


Abstract
Clinical studies have revealed that plasma concentrations of midazolam after oral administration are greatly increased by coadministration of erythromycin and clarithromycin, whereas azithromycin has little effect on midazolam concentrations. Several macrolide antibiotics are known to be mechanism-based inhibitors of CYP3A, a cytochrome P450 isoform responsible for midazolam hydroxylation. The aim of the present study was to quantitatively predict in vivo drug interactions in humans involving macrolide antibiotics with different inhibitory potencies based on in vitro studies. α- and 4-Hydroxylation of midazolam by human liver microsomes were evaluated as CYP3A-mediated metabolic reactions, and the effect of preincubation with macrolides was examined. The hydroxylation of midazolam was inhibited in a time- and concentration-dependent manner following preincubation with macrolides in the presence of NADPH, whereas almost no inhibition was observed without preincubation. The kinetic parameters for enzyme inactivation (K′app and kinact) involved in midazolam α-hydroxylation were 12.6 μM and 0.0240 min–1, respectively, for erythromycin, 41.4 μM and 0.0423 min–1, respectively, for clarithromycin, and 623 μM and 0.0158 min–1, respectively, for azithromycin. Similar results were obtained for the 4-hydroxylation pathway. These parameters and the reported pharmacokinetic parameters of midazolam and macrolides were then used to simulate in vivo interactions based on a physiological flow model. The area under the concentration-time curve (AUC) of midazolam after oral administration was predicted to increase 2.9- or 3.0-fold following pretreatment with erythromycin (500 mg t.i.d. for 5 or 6 days, respectively) and 2.1- or 2.5-fold by clarithromycin (250 mg b.i.d. for 5 days or 500 mg b.i.d. for 7 days, respectively), whereas azithromycin (500 mg o.d. for 3 days) was predicted to have little effect on midazolam AUC. These results agreed well with the reported in vivo observations.
Macrolide antibiotics have now been used to treat a variety of infectious diseases for several decades and are often coadministered with other drugs. Like ketoconazole, erythromycin has been reported to cause severe cardiac toxicity with terfenadine when coadministered in humans (Food and Drug Administration, 1990). Honig et al. (1994) have reported that the plasma concentrations of terfenadine, which is undetectable when given alone (60 mg b.i.d. for 7 days), increased to detectable levels following coadministration of erythromycin (500 mg t.i.d. for 7 days). This interaction has been shown to be caused by the inhibitory effect of erythromycin on CYP3A, a cytochrome P450 isoform responsible for terfenadine metabolism (Jurima-Romet et al., 1994).
The degree of inhibition of CYP3A varies among the macrolides: the area under the concentration-time curve (AUC) of orally administered midazolam, a substrate of CYP3A, was reported to increase by a factor of 3.8 or 3.6 after pretreatment with erythromycin (500 mg t.i.d. for 5 days) (Zimmermann et al., 1996) or clarithromycin (250 mg b.i.d. for 5 days) (Yeates et al., 1996), respectively. On the other hand, a relatively small increase, by a factor of 1.5 and 1.2, has been reported in the case of pretreatment with roxithromycin (300 mg o.d. for 6 days) (Backman et al., 1994) and azithromycin (500 mg o.d. for 3 days) (Yeates et al., 1996), respectively.
These interactions are based on a “mechanism-based inhibition” (Silverman, 1988), which differs from competitive or noncompetitive inhibition. CYP3A demethylates the macrolide to a nitrosoalkane which then forms a stable, inactive complex with P450 (Periti et al., 1992). In such a case, the inhibitory effect remains after elimination of the inhibitor from plasma or tissues, which may lead to more serious toxicity compared with the case of reversible inhibition.
We have already succeeded in making quantitative predictions of in vivo 5-fluorouracil/sorivudine and triazolam/erythromycin interactions, both involving mechanism-based inhibition of metabolic enzymes, based on a physiologically based pharmacokinetic model and in vitro data (Kanamitsu et al., 2000a,b; Ito et al., 1998). In the present study, using a similar methodology, an attempt was made to predict the degree of in vivo drug interactions in humans involving macrolides with different inhibitory potencies based on in vitro metabolic inhibition studies.
Materials and Methods
Chemicals and Reagents. Midazolam, α-hydroxy (OH) midazolam, and 4-OH midazolam were generously donated by Nippon Roche K.K. (Tokyo, Japan), etizolam by Welfide Corp. (Osaka, Japan), clarithromycin by Taisho Pharmaceutical Co., Ltd. (Tokyo, Japan), and azithromycin by Pfizer, Inc. (Groton, CT). Erythromycin and EDTA-2Na were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). NADP, glucose 6-phosphate and glucose-6-phosphate dehydrogenase were obtained from Roche Diagnostics (Mannheim, Germany). Acetonitrile, methanol, and other reagents of analytical grade were purchased from Kanto Chemical Co. (Tokyo, Japan). Pooled human liver microsomes (H161) were a gift from BD Gentest (Woburn, MA).
