem. Most of the parameters we have obtained agree with literature reports. In depth evaluation of each mechanism revealed the significance of each mechanism of inactivation/inhibition by both parent drug and metabolites on diltiazem-induced loss of CYP3A. Among different mechanisms, TDI by MA appears to be the major contributor, which is further supported by results from hepatocyte experiments.

A, effect of diltiazem incubation on CYP3A activity in cryopreserved human hepatocytes at the end of 1-h incubation. Open square, observed CYP3A activities (mean ± S.D., n = 3–4 individual experiments); dotted line, remaining CYP3A activity predicted using microsomal TDI parameters of diltiazem according to eq. 2. B and C, time courses of total MA formation (B) and MD formation (C) in hepatocytes incubated with diltiazem. (Legends show nominal diltiazem concentrations.)
Hepatocyte is a more integrated system than liver microsomes and has been used to predict metabolic clearance and drug interactions in vivo in pharmaceutical industries (McGinnity et al., 2006; Lu et al., 2006, 2007). Given its unique position along the hierarchy between liver microsomes and in vivo, hepatocytes can be used to test the predictability of parameters obtained using liver microsomes. We have demonstrated that the discrepancies between the predicted inactivation using microsomal kinetic parameters and those observed in hepatocytes can be partially explained by factors governing the accessibility of inactivators to CYP3A in hepatocytes (Zhao et al., 2005). These factors include, but are not limited to, nonspecific binding, metabolic consumption, and active transport. Results from this work demonstrate that sequentially formed metabolites are another important factor to consider when discrepancies are observed between HLMs and hepatocytes.
In hepatocytes incubated with 10 and 20 μM diltiazem, MA concentration increased to 0.88 and 1.44 μM, respectively, after 60 min (Fig. 4B), concentrations approaching its microsomal KI,u,ave and Ki values (Tables 3 and 5). At 20 μM diltiazem, MD concentration was 74 nM at 60 min (Fig. 4C), which is approximately 30% of its microsomal competitive Ki value (Table 5). The values of fc are lower than unity for both MA and MD (Table 7), implicating possible entrapment of metabolites inside the cells upon formation. Preliminary calculation of hepatocyte volume in the initial incubation (primary incubation) with diltiazem suggests that the cells account for less than 5% of total incubation volume (data not shown). Assuming fc of 0.6, the small cell volume results in approximately 10-fold higher cellular concentrations for MA and MD than those observed in Fig. 4, B and C. Under our experimental condition, the activity study (secondary incubation with midazolam) was carried out after the cell pellets were washed twice with fresh medium to achieve at least a 20-fold dilution of the primary incubation. At this stage, the competitive inhibition is expected to become less important if the washing caused similar dilution of intracellular levels of metabolites, especially for hepatocytes incubated with lower concentrations of diltiazem (<4 μM). Thus, TDI by MA was probably the major cause of decreased CYP3A activity described in Fig. 4A.
Theoretically, the time-dependent loss of CYP3A in hepatocytes can be modeled if the following information is available: microsomal parameters of metabolite formation (i.e., Km, Vmax), inhibition/inactivation (Ki, KI, and kinact), and the contribution of each inhibitory mechanism for the parent and each of its sequential metabolites. For example, if MA-induced TDI is the predominant mechanism, a model consisting of the loss of CYP3A depicted by eqs. 1 and 2, and by a response relationship relating CYP3A activity loss as a function of MA levels, can be established. However, the selection of inhibitor concentration and the contribution of reversible inhibition will significantly complicate the model. Since MA is formed from diltiazem in the active site inside the cell, it likely causes inactivation before appearing in the extracellular space. Thus, a kinetic profile of MA based on its Iu is not appropriate. Similarly, cellular accumulation of MD may result in an appreciable concentration to cause reversible inhibition of CYP3A, at higher diltiazem concentrations. The trapping effect of MA and MD in hepatocytes incubated with diltiazem is not understood. This poor partitioning may be due to nonspecific binding to the cellular compartment or active transport process(es). Nonetheless, if the accumulation of MA and MD is true in vivo, predicting diltiazem-induced clinical drug interaction would be more complicated.
In this study we focused on the inhibitory effect of diltiazem and its N-desmethyl metabolites MA and MD. Diltiazem also undergoes O-demethylation and deacetylation (“other pathways” in Fig. 1; Homsy et al., 1995; Murray and Butler, 1996; Sutton et al., 1997). O-Desmethyl diltiazem did not inhibit CYP3A in HLMs (Sutton et al., 1997). Although no report on the inhibitory effects of deacetylated metabolites on human CYP3A is available, a study conducted in rats demonstrated that metabolites associated with deacetylation and O-demethylation were an ineffective inhibitor of CYP3A2-mediated 6β-hydroxylation of testosterone, and MA and MD were more potent competitive inhibitors of CYP3A2 than diltiazem (Murray and Butler, 1996). These findings appear to support the hypothesis that metabolites other than MA and MD do not inhibit human CYP3A, although it needs to be confirmed when these metabolites are available.
