


Abstract
Macrolides may cause severe drug interactions due to the inhibition of metabolizing enzymes. Transporter-mediated uptake of drugs into cells [e.g., by members of the human organic anion transporting polypeptide (OATP) family] is a determinant of drug disposition and a prerequisite for subsequent metabolism. However whether macrolides are also inhibitors of uptake transporters, thereby providing an additional mechanism of drug interactions, has not been systematically studied. The human OATP family members OATP1B1 and OATP1B3 mediate the uptake of endogenous substances and drugs such as antibiotics and HMG-CoA reductase inhibitors (statins) into hepatocytes. In this study we investigated the potential role of these uptake transporters on macrolide-induced drug interactions. By using sulfobromophthalein (BSP) and the HMG-CoA reductase inhibitor pravastatin as substrates, the effects of the macrolides azithromycin, clarithromycin, erythromycin, and roxithromycin and of the ketolide telithromycin on the OATP1B1- and OATP1B3-mediated uptake were analyzed. These experiments demonstrated that the OATP1B1- and OATP1B3-mediated uptake of BSP and pravastatin can be inhibited by increasing concentrations of all macrolides except azithromycin. The IC50 values for the inhibition of OATP1B3-mediated BSP uptake were 11 μM for telithromycin, 32 μM for clarithromycin, 34 μM for erythromycin, and 37 μM for roxithromycin. These IC50 values were lower than the IC50 values for inhibition of OATP1B1-mediated BSP uptake (96–217 μM). These macrolides also inhibited in a concentration-dependent manner the OATP1B1- and OATP1B3-mediated uptake of pravastatin. In summary, these results indicate that alterations of uptake transporter function by certain macrolides/ketolides have to be considered as a potential additional mechanism underlying drug-drug interactions.
Macrolide antibiotics (e.g., erythromycin and clarithromycin) can cause severe drug interactions by increasing plasma concentrations of simultaneously administered compounds. The major mechanism underlying these drug interactions is believed to be inhibition of the major drug metabolizing enzyme CYP3A4 in small intestine and liver (Wrington and Thummel, 2000; Ito et al., 2003; Polasek and Miners, 2006).
Published data indicate that certain macrolides are also inhibitors of the apically/luminally localized drug efflux pump P-glycoprotein (Kim et al., 1999; Marzolini et al., 2004; Eberl et al., 2005). By inhibition of P-glycoprotein function they increase drug absorption from the gut lumen and decrease biliary elimination and renal secretion of concomitantly administered drugs such as the cardiac glycoside digoxin (Rengelshausen et al., 2003). This in turn leads to increased drug concentrations and drug toxicity.
Newly recognized, additional determinants of drug disposition are uptake transporters of the OATP (SLCO) family (Hagenbuch and Meier, 2004; König et al., 2006). Members of the OATP family transport a wide range of drugs including HMG-CoA reductase inhibitors (cerivastatin, fluvastatin, pitavastatin, pravastatin, and rosuvastatin), benzylpenicillin, digoxin, fexofenadine, methotrexate, and rifampicin (Hagenbuch and Meier, 2003; König et al., 2006). OATP1B1 and OATP1B3 are expressed in the basolateral membrane of hepatocytes and mediate the uptake of endogenous substances and drugs from the portal venous blood into the liver. The importance of uptake transporters for drug disposition has been demonstrated by analyzing genetic alterations in the SLCO1B1 gene encoding human OATP1B1. Several polymorphisms or haplotypes have been associated with reduced drug uptake activity in vitro (Tirona et al., 2001; Michalski et al., 2002; Iwai et al., 2004; Kameyama et al., 2005). Furthermore, it has been shown in vivo that the base pair exchange T521C, resulting in an amino acid exchange Val174Ala, was related to increased drug concentrations [e.g., for atrasentan (Katz et al., 2006), fexofenadine (Niemi et al., 2005b), pitavastatin (Chung et al., 2005), pravastatin (Nishizato et al., 2003; Niemi et al., 2006), simvastatin acid (Pasanen et al., 2006), repaglinide (Niemi et al., 2005a), and rosuvastatin (Lee et al., 2005)].
Because alterations in the OATP1B1 protein can be associated with a change in transport activity for certain drugs, uptake transporters may also be a mechanism for drug-drug interactions. For instance, it has been demonstrated that the macrolides clarithromycin and erythromycin significantly increase pravastatin plasma concentrations (Jacobson, 2004; Pravastatin (Pravasin) product information, Bristol-Myers Squibb, Munich, Germany, 2005). Because pravastatin is not metabolized by cytochrome P450 enzymes, uptake transporters may account for this drug-drug interaction. Despite the increasingly recognized role of OATP uptake transporters for drug disposition, it has not been systematically studied whether macrolides are inhibitors of the uptake of concomitantly administered drugs mediated by OATPs, thereby providing a new additional mechanism of macrolide-induced drug interactions.
Therefore, using HEK293 cells stably expressing the human uptake transporters OATP1B1 or OATP1B3, we tested in the present study the influence of macrolide antibiotics on the OATP1B1- and OATP1B3-mediated uptake of organic anions and drugs.