Midazolam Metabolism by Pooled Human Liver Microsomes. After 5 min of preincubation, at 37°C, of a reaction mixture (0.72 ml) consisting of 0.1 mg/ml human liver microsomes and an NADPH-generating system (0.33 mM NADP, 8 mM glucose 6-phosphate, 0.1 U/ml glucose-6-phosphate dehydrogenase, 6 mM MgCl2) in 100 mM potassium phosphate buffer (pH 7.4) containing 0.1 mM EDTA, enzyme reactions were initiated by adding 80 μl of midazolam in 20% acetone solution. After incubation at 37°C in a shaking water bath for 3 min, the reaction was terminated by transferring the 600-μl aliquot to another tube containing 800 μl of ice-cold 100 mM Na2CO3 and 4 ml of ethyl acetate followed by vortex mixing for extraction as described below. The final midazolam concentration ranged from 2.5 to 320 μM, and the linearity of metabolism had been confirmed under the above conditions in terms of both protein concentration and incubation time. Data are presented as the means ± S.D. of triplicate experiments.
Inhibition of Midazolam Metabolism by Macrolides. After 5 min of preincubation at 37°C of a reaction mixture (0.64 ml) consisting of human liver microsomes and an NADPH-generating system in potassium phosphate buffer containing EDTA as described above, 80 μl of macrolide solution (10% acetone solution for erythromycin and clarithromycin; 20% acetone solution for azithromycin) was added and then preincubated further at 37°C for 0, 5, 10, or 20 min. Then, 80 μl of midazolam in 20% acetone solution was added and incubated at 37°C for another 3 min. The enzyme reaction was terminated as described above. The final concentration of midazolam was set at 200 μM, whereas that of erythromycin and clarithromycin ranged from 5 to 100 μM, and that of azithromycin ranged from 50 to 1000 μM. Data are presented as means ± S.E. of three determinations.
Quantification of Midazolam Metabolites by HPLC. α- and 4-OH Midazolam in the incubation mixture were determined by an HPLC-UV detection method. One hundred microliters of 2 μg/ml etizolam (methanol solution) was added to the extraction mixture as an internal standard and centrifuged at 1500g for 10 min after vortex mixing. Three milliliters of supernatant was evaporated to dryness under a gentle stream of nitrogen. The residues were reconstituted with 300 μl of HPLC mobile phase as described below, and 50 μl was injected into the HPLC column. The HPLC system consisted of a model LC-10AD pump (Shimadzu Ltd., Kyoto, Japan), a model SIL-10A sample injector (Shimadzu), a model SPD-10A UV absorbance detector (Shimadzu) set at 220 nm, and a Mightysil RP-18 reversed-phase column (150 × 4.6 mm inner diameter, Kanto Chemical Co., Tokyo, Japan). The mobile phase consisted of a 12/5/7 (v/v) mixture of 10 mM potassium phosphate buffer (pH 7.4), methanol, and acetonitrile delivered at 1.0 ml/min. All chromatograms were recorded using a model C-R4 Chromato-Integrator (Shimadzu), and α- and 4-OH midazolam were quantified based on their peak areas.
Kinetic Analysis of Midazolam Metabolism by Pooled Human Liver
Microsomes. The kinetic parameters (Vmax and
Km) for midazolam α- and 4-hydroxylation by human
liver microsomes were determined by the nonlinear least-squares regression
program MULTI (Yamaoka et al.,
1981) according to the following equation:
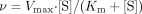 where v, Vmax, and Km represent the
metabolic rate, the maximum metabolic rate, and the Michaelis constant,
respectively. The intrinsic clearance (CLint) for each metabolic
pathway was obtained as the ratio of Vmax and
Km.
where v, Vmax, and Km represent the
metabolic rate, the maximum metabolic rate, and the Michaelis constant,
respectively. The intrinsic clearance (CLint) for each metabolic
pathway was obtained as the ratio of Vmax and
Km.
Kinetic Analysis of Enzyme Inactivation by Macrolides. Kinetic
parameters for enzyme inactivation were obtained as reported elsewhere
(Ito et al., 1998). The
logarithm of the remaining enzymatic activity (formation rate of α- or
4-OH midazolam) was plotted against the preincubation time, and the apparent
inactivation rate constant (kobs) was determined from the
slope of the initial linear phase. Then, the value of kobs
was plotted against the macrolide concentration ([I]), and the parameters
(kinact, K′app, and
kd) were obtained by the nonlinear least-squares
regression method (MULTI) according to the following equation
(Waley, 1985;
Silverman, 1988):
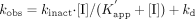 where kinact, K′app, and
kd represent the maximum inactivation rate constant, the
apparent dissociation constant between the enzyme and the macrolides, and the
spontaneous in vitro degradation rate constant of the enzyme in the absence of
the inhibitor, respectively.
where kinact, K′app, and
kd represent the maximum inactivation rate constant, the
apparent dissociation constant between the enzyme and the macrolides, and the
spontaneous in vitro degradation rate constant of the enzyme in the absence of
the inhibitor, respectively.