Diltiazem showed different inhibition/inactivation kinetics toward CYP3A4 and CYP3A5 (McConn et al., 2004). Because both CYP3A isoforms catalyze 1′-hydroxylation reaction of midazolam (Williams et al., 2002; Huang et al., 2004), one cannot disregard the impact that differential expression of these two CYP3A isoforms in HLMs and hepatocytes may have on the interpretation of the results presented in this study. We have chosen gender-pooled HLMs (n = 59) and hepatocytes (n = 5) to minimize this impact as well as the influence by large interindividual variability known to the human CYP3A family (Kuehl et al., 2001). Although beyond the scope of this article, studies of the inhibition/inactivation by diltiazem in CYP3A4 and CYP3A5 Supersomes should be performed to further understand the mechanism. For example, if quantitative prediction were to be performed in hepatocytes using HLM data, caution should be taken because of the differences in inhibition/inactivation kinetics by diltiazem and its metabolites and the kinetics of 1-hydroxymidazolam formation between CYP3A4 and CYP3A5.
Impact of Sequential Metabolism on the Prediction of Drug Interaction Using Microsomal Parameters of Parent Compound. The second objective of this study was to investigate the impact of sequential metabolism of inhibitor on the validity of using microsomal kinetic parameter of the parent compound to predict its inactivation potential in vivo. The results presented in this article suggest that prediction of in vivo drug interaction by using microsomal kinetic parameters (i.e., KI and kinact) obtained under one experimental condition may be inadequate.
Because TDI has been recognized as an important mechanism to cause drug interactions, pharmaceutical industries have implemented methodologies to evaluate the potential for new chemical entities to cause time-dependent inhibition (Favreau et al., 1999; Lim et al., 2005). The advancement of automation in drug discovery and development allows companies to obtain more information when screening TDI. Compounds can be tested for TDI by monitoring the time-dependent change in inhibition potency, i.e., the value of inhibitor concentration causing 50% of enzyme inhibition (“IC50 shift” method) at early discovery stage, or by definitive TDI studies at a later development stage to obtain kinact and KI, as described by Ghanbari et al. (2006). These TDI parameters are then used to predict in vivo drug interactions. Our findings that diltiazem-induced TDI in HLMs is dependent on protein concentration clearly demonstrated that kinetic parameters obtained from a microsomal study under one condition can be misleading. For diltiazem, increased TDI of CYP3A is associated with loss of parent drug and simultaneous accumulation of sequential metabolites MA and MD (Fig. 3). Thus, if definitive TDI experiments were performed for diltiazem using different microsomal protein concentrations, one would expect to obtain different kinact values (depending on reaction time and the extent of metabolite formation) and possibly different KI values (lower Iu,ave of diltiazem at higher protein concentration), whereas data collected from the condition with the lowest protein concentration may implicate diltiazem as a remote inactivator, resulting in false negative.
In conclusion, we have demonstrated that TDI by MA is the main contributor responsible for the loss of CYP3A activity in microsomes and hepatocytes incubated with diltiazem. Given the complexity of inhibition/inactivation mechanisms by diltiazem and its metabolites, caution should be taken when predicting inhibition/inactivation using microsomal kinetic parameters derived from parent drug only and/or from one set of conditions.
Acknowledgments
We acknowledge David Neul (Pfizer La Jolla, San Diego, CA) for providing human cryopreserved hepatocytes and Tanabe Seiyaku Co. Ltd. for providing N-desmethyl metabolites of diltiazem for the studies reported in the article. We also thank Dr. Ellen Wu (Pfizer La Jolla, San Diego, CA) for critical review of the manuscript.
Footnotes
-
This work was partially supported by National Institutes of Health GM32165.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.013847.
-
ABBREVIATIONS: MA, N-desmethyl diltiazem; HLM, human liver microsome; fc, free fraction of inactivator/inhibitor in hepatocyte incubation; Iu, free inactivator/inhibitor concentration in hepatocyte incubation medium; [I]u,ave, microsomal inactivator concentration corrected for both percentage free and metabolic consumption by time-averaged factor; Ki, reversible inhibition constant; kinact, maximum inactivation rate constant; KI, apparent inactivation constant; KI,u,ave, KI obtained using [I]u,ave; λ, apparent inactivation rate constant; MD, N,N-didesmethyl diltiazem; TDI, time-dependent inactivation.
- Received November 9, 2006.
- Accepted February 7, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}