Materials and Methods
Chemicals and Antibodies. [3H]Sulfobromophthalein ([3H]BSP; 7585 GBq/mmol) was obtained from Hartmann Analytic (Braunschweig, Germany). Unlabeled sulfobromophthalein, erythromycin, and poly-d-lysine hydrobromide were purchased from Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany). Unlabeled pravastatin sodium salt was obtained from Tocris Cookson Inc. (Ellisville, MO). Unlabeled azithromycin, clarithromycin, and roxithromycin were obtained from Chemos GmbH (Regenstauf, Germany). Unlabeled telithromycin was obtained after extraction of Ketek tablets (Sanofi-Aventis Deutschland GmbH, Bad Soden, Germany) using ethyl acetate and crystallization from ethyl acetate/hexane (8:2, v/v). Purity was assayed by HPLC-UV to be >99%.
The polyclonal antibodies pESL (König et al., 2000b) and pSKT (König et al., 2000a) were raised in rabbits against human OATP1B1 and OATP1B3, respectively. Both were kind gifts of Professor Dr. D. Keppler (German Cancer Research Center, Heidelberg, Germany). Horseradish peroxidase-conjugated goat anti-rabbit IgG was obtained from Amersham (GE Healthcare Europe GmbH, Munich, Germany).
Methanol (hypergrade quality), n-hexane (pro analysis), acetonitrile (hypergrade quality), and acetic acid (supra pure quality) were purchased from Merck KGaA (Darmstadt, Germany). Diethyl ether (99.8% purity), ammonium acetate (pro analysis), and ibuprofen were obtained from Sigma-Aldrich Chemie GmbH.
Cell Culture and Transfection. Human embryonic kidney (HEK293) cells were cultured in minimum essential medium; containing 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C and 5% CO2. The cells were routinely subcultivated by trypsinization using trypsin (0.05%)-EDTA (0.02%) solution. All cell culture media supplements were obtained from Invitrogen GmbH (Karlsruhe, Germany). HEK293 cells were transfected with the respective plasmid pcDNA3.1(+)-OATP1B1 (König et al., 2000b) and pcDNA3.1/Hygro(–)-OATP1B3 (Cui et al., 2001a) using Effectene transfection reagent (QIAGEN GmbH, Hilden, Germany). Plasmids were a generous gift of Professor Dr. D. Keppler (Heidelberg, Germany). After geneticin (for OATP1B1, 800 μg/ml) or hygromycin (for OATP1B3, 250 μg/ml) selection, single colonies were characterized for SLCO1B1 (encoding human OATP1B1) and SLCO1B3 (encoding human OATP1B3) mRNA and OATP1B1 or OATP1B3 protein expression by real-time polymerase chain reaction and immunoblot analysis. Vector transfected HEK-control cells were established by the same method using the respective expression plasmid without insert for transfection.
For BSP uptake and immunoblot experiments HEK cells were seeded in Petri dishes or six-well plates (coated with 0.1 mg/ml poly-d-lysine; PS plate, six-well, Greiner Bio-One, Frickenhausen, Germany), respectively, at an initial density of 125,000 cells (OATP1B1) and 80,000 cells (OATP1B3) per square centimeter of growth area. For pravastatin uptake HEK cells were seeded in poly-d-lysine (0.1 mg/ml)-coated 12-well plates (Cell Culture Multiwell Plate CELLSTAR; Greiner Bio-One) at an initial density of 700,000 cells/well.
The cells (HEK-control, HEK-OATP1B1, and HEK-OATP1B3) were grown to confluence for 3 days and induced with 10 mM sodium butyrate (Merck KGaA) for 24 h before the uptake and immunoblot experiments to obtain higher levels of the recombinant proteins (Cui et al., 1999).
Immunoblot Analysis. Pelleted HEK293 cells expressing the respective protein were resuspended in protein storage buffer (100 mM Tris-HCl and 1 mM EDTA, pH 7.4) containing protease inhibitors (mini-complete protease inhibitor cocktail tablets, Roche Diagnostics-Applied Science, Mannheim, Germany), homogenized, and sonicated. Protein concentrations were determined by bicinchonic acid assay (BCA Protein Assay Kit; Pierce, Rockford, IL). Twenty micrograms of total protein was diluted with Laemmli buffer and incubated at 95°C for 5 min before separation on 4% stacking and 10% resolving SDS-polyacrylamide gels. Immunoblotting was performed using a tank blotting system from Bio-Rad (Munich, Germany) and enhanced chemiluminescence detection (PerkinElmer Life Sciences GmbH, Rodgau-Jügesheim, Germany). The primary antibodies pESL and pSKT were diluted 1:5000 in TPBS (Dulbecco's phosphate-buffered saline, pH 7.4, and 0.1% Tween 20). The secondary antibody was a horseradish peroxidase-conjugated goat anti-rabbit IgG from Amersham (GE Healthcare Europe GmbH) used at a 1:10,000 dilution. Human liver samples and vector-transfected HEK293 cells served as positive and negative controls, respectively.
Uptake Assays. Before the uptake experiments were started, the cells were washed with prewarmed (37°C) uptake buffer (142 mM NaCl, 5 mM KCl, 1 mM K2HPO4, 1.2 mM MgSO4, 1.5 mM CaCl2, 5 mM glucose, and 12.5 mM HEPES, pH 7.3). The [3H]BSP was dissolved in uptake buffer, and unlabeled BSP was added to the final concentration of 0.05 μM and 1 μM BSP for studies with HEK-OATP1B1 and HEK-OATP1B3 cells, respectively. To characterize the macrolides as inhibitors, they were added in increasing concentrations (up to 500 μM). The cells were incubated with the test solution at 37°C for 10 min as described previously (Michalski et al., 2002; Letschert et al., 2004). Subsequently, the cells were washed three times with ice-cold uptake buffer. After the cells were lysed with 0.2% SDS, the intracellular accumulation of radioactivity was calculated by liquid scintillation counting (PerkinElmer Life Sciences GmbH) and the appropriate protein concentration was determined by bicinchonic acid assay (BCA Protein Assay Kit).