Quantitative Prediction of the in Vivo Midazolam/Macrolide
Interaction. The differential equations for active and inactive CYP3A in
the liver (Eact and Einact, respectively) can be
described as follows:
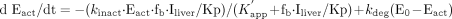
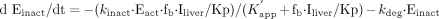 where kdeg, Kp, fb, Iliver, and
E0 represent the degradation rate constant (turnover rate constant)
of CYP3A, liver-to-blood concentration ratio of macrolides, unbound fraction
of macrolides in blood, macrolide concentration in the liver, and total
concentration of CYP3A, respectively. The initial conditions (at t =
0) are Eact = E0 and Einact = 0. In the
absence of macrolides, the CYP3A content in the liver is at steady state and
the degradation rate (kdeg · E0) is
equal to the synthesis rate, which was assumed to be unaffected by macrolides.
It was also assumed in the above equations that the kdeg
for the inactive enzyme is the same as that for the active enzyme.
where kdeg, Kp, fb, Iliver, and
E0 represent the degradation rate constant (turnover rate constant)
of CYP3A, liver-to-blood concentration ratio of macrolides, unbound fraction
of macrolides in blood, macrolide concentration in the liver, and total
concentration of CYP3A, respectively. The initial conditions (at t =
0) are Eact = E0 and Einact = 0. In the
absence of macrolides, the CYP3A content in the liver is at steady state and
the degradation rate (kdeg · E0) is
equal to the synthesis rate, which was assumed to be unaffected by macrolides.
It was also assumed in the above equations that the kdeg
for the inactive enzyme is the same as that for the active enzyme.
The differential equations for midazolam (S) and macrolides (I) can be expressed as follows according to the perfusion model (Fig. 1):

Physiological model for the description of the time-profiles of midazolam and macrolide concentrations.
For midazolam:
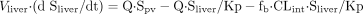
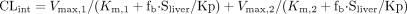

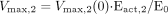
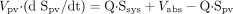
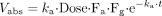

For macrolides:

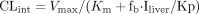
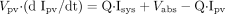

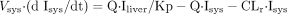 where Vliver and Vpv represent the
volume of liver and portal vein, respectively; Vsys
represents the volume of distribution in the central compartment;
Sliver represents the concentration in the liver; Spv
and Ipv represent the concentration in the portal vein;
Ssys and Isys represent the concentration in the central
compartment; Q represents the blood flow rate; CLr represents the
renal clearance; CLint represents the intrinsic metabolic
clearance; Km represents the Michaelis constant;
Km,1 and Km,2 represent the
Km for α- and 4-hydroxylation of midazolam,
respectively; Vmax represents the maximum rate of
metabolism; Vmax,1 and Vmax,2
represent the Vmax for α- and 4-hydroxylation of
midazolam, respectively; Eact,1 and Eact,2 represent the
Eact for α- and 4-hydroxylation of midazolam, respectively;
Vabs represents the absorption velocity;
ka represents the first-order absorption rate constant;
Fa represents the fraction absorbed from the gastrointestinal
tract; and Fg represents the intestinal availability. In the case
of intravenous administration of midazolam, the absorption term in eq. 9
(Vabs) was deleted, and the dose (nmol) was used as the
initial value of Vsys · Ssys.
where Vliver and Vpv represent the
volume of liver and portal vein, respectively; Vsys
represents the volume of distribution in the central compartment;
Sliver represents the concentration in the liver; Spv
and Ipv represent the concentration in the portal vein;
Ssys and Isys represent the concentration in the central
compartment; Q represents the blood flow rate; CLr represents the
renal clearance; CLint represents the intrinsic metabolic
clearance; Km represents the Michaelis constant;
Km,1 and Km,2 represent the
Km for α- and 4-hydroxylation of midazolam,
respectively; Vmax represents the maximum rate of
metabolism; Vmax,1 and Vmax,2
represent the Vmax for α- and 4-hydroxylation of
midazolam, respectively; Eact,1 and Eact,2 represent the
Eact for α- and 4-hydroxylation of midazolam, respectively;
Vabs represents the absorption velocity;
ka represents the first-order absorption rate constant;
Fa represents the fraction absorbed from the gastrointestinal
tract; and Fg represents the intestinal availability. In the case
of intravenous administration of midazolam, the absorption term in eq. 9
(Vabs) was deleted, and the dose (nmol) was used as the
initial value of Vsys · Ssys.
The following assumptions were made in the above mass-balance equations.
-
Midazolam is administered intravenously or orally and macrolides are administered orally.
-
Midazolam is eliminated only by the liver.
-
The distribution of midazolam and macrolides in the liver rapidly reaches equilibrium, and the unbound concentrations in the hepatic vein are equal to those in the liver at equilibrium (well stirred model).
-
Only the unbound molecule in the liver is subject to metabolism.
-
The contribution of CYP3A to the total elimination of macrolides in the liver is small (i.e., the elimination of a macrolide itself is not altered by inactivation of CYP3A).
-
Gastrointestinal absorption can be described by a first-order rate constant.
The pharmacokinetic parameters of midazolam and macrolides were determined from data in the literature (Tables 1 and 2). Using the program STELLA II (High Performance Systems, Inc., Hanover, NH), and kinetic parameters for CYP3A inactivation obtained in in vitro studies, the above differential equations were numerically solved to simulate the time courses of the macrolide concentration in blood, the active CYP3A content in the liver (Eact), and midazolam concentration in blood. According to clinical reports, the dosing schedules were assumed as follows: in the case of intravenous administration of midazolam, erythromycin (500 mg = 681 μmol, t.i.d. for 6 days) followed by midazolam (9.67 μmol) (Olkkola et al., 1993) or clarithromycin (500 mg = 668 μmol, b.i.d. for 7 days), followed by midazolam (9.82 μmol) (Gorski et al., 1998). In the case of oral administration of midazolam, erythromycin (500 mg = 681 μmol, t.i.d. for 5 or 6 days) followed by midazolam (15 mg = 46 μmol) (Olkkola et al., 1993; Zimmermann et al., 1996), clarithromycin (250 mg = 334 μmol, b.i.d. for 5 days) followed by midazolam (15 mg = 46 μmol) (Yeates et al., 1996), clarithromycin (500 mg = 668 μmol, b.i.d. for 7 days) followed by midazolam (4 mg = 12 μmol) (Gorski et al., 1998), or azithromycin (500 mg = 637 μmol, o.d. for 3 days) followed by midazolam (15 mg = 46 μmol) (Yeates et al., 1996; Zimmermann et al., 1996). The AUCs from time 0 to infinity of the simulated midazolam concentration profiles were compared with the reported values.
Pharmacokinetic parameters of midazolam and enzyme turnover used in the simulation
Pharmacokinetic parameters of macrolides used in the simulation
Results
In Vitro Metabolism of Midazolam by Human Liver Microsomes. The metabolism of midazolam by human liver microsomes followed Michaelis-Menten kinetics with the kinetic constants summarized in Table 3. The Km value for 4-hydroxylation of midazolam was about 3.5-fold higher than that for the α-hydroxylation pathway, whereas the Vmax was greater for α-hydroxylation. Consequently, the CLint (Vmax/Km ratio) for the α- and 4-hydroxylation pathways was 84.8% and 15.2%, respectively, of the total CLint, indicating that α-hydroxylation is the major metabolic pathway of midazolam. This result is consistent with the previous finding by Gorski et al. (1994).
Kinetic parameters for midazolam hydroxylation by human liver microsomes
Inhibition of Midazolam Metabolism by Macrolide Antibiotics.Figure 2 shows the effect of macrolide concentration and preincubation time on midazolam metabolism by human liver microsomes. Midazolam metabolism was not inhibited without preincubation, even if the macrolide concentration was increased. The degree of inhibition depended on the preincubation time and the macrolide concentration. α-Hydroxylation of midazolam by human liver microsomes was reduced to 43.1%, 39.4%, and 67.5% of the control value following a 20-min preincubation in the presence of 100 μM erythromycin, 100 μM clarithromycin, and 1000 μM azithromycin, respectively. Similar results were obtained for the 4-hydroxylation pathway.