For experiments with pravastatin as substrate, 50 μM pravastatin was dissolved in the uptake buffer. In addition, a 10 or 100 μM concentration of each macrolide was added. The uptake assay was performed as described above except for the determination of the intracellular pravastatin accumulation: after the cells were lysed with 0.2% SDS, the amount of intracellular pravastatin was determined by LC/MS/MS.
LC/MS/MS Assay for Pravastatin. Samples were prepared by adding 100 μl of internal standard solution (200 ng/ml ibuprofen in eluent) to 100 μl of the cell lysates. The injected volume was 30 μl. LC/MS/MS analysis was performed using a Sciex API 4000 (Applied Biosystems, Toronto, ON, Canada) triple quadrupole mass spectrometer equipped with an atmospheric pressure ionization Turbo Ion Spray interface coupled with a two-position actuator control module (VICI Valco Instrument Co. Inc., Houston, TX) to separate the cell lysate salts. The HPLC system was an Agilent Series 1100 (Agilent Technologies Deutschland GmbH, Böblingen, Germany). The HPLC column used was a Luna 3 μ CN 100 Å (100 × 2.0 mm) with a precolumn VS (Cyano, 4 mm length × 2 mm i.d.) purchased from Phenomenex Ltd. Deutschland (Aschaffenburg, Germany). A mixture of 12 mM ammonium acetate and methanol (50:50, v/v) was used as the mobile phase. The flow rate was set at 0.2 ml/min. The retention time of pravastatin was 1.2 min and of the internal standard was 1.4 min. The peak area ratio of pravastatin to the internal standard was calculated using Analyst 1.4.2 software (Applied Biosystems). The lower limit of quantification was 0.5 ng/ml. A calibration curve was constructed using 1/X-weighted linear regression between spiked cell lysate concentrations and the measured ratios. The calibration curves were linear over the range 0.5 to 30 ng/ml with the mean correlation coefficients (n = 7 analytical runs) between 0.9967 and 0.9989. Cell lysate calibration standards (0.5, 1.0, 2.5, 5.0, 10.0, 15.0, 20.0, 25.0, and 30.0 ng/ml), quality controls, and blank and double blank samples were prepared in the same manner. The intraday coefficient of variation was 2.14% at 0.5 ng/ml, 1.60% at 1 ng/ml, 6.93% at 10 ng/ml, and 5.32% at 30 ng/ml (n = 4 to 5).
Data Analysis. The OATP1B1- and OATP1B3-mediated net uptakes were obtained by subtracting the uptake in vector-transfected cells from that in OATP1B1- and OATP1B3-expressing cells. The percentage of uptake inhibition was calculated from control experiments in the absence of macrolides (100% uptake). The corresponding IC50 values for inhibition of OATP1B1- and OATP1B3-mediated BSP uptake were calculated by fitting the data to a sigmoidal dose-response regression curve (Prism 4.01 2004; GraphPad Software, San Diego, CA). The IC50 value is the concentration at which half of the substrate uptake was inhibited.

Characterization of stably transfected HEK293 cells. A, immunoblot analysis of HEK-OATP1B1 and HEK-OATP1B3 cells. Cell homogenates (20 μg) were separated by SDS-polyacrylamide gel electrophoresis. The left blot shows the expression of OATP1B1, detected by the polyclonal antibody pESL (diluted 1:5000). The expression of OATP1B3 is shown on the right blot, using polyclonal antibody pSKT (diluted 1:5000). A human liver sample (Human liver, 10 μg) and vector-transfected HEK293 cells (Control) served as positive and negative controls. B, intracellular [3H]BSP accumulation in HEK-OATP1B1 (OATP1B1), HEK-OATP1B3 (OATP1B3), and HEK-control (Control) cells after a 10-min incubation with 0.05 and 1 μM BSP, respectively. Data are shown as mean values ± S.E. (n = 6–20). Error bars in control cells are within the borders of the bars.
Statistical Analysis. The experiments were repeated at least four times. All data are presented as means ± S.E. Multiple comparisons were analyzed by analysis of variance with subsequent Dunnett's or Tukey's multiple comparison tests by using Prism 4.01 2004. A value of P < 0.05 was required for statistical significance.
Results
BSP Uptake in HEK-OATP1B1 and HEK-OATP1B3 Cells. The prerequisite for analyzing the inhibitory potency of drugs on OATP1B1- and OATP1B3-mediated uptake is the availability of stably transfected cells expressing the recombinant protein in high amounts. Therefore, HEK293 cells were stably transfected with SLCO1B1 cDNA and the SLCO1B3 cDNA and selected for a high expression of the respective uptake transporter. The protein expression of the selected cell clones has been analyzed using the OATP1B1-specific antibody pESL (König et al., 2000a) and the OATP1B3-specific antibody pSKT (König et al., 2000b). This analysis demonstrated high protein expression in the HEK-OATP1B1 and HEK-OATP1B3 cells (Fig. 1A).