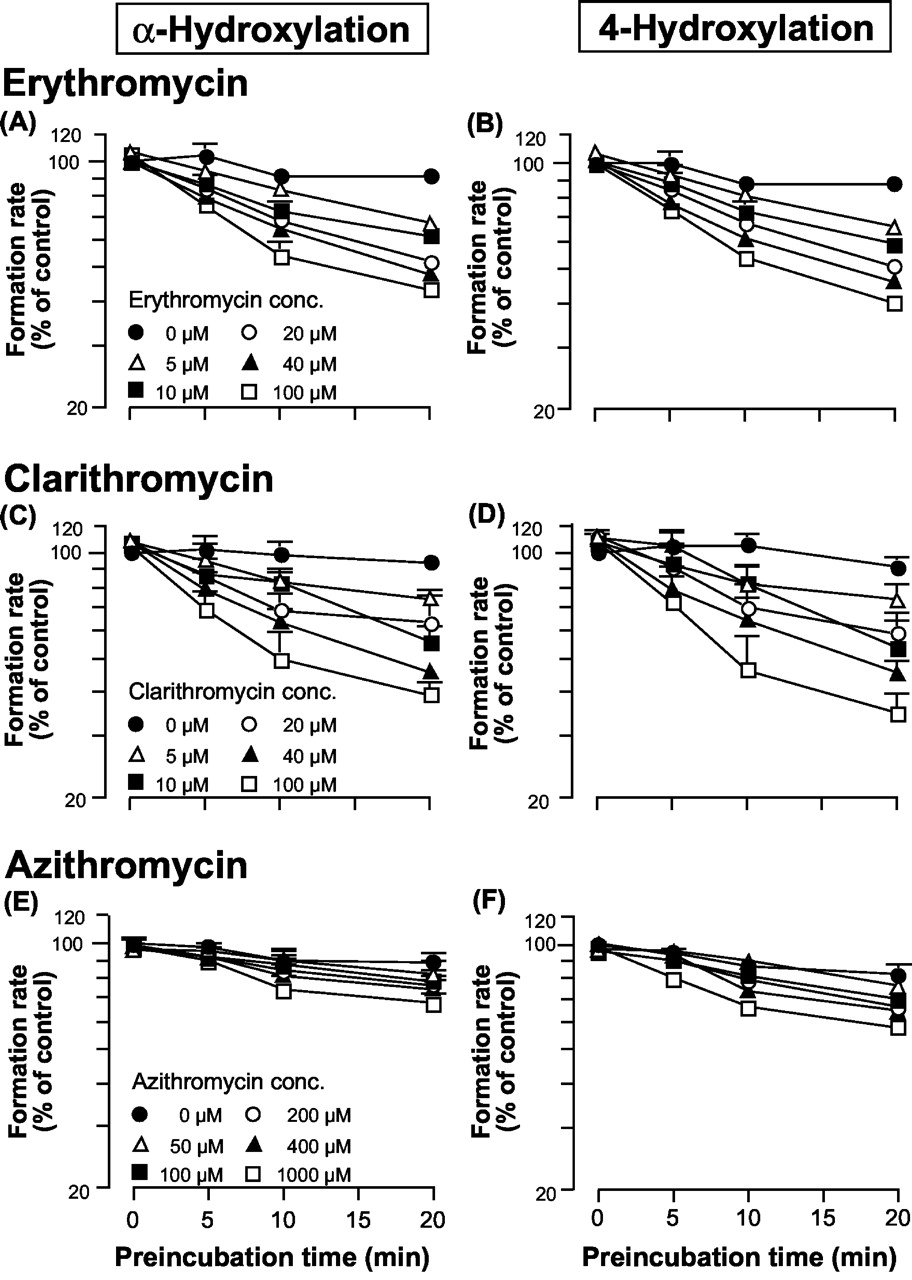
Inhibitory effect of macrolides on midazolam hydroxylation by human liver microsomes.
The y-axis represents the formation rate of α-OH midazolam (A, C, and E) or 4-OH midazolam (B, D, and F). The control activities were A, 3.77; B, 1.63; C, 4.52; D, 1.94; E, 3.14; and F, 1.38 nmol/min/mg protein, respectively.
The calculated kinetic parameters for CYP3A inactivation are summarized in Table 4. The data points of the 0- to 10-min preincubation were considered to reflect the initial inactivation rate and were used to estimate the values of kobs. For each macrolide, the obtained values of both K′app and kinact were almost identical for both hydroxylation pathways of midazolam.
Kinetic parameters for inhibition of midazolam hydroxylation by macrolides
Quantitative Prediction of the Midazolam/Macrolide Interaction. Concentration profiles of midazolam and macrolides simulated by the kinetic parameters in Tables 1 and 2 were compared with the reported profiles (Birkett et al., 1990; Foulds et al., 1990; Chu et al., 1992; Olkkola et al., 1993). Figure 3 shows the concentration profiles of midazolam in blood after a single intravenous or oral administration and those of macrolides after a single oral administration. In all cases, the simulated and reported profiles were comparable. Furthermore, the simulated profiles were constant after each dose following repeated administration (Figs. 4 and 5), which was also consistent with the previous findings that no accumulation was observed for each macrolide following repeated administration (Smith et al., 1953; Suwa et al., 1988; Foulds et al., 1990). These findings indicate the validity of the pharmacokinetic parameters used in the present simulation.

Comparison between observed (closed circles) and simulated (dashed line) concentration profiles of midazolam and macrolides.
A and B, midazolam concentration profiles in blood after single intravenous or oral administration of midazolam (3 or 15 mg) (Olkkola et al., 1993). C, erythromycin concentration profile in blood after single oral administration of erythromycin (250 mg) (Birkett et al., 1990). D, clarithromycin concentration profile in blood after single oral administration of clarithromycin (200 mg) (Chu et al., 1992). E, azithromycin concentration profile in blood after single oral administration of azithromycin (500 mg) (Foulds et al., 1990).

Simulated profiles of erythromycin concentration in blood (1), active CYP3A content in the liver (2), and midazolam concentration in blood [(3) and (4)].
In (2), (3), and (4), solid line represents control and dashed line represents +erythromycin.

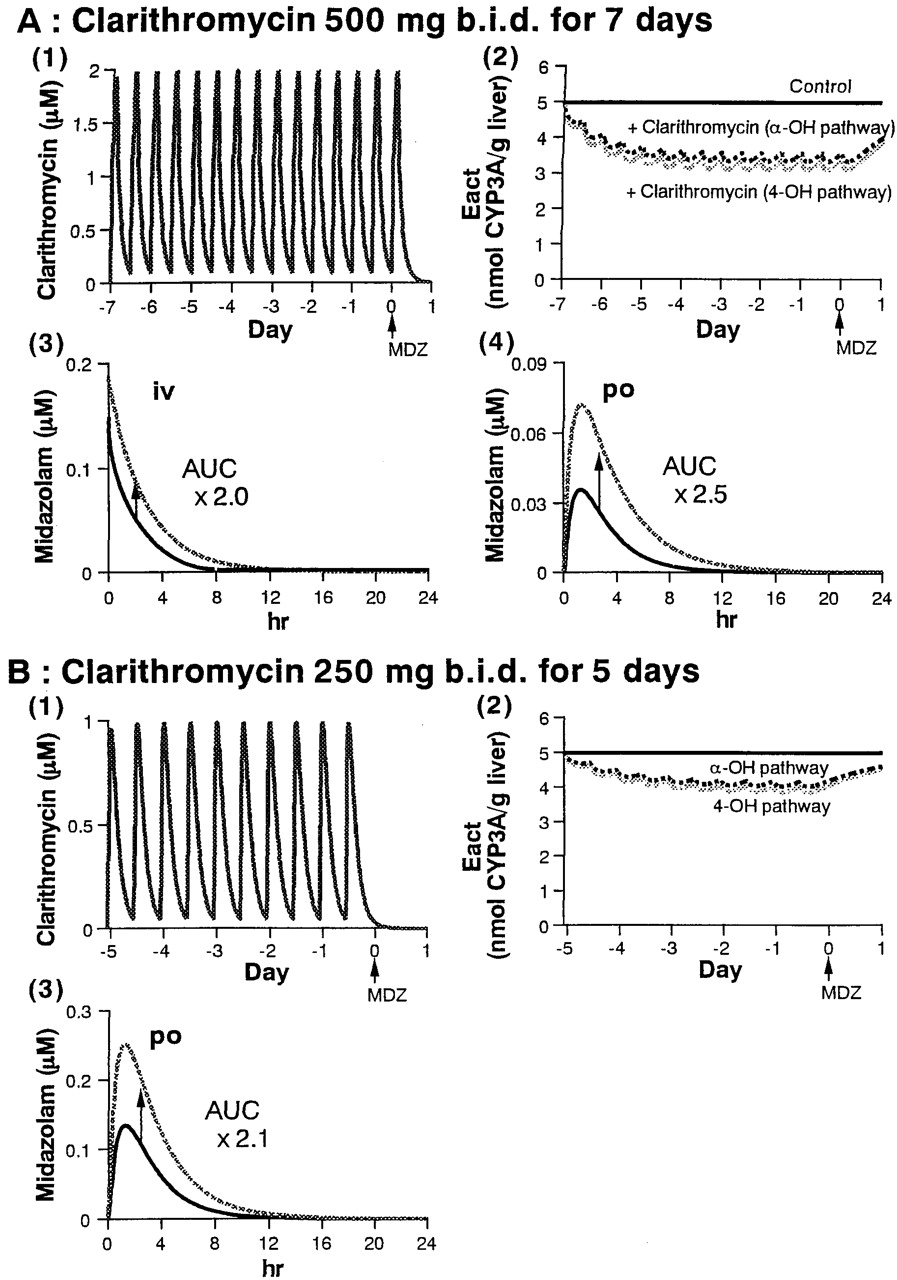
Simulated profiles of clarithromycin concentration in blood (1), active CYP3A content in the liver (2), and midazolam concentration in blood [(3) and (4)].
In (2), (3), and (4), solid line represents control and dashed line represents +clarithromycin.
Figures 4 and 5 also show the simulated effects of erythromycin and clarithromycin, respectively, on the active CYP3A content in the liver and midazolam concentrations in blood. The results are also summarized in Table 5. Following administration of erythromycin (500 mg t.i.d. for 6 days), the active CYP3A was predicted to fall gradually, and a maximum 45% inactivation of CYP3A involved in both hydroxylation pathways of midazolam was predicted after 3 days of administration of erythromycin, with no further inactivation thereafter [Fig. 4A, (2)]. The concentration of midazolam in blood was predicted to increase following administration of erythromycin, and the predicted AUC increase compared with control group was 1.7- and 3.0-fold after intravenous and oral administration of midazolam, respectively [Fig. 4A, (3) and (4)]. In another case of erythromycin administration (500 mg t.i.d. for 5 days), a 2.9-fold increase was predicted in the AUC of midazolam after oral administration [Fig. 4B, (3)].