Uptake mediated by OATP1B1 or OATP1B3 was analyzed using the prototypic tritium-labeled substrate BSP. BSP was shown to be a high-affinity substrate for both OATP1B1 and OATP1B3 with Km values of 140 nM (Cui et al., 2001b) and 3.3 μM (König et al., 2000a), respectively. The uptake experiments (Fig. 1B) demonstrated that HEK-OATP1B1 cells as well as HEK-OATP1B3 cells were able to mediate BSP uptake into cells. The net uptake rates were 2.1 pmol × mg of protein–1 × min–1 for HEK-OATP1B1 cells and 10.8 pmol × mg of protein–1 × min–1 for HEK-OATP1B3 cells.
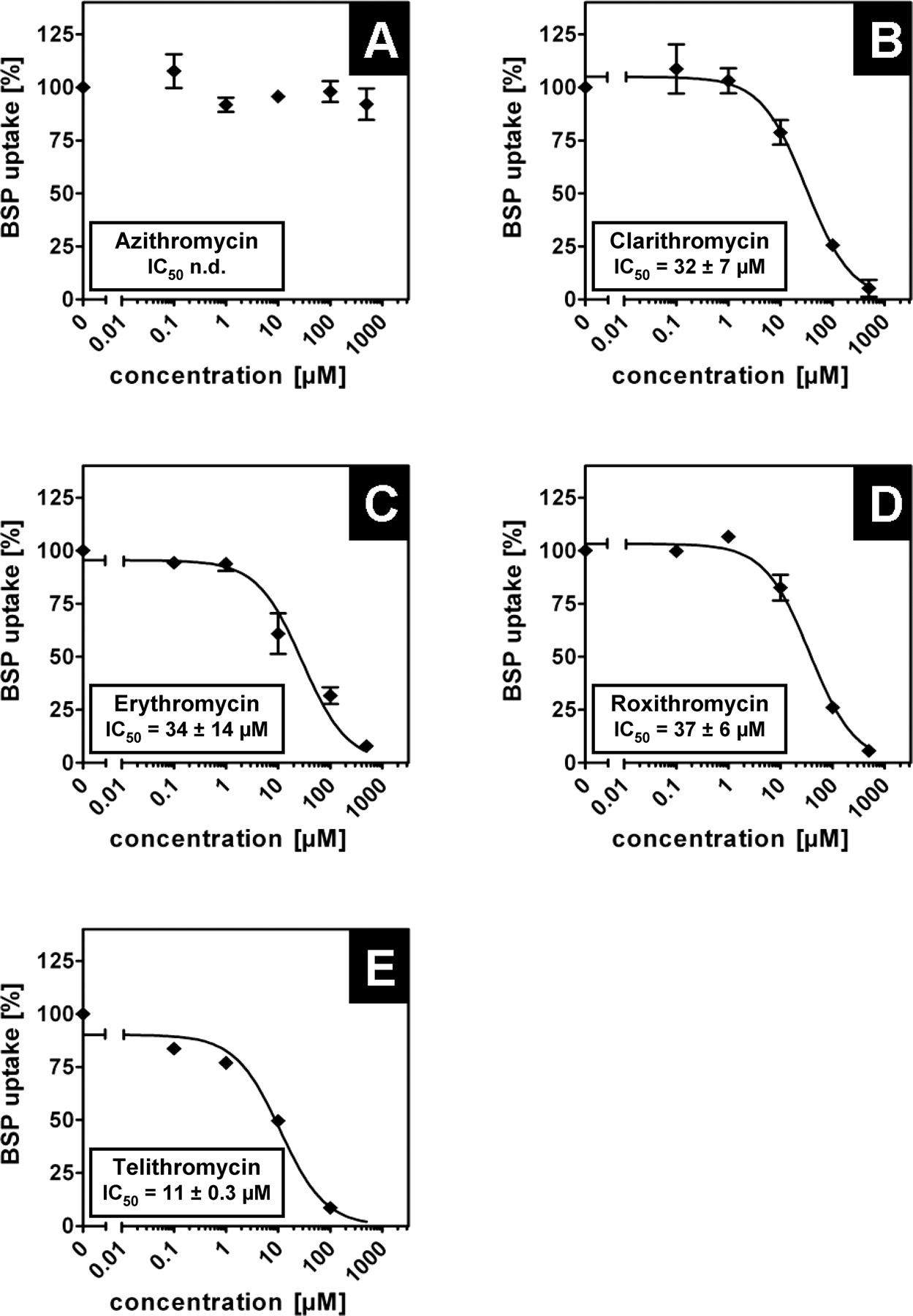
Inhibition of OATP1B1-Mediated BSP Uptake by Macrolides. Uptake experiments have been carried out as described with addition of different concentrations of the respective macrolide. Interestingly, all investigated macrolides except azithromycin showed a clear dose-dependent inhibition of OATP1B1-mediated BSP uptake into HEK-OATP1B1 cells (Fig. 2). Azithromycin has been analyzed up to a concentration of 500 μM and only at this high concentration was a slight decrease in BSP uptake observed (Fig. 2A). Erythromycin also has a high IC50 value of 217 ± 19 μM (Fig. 2C), whereas clarithromycin, telithromycin, and roxithromycin have IC50 values of 96 ± 5, 121 ± 19, and 153 ± 4 μM, respectively (Fig. 2, B, D, and E). Taken together, clarithromycin, erythromycin, roxithromycin, and telithromycin were identified as inhibiting the OATP1B1-mediated BSP transport.