Prediction of the increase in midazolam AUC by macrolides
Following administration of clarithromycin (500 mg b.i.d. for 7 days), a maximum of 34 and 38% inactivation of the CYP3A concerned with the α- and 4-hydroxylation pathway, respectively, of midazolam was predicted after 3 days of administration, with no further inactivation thereafter [Fig. 5A, (2)]. The predicted increase in midazolam AUC was 2.0- and 2.5-fold after intravenous and oral administration, respectively [Fig. 5A, (3) and (4)]. In another case of clarithromycin administration (250 mg b.i.d. for 5 days), a maximum of 20 and 23% inactivation of the CYP3A concerned with α- and 4-hydroxylation pathway, respectively, of midazolam was predicted after 3 days of administration, with no further inactivation thereafter [Fig. 5B, (2)]. A 2.1-fold increase was predicted in the AUC of midazolam after oral administration [Fig. 5B, (3)].
In the case of azithromycin administration (500 mg o.d. for 3 days), only 1% of the CYP3A concerned with both hydroxylation pathways was predicted to be inactivated after 2 days of administration, with no further inactivation thereafter (data not shown). Almost no change was predicted in the AUC of midazolam after oral administration (Table 5).
Discussion
Various types of macrolide antibiotics are now being used in clinical practice. It has been demonstrated that macrolides inhibit the metabolic enzyme CYP3A by forming an inactive complex in vitro (Periti et al., 1992). They are also reported to cause many in vivo interactions with drugs that are substrates of CYP3A, and the severity of such interactions varies from macrolide to macrolide (Periti et al., 1992; Westphal, 2000). Whereas erythromycin and clarithromycin markedly affect the AUC of midazolam, a CYP3A substrate, in humans, the effect of azithromycin is reported to be small (Backman et al., 1994; Yeates et al., 1996; Zimmermann et al., 1996). Periti et al. (1992) have classified troleandomycin and erythromycin, 14-carbon member ring macrolides, as belonging to the first group with a high drug interaction potential; clarithromycin and roxithromycin (14-carbon), midecamycin and josamycin (16-carbon) belong to the second group, with a low interaction potential; and azithromycin (15-carbon), rokitamycin, and spiramycin (16-carbon) represent the third group with no interaction potential.
The inhibitory effects of macrolides on CYP3A have been analyzed based on competitive or noncompetitive inhibition (Echizen et al., 1993; Wrighton and Ring, 1994; von Moltke et al., 1996; Thummel and Wilkinson, 1998). On the other hand, complex formation with P450 is also reported to be involved in the inhibition by macrolides (Murray, 1987; Periti et al., 1992). Tinel et al. (1989) evaluated the complex formation potential of macrolides using the liver microsomes from dexamethasone-treated rats and reported that the rate of complex formation was highest for troleandomycin, followed by erythromycin, and that the rates for clarithromycin and roxithromycin were lower than those for erythromycin. Using human liver microsomes, Yamazaki and Shimada (1998) have reported similar results, showing that the complex formation potential is greatest for troleandomycin, followed by erythromycin, with roxithromycin having the lowest potential.
In the prediction of in vivo drug interactions involving this type of enzyme inhibition from in vitro studies, the exposure time of the enzyme to the inhibitor should be taken into account as well as the turnover rate of the enzyme. We have already succeeded in quantitatively predicting the triazolam/erythromycin interaction in humans based on a physiologically based pharmacokinetic model taking the type of inhibition into consideration (Kanamitsu et al., 2000b). Yamano et al. (2001) have also presented a successful prediction of interaction between midazolam and erythromycin using a similar model. In the present study, a similar model was applied to the interaction between midazolam and macrolides with different inhibitory potentials.
At first, we tried to predict the increase in midazolam AUC assuming competitive inhibition of CYP3A by erythromycin and clarithromycin (Table 6). Gascon and Dayer (1991) have reported the inhibition constant (Ki) of erythromycin on midazolam α-hydroxylation by human liver microsomes, assuming competitive inhibition. The maximum unbound concentration of erythromycin at the inlet to the liver (Iin,u) was estimated by Iin,u = (Imax + ka Dose Fa/Q) × fb, where Imax is the maximum concentration in the systemic circulation (Ito et al., 1998). Since midazolam is eliminated from human body predominantly via CYP3A-mediated metabolism (Smith et al., 1981), the AUC increase by erythromycin was predicted by 1 + Iin,u/Ki (Ito et al., 1998). The increase in the midazolam AUC produced by clarithromycin was also predicted in the same way, except that the IC50 value reported by Gascon and Dayer (1991) was used instead of the Ki, which was not available for clarithromycin. As shown in Table 6, almost no increase was predicted in the AUC of midazolam, indicating that the reported 3.6- to 7.0-fold increase in vivo (Olkkola et al., 1993; Yeates et al., 1996; Zimmermann et al., 1996; Gorski et al., 1998) cannot be explained by competitive inhibition of the enzyme.
Prediction of the increase in midazolam AUC assuming a competitive inhibition of CYP3A by erythromycin or clarithromycin
In the present in vitro studies using human liver microsomes, midazolam metabolism was not inhibited without preincubation, even if the concentration of the macrolides was increased, and the degree of inhibition depended on the preincubation time and the macrolide concentration (Fig. 2). These findings indicate that the inhibitory effect of macrolides on midazolam metabolism is predominantly caused by mechanism-based inhibition of CYP3A, with little contribution from competitive inhibition. The estimated values of K′app and kinact for each macrolide were almost the same for both midazolam hydroxylation pathways (Table 4), and the values for erythromycin were comparable to those for triazolam metabolism reported by Kanamitsu et al. (2000b).
As shown in Table 5, using the kdeg of 0.0005 min–1, the increase in midazolam AUC was slightly underestimated. The ratio of the predicted and reported increase in midazolam AUC was between 0.6 and 0.8 except for the case of oral administration of midazolam after clarithromycin treatment (500 mg b.i.d. for 7 days) (Gorski et al., 1998), in which case the in vivo interaction was significantly underestimated. One of the reasons for this may be incorrect estimation of the observed AUC due to the lack of midazolam concentration data in the elimination phase.
The average turnover rate constant (kdeg) of rat P450 (0.0005 min–1) (Shiraki and Guengerich, 1984) was used in the present simulation because the corresponding value for human CYP3A has not been reported. When the minimum reported value of kdeg (0.00033 min–1) was used, almost the same results were obtained for clarithromycin and azithromycin, whereas the predicted degree of in vivo interaction was increased in the case of erythromycin, compared with using the kdeg of 0.0005 min–1 (Table 5). Once an enzyme is inactivated in vivo by a mechanism-based inhibitor such as macrolides, the recovery of the metabolic activity depends solely on the synthesis of the enzyme. Thus, the turnover rate of the enzyme is one of the most important parameters in the prediction of interactions involving mechanism-based inhibition, and in the present study, this was found to affect the results in some cases. In cases where the turnover rate of the human enzyme is unavailable, it seems to be important to alter this parameter to some extent in the simulation, referring to animal data, to predict the range of the interaction.
In addition, the liver-to-blood concentration ratio (Kp) of midazolam and macrolides was assumed to be 1 in the present prediction because this cannot be measured in humans. To examine the effect of Kp on the prediction, the Kp of midazolam was changed to 0.1 and 10, whereas that of the macrolides was fixed at 1, and the same simulation was conducted using the values of Vd and CLint of midazolam redetermined to fit its concentration profile in blood. In another case, the Kp of macrolides was changed to 10, with that of midazolam being fixed at 1 and the simulation was conducted in a similar manner. As a result, the predicted increase in midazolam AUC was 2.8- to 3.0-fold for erythromycin (500 mg t.i.d. for 6 days), 2.8- to 2.9-fold for erythromycin (500 mg t.i.d. for 5 days), 2.3- to 2.5-fold for clarithromycin (500 mg b.i.d. for 7 days), 2.1-fold for clarithromycin (250 mg b.i.d. for 5 days), and 1.0- to 1.1-fold for azithromycin (500 mg o.d. for 3 days), suggesting that the value of Kp has little impact on the prediction.
Although only the interaction involving the hepatic enzyme has been evaluated in the present in vitro study, it has been reported that CYP3A is also present in the small intestine (Paine et al., 1997) and that midazolam is metabolized by human intestinal microsomes in vitro (Thummel et al., 1996). In vivo human studies have revealed that the hepatic and intestinal availability of midazolam is 0.74 and 0.42, respectively (Gorski et al., 1998), demonstrating that there is also significant intestinal metabolism of midazolam in vivo. Furthermore, the intestinal availability (Fg) of midazolam is reported to increase approximately 2-fold by pretreatment with either erythromycin or clarithromycin (Olkkola et al., 1993; Gorski et al., 1998), indicating an interaction involving intestinal first-pass metabolism of midazolam. In the present study, the reported values of midazolam Fg, with and without pretreatment with macrolides, were used in the prediction. However, in the case of interaction with azithromycin, the control Fg value was used as a constant value assuming the absence of an interaction involving intestinal metabolism, because of lack of any reported value of Fg after pretreatment with azithromycin. The effects of 10 min of preincubation with erythromycin (100 μM), clarithromycin (100 μM), and azithromycin (1000 μM) on midazolam metabolism were similar between liver and jejunal microsomes from the same three donors, the degree of inhibition by azithromycin being much smaller