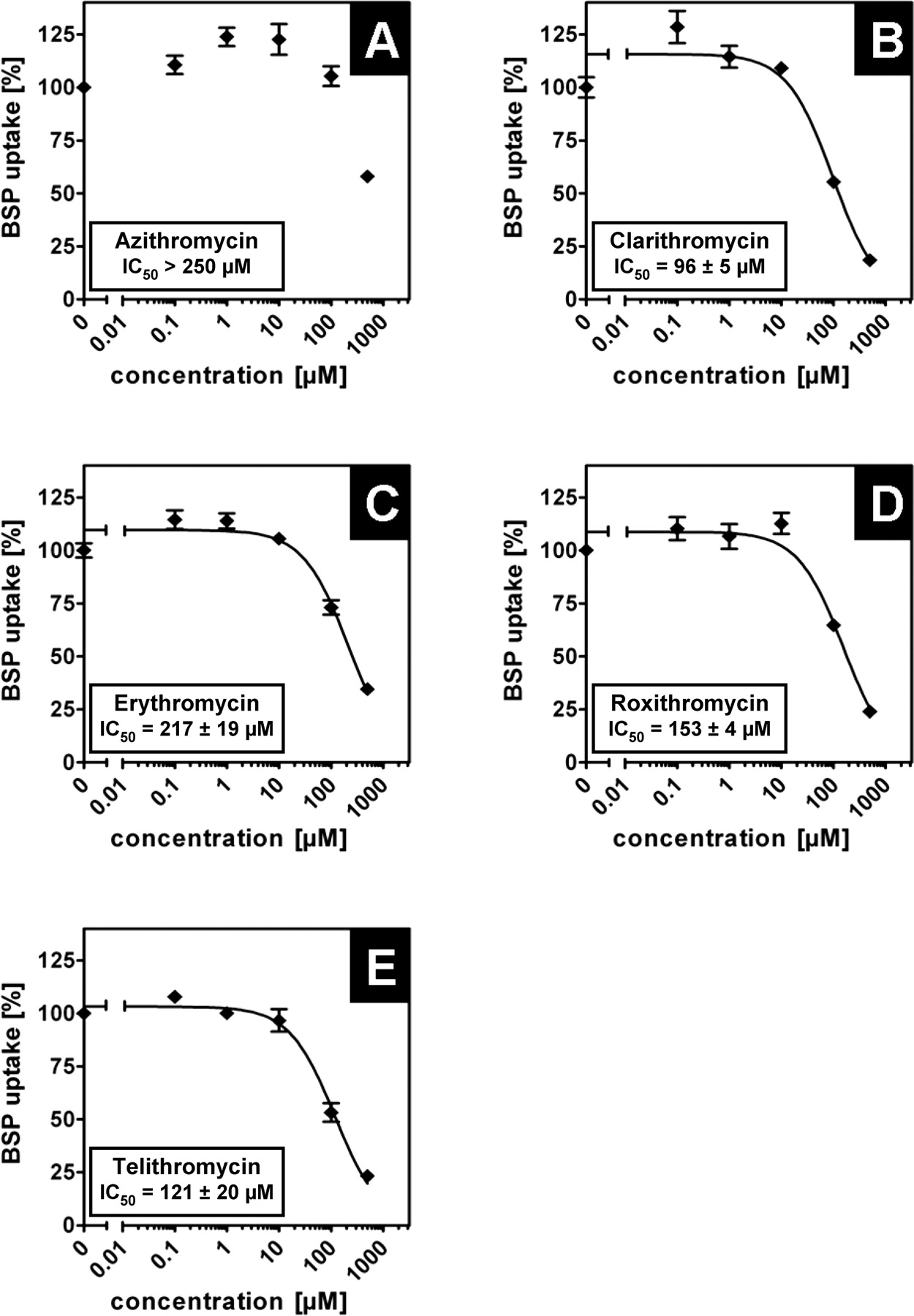
Inhibition of OATP1B3-Mediated BSP Uptake by Macrolides. A similar experimental setup was used to analyze the inhibitory effect of macrolides on OATP1B3-mediated BSP uptake. As shown for the inhibition of OATP1B1-mediated uptake, azithromycin did not inhibit the uptake mediated by the OATP1B3 protein (Fig. 3A). All other macrolides investigated inhibited the OATP1B3-mediated BSP uptake (Fig. 3, B–E). Telithromycin was a potent inhibitor for OATP1B3-mediated uptake with an IC50 value of 11 ± 0.3 μM (Fig. 3E). The macrolides erythromycin, clarithromycin, and roxithromycin showed inhibitory potency with IC50 values of 34 ± 14, 32 ± 7, and 37 ± 6 μM, respectively (Fig. 3, B–D). Interestingly, the calculated IC50 values for clarithromycin, erythromycin, roxithromycin, and telithromycin were determined to be lower than the respective IC50 values for OATP1B1-mediated uptake.
OATP1B1- and OATP1B3-Mediated Pravastatin Uptake and Inhibition by Macrolides. To test whether macrolides are also inhibitors for the OATP1B1- and OATP1B3-mediated uptake of pravastatin, we performed pravastatin uptake and inhibition experiments. Pravastatin is a known substrate for OATP1B1 (Km value of 34 μM) (Hsiang et al., 1999). We confirmed that pravastatin is transported by OATP1B1 with a significantly higher uptake in HEK-OATP1B1 cells (4.5 pmol × mg of protein–1 × min–1) compared with HEK-control cells (0.8 pmol × mg of protein–1 × min–1) (Fig. 4A). Furthermore, we could demonstrate for the first time that pravastatin is also a substrate for OATP1B3 (Fig. 4B). The uptake experiments also demonstrated a significantly higher uptake in HEK-OATP1B3 cells in comparison with HEK-control cells (3.1 pmol × mg of protein–1 × min–1 versus 0.3 pmol × mg of protein–1 × min–1) (Fig. 4B).

Inhibition of BSP uptake by macrolides in HEK-OATP1B1 cells. Inhibitory effect of (A) azithromycin, (B) clarithromycin, (C) erythromycin, (D) roxithromycin, and (E) telithromycin on OATP1B1-mediated BSP (0.05 μM) uptake after a 10-min incubation. The OATP1B1-mediated uptake was obtained by subtracting the uptake in vector-transfected cells from that in OATP1B1-expressing cells. IC50 values were calculated by fitting the data to a sigmoidal dose-response regression curve. OATP1B1-mediated BSP uptake is shown as the percentage of uptake without macrolides. Each value is the mean value ± S.E. (n = 4).
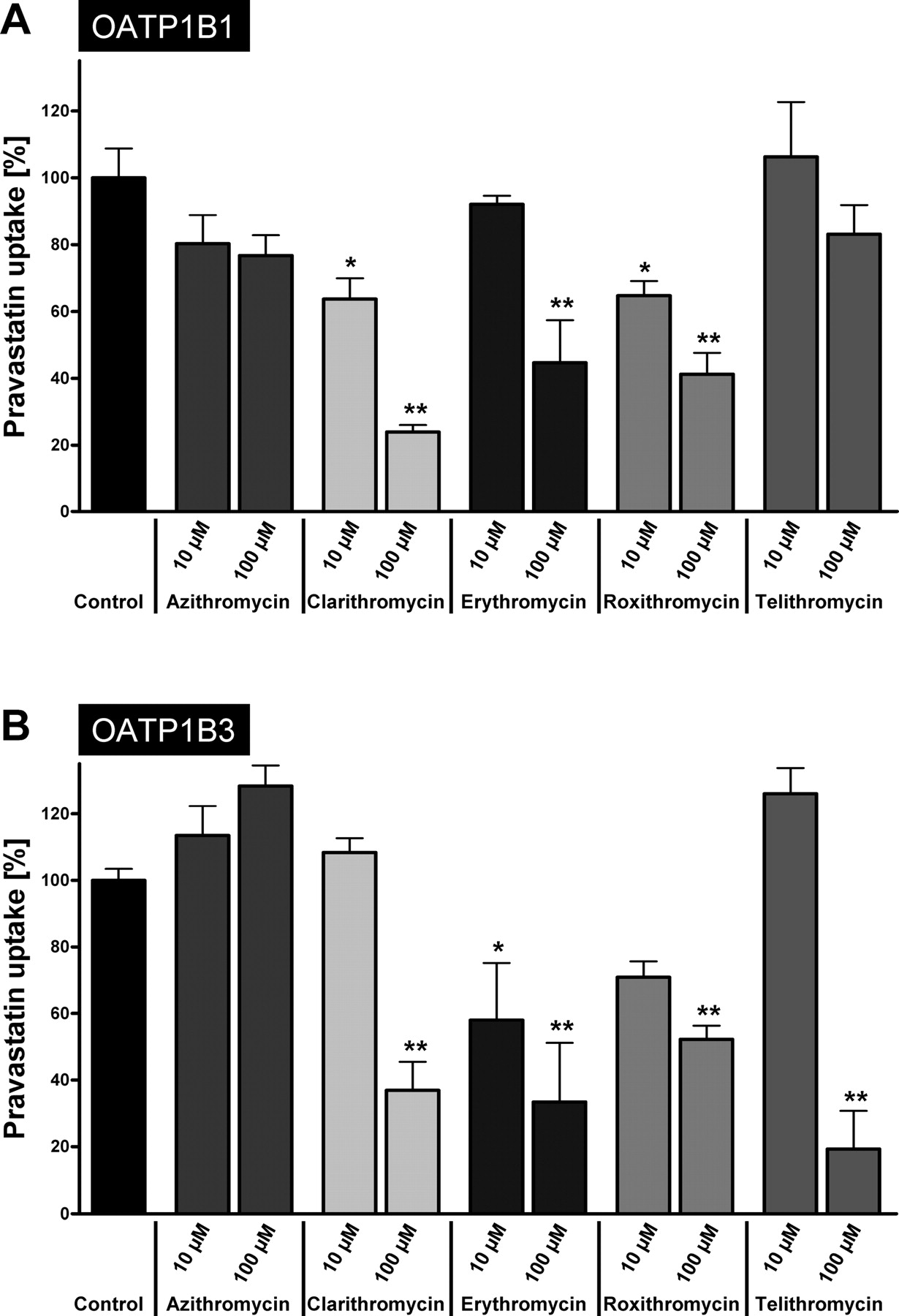
Clarithromycin, erythromycin, and roxithromycin significantly inhibited the uptake of pravastatin in HEK-OATP1B1 cells (Fig. 5A). Addition of 10 μM clarithromycin or roxithromycin resulted in reduced intracellular accumulation of pravastatin to 64% and 65% compared with the control experiments without macrolides (Fig. 5A). Clarithromycin (100 μM), a potent inhibitor of the OATP1B1-mediated BSP uptake, led to a reduction to 24% intracellular accumulation of pravastatin compared with the control experiments. As for BSP, azithromycin did not inhibit the transporter-mediated uptake of pravastatin. In contrast, telithromycin, which was a moderate inhibitor for BSP uptake, did not significantly affect the uptake of pravastatin (Fig. 5A) and showed a moderate uptake inhibition only at the high concentration of 100 μM.
At low macrolide concentrations (10 μM), erythromycin and roxithromycin inhibited OATP1B3-mediated pravastatin uptake. The addition of 100 μM clarithromycin, erythromycin, roxithromycin, and telithromycin reduced the pravastatin uptake to 37%, 36%, 52%, and 19%, respectively (Fig. 5B). Interestingly, telithromycin did not inhibit the uptake of pravastatin at the low concentration, whereas the higher concentration significantly inhibited the uptake. Moreover, a slight but not significant transport activation by low clarithromycin and telithromycin concentrations could be observed. In accordance with the BSP inhibition assay, azithromycin was the only macrolide showing no inhibition among the macrolides tested (Fig. 5B).
Discussion
In this study we focused on the analysis of the interaction of several macrolide/ketolide antibiotics with the transport of the organic anion BSP and the HMG-CoA reductase inhibitor pravastatin mediated by the hepatocellular uptake transporters OATP1B1 and OATP1B3. Using newly established HEK cells recombinantly expressing human OATP1B1 and OATP1B3 (Fig. 1), we found a concentration-dependent inhibition of BSP uptake both in HEK-OATP1B1 and HEK-OATP1B3 cells for all macrolides (except for azithromycin) and for the ketolide telithromycin. IC50 values were considerably smaller for the uptake inhibition of OATP1B3 than for OATP1B1 (Figs. 2 and 3). In addition to the inhibition studies with the prototypic substrate BSP, we have investigated the influence of macrolides and the ketolide telithromycin on the uptake of the HMG-CoA reductase inhibitor pravastatin. We demonstrated OATP1B1-mediated pravastatin uptake into HEK-OATP1B1 cells as described earlier (Hsiang et al., 1999). Furthermore to the best of our knowledge, we determined for the first time that pravastatin uptake is mediated by the second major hepatocyte OATP family member OATP1B3 (Fig. 4). This transporter-mediated pravastatin uptake could be inhibited by coadministration of clarithromycin, erythromycin, and roxithromycin. In the case of OATP1B3, telithromycin was a potent inhibitor for BSP uptake; however, a strong inhibition of pravastatin uptake was observed only at high concentration (100 μM) of telithromycin. Clarithromycin, erythromycin, and roxithromycin inhibited both OATP1B1- and OATP1B3-mediated BSP and pravastatin uptake. On the other hand, azithromycin had no effect on BSP or pravastatin uptake.
Inhibition of BSP uptake by macrolides in HEK-OATP1B3 cells. Inhibitory effect of (A) azithromycin, (B) clarithromycin, (C) erythromycin, (D) roxithromycin, and (E) telithromycin on OATP1B3-mediated BSP (1 μM) uptake after a 10-min incubation. The OATP1B3-mediated uptake was obtained by subtracting the uptake in vector-transfected cells from that in OATP1B3-expressing cells. IC50 values were calculated by fitting the data to a sigmoidal dose-response regression curve. OATP1B3-mediated BSP uptake is shown as the percentage of uptake without macrolides. Each value is the mean value ± S.E. (n = 4).

Pravastatin uptake by HEK-OATP1B1 and HEK-OATP1B3 cells. A, intracellular pravastatin accumulation in HEK-OATP1B1 and HEK-control (Control) cells after a 10-min incubation with pravastatin (50 μM). B, intracellular pravastatin accumulation in HEK-OATP1B3 and HEK-control (Control) cells after a 10-min incubation with pravastatin (50 μM). Each value is the mean value ± S.E. (n = 4–6). ***, P < 0.001 versus control.
OATP1B1 and OATP1B3 are expressed predominantly in the basolateral membrane of human hepatocytes mediating the uptake of endogenous substances as well as several xenobiotics and drugs. Both transporters share an overlapping substrate spectrum. Important drugs that are taken up by OATP1B1 and OATP1B3 are several HMG-CoA reductase inhibitors such as fluvastatin and pitavastatin, the antibiotic rifampicin, and the endothelin receptor antagonist BQ123 (König et al., 2006). Furthermore, transport of cerivastatin (Shitara et al., 2003), pravastatin (Hsiang et al., 1999), and rosuvastatin (Schneck et al., 2004) has been shown for OATP1B1, whereas OATP1B3 is able to mediate the uptake of digoxin (Kullak-Ublick et al., 2001). Because of these substrate spectra and their localization between portal venous blood and important drug-metabolizing enzymes (e.g., CYP3A4) expressed in hepatocytes, uptake transporters are increasingly being recognized as important factors in the directed elimination of drugs out of the body. Their presence can be a prerequisite for substances to enter hepatocytes and get metabolized before their elimination over the canalicular membrane into bile. Modification of uptake rates, e.g., by drug competition, therefore, may cause drug-drug interactions by lowering the uptake rate of one drug followed by increased blood concentrations due to reduced hepatic metabolism and/or decreased biliary elimination.
Inhibition of cytochrome P450 isoenzymes is one established mechanism of drug-drug interactions. Multiple studies have demonstrated that macrolides are potent inhibitors of CYP3A4 and therefore can increase the plasma concentrations of coadministered drugs that are CYP3A4 substrates (Niemi et al., 2001). Drug interactions have also been reported between macrolides and some HMG-CoA reductase inhibitors. Clarithromycin, for example, increases the plasma concentrations of concomitantly administered simvastatin, atorvastatin, and pravastatin (Jacobson, 2004). For simvastatin and atorvastatin, this drug interaction can be explained by the inhibition of CYP3A4, which predominantly metabolizes these statins. Interestingly, pravastatin is one of the statins that is not metabolized by cytochromes in humans and that is excreted almost unchanged into bile or to a small extent into urine (Jacobson, 2004). In this case, an interaction of drug-transporting proteins, located in the basolateral hepatocyte membrane, may account for the increased plasma concentration. The data presented in this article confirmed these in vivo analyses of an interaction between clarithromycin and pravastatin. Clarithromycin inhibited in a dose-dependent manner the OATP1B1- as well as the OATP1B3-mediated pravastatin uptake in vitro. Therefore, transporter inhibition could be the underlying mechanism of this pharmacokinetic drug-drug interaction. In accordance with our findings, Hirano et al. (2006) very recently demonstrated that both clarithromycin and erythromycin were inhibitors for the uptake of pitavastatin, an established OATP1B1 substrate with Ki values of 8.3 and 11.4 μM, respectively.

Inhibition of the pravastatin uptake by macrolides. Inhibitory effect using 10 and 100 μM of the macrolides azithromycin, clarithromycin, erythromycin, roxithromycin, and telithromycin on (A) OATP1B1- and (B) OATP1B3-mediated pravastatin (50 μM) uptake after a 10-min incubation. The transporter-mediated uptake was obtained by subtracting the uptake in vector-transfected cells from that in transporter-expressing cells. OATP-mediated pravastatin uptake is shown as the percentage of uptake without macrolides. Each value is the mean value ± S.E. (n = 4–6). *, P < 0.05; **, P < 0.01 versus control.
Interestingly, an in vivo interaction between rosuvastatin, a recently established HMG-CoA reductase inhibitor, and erythromycin does not appear (Cooper et al., 2003). Rosuvastatin is a substrate of several OATP-family members (OATP1B1, OATP1B3, OATP2B1, OATP1A2) and also of the sodium dependent bile salt transporter NTCP (Ho et al., 2006; Schneck et al., 2004) and therefore, the uptake inhibition of OATP1B1 or OATP1B3 could be compensated by transport via alternative transporting proteins.
Published Ki values for inhibition of the metabolizing enzyme CYP3A4 by clarithromycin, erythromycin, roxithromycin, and telithromycin are 30 μM, 13 μM, 72 μM, and 58 μM, respectively [Aventis Pharmaceuticals, Ketek (telithromycin): Briefing Document for the FDA Anti-Infective Drug Products Advisory Committee Meeting, http://www.fda.gov/ohrms/dockets/ac/01/briefing/3746b_01_aventis.pdf; Polasek and Miners, 2006]. Interestingly, the determined IC50 values for macrolide-induced OATP1B3 inhibition are in the same concentration range. In addition, azithromycin, which has a high Ki value for CYP3A4 (Polasek and Miners, 2006) is the only macrolide showing neither uptake inhibition of OATP1B1- nor OATP1B3-mediated uptake.
As drugs reach the portal vein directly after intestinal absorption, the drug concentration in portal venous blood is higher than in the systemic circulation. For calculation of the predicted maximum drug concentration at the inlet to the liver we used the method of Ito et al. (1998) taking into account the maximum plasma concentration in the systemic circulation, the single dosage, the absorbed fraction of the macrolide, the absorption rate and the hepatic blood flow rate (Table 1) (Ito et al., 1998). For clarithromycin, erythromycin, roxithromycin, and telithromycin the predicted portal venous concentrations are in the same range as the determined IC50 values for inhibition of OATP1B3-mediated uptake. We therefore conclude that inhibition of drug transporters by macrolides/ketolides could be an additional mechanism for clinical relevant drug-drug interactions. Further studies are necessary to gain more inside into the molecular nature of this inhibition mechanism.
Comparison of pharmacokinetic data of macrolides in humans with IC50 values of OATP1B1 and OATP1B3 inhibition obtained in the present study
IC50 data are derived from the measurements shown in Figs. 2 and 3 (each IC50 value is calculated as the mean values of four experiments ± S.E.). Superior letters link data with references: data for dosages and cmax in the second and third columns are from the References in the last column.
Taken together our data demonstrate that macrolides/ketolides can inhibit uptake of organic anions and drugs mediated by the OATP family members OATP1B1 and OATP1B3. This modification of uptake rates is a new mechanism of drug-drug interactions in addition to the hitherto known mechanism of drug-drug interactions due to the modification of metabolizing enzymes and efflux transporters. Based on our findings it is therefore of importance to gain more knowledge on the modification of uptake transporter function as additional mechanism underlying drug-drug interactions.
Acknowledgments
We thank C. Hoffmann and B. Endress for excellent technical assistance. We thank Professor Dr. D. Keppler (German Cancer Research Center, Heidelberg, Germany) for providing the polyclonal antibodies pESL and pSKT and the plasmids pcDNA3.1(+)-OATP1B1 and pcDNA3.1/Hygro(–)-OATP1B3.
Footnotes
-
This work was supported by Grants DFG Ko 2120/1-3 and Fr 1298/2-4 of the Deutsche Forschungsgemeinschaft.
-
A.S. and S.E. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.014407.
-
ABBREVIATIONS: OATP, organic anion transporting polypeptide; BSP, sulfobromophthalein; HPLC, high-performance liquid chromatography; LC, liquid chromatography; MS/MS, tandem mass spectrometry.
- Received December 18, 2006.
- Accepted February 8, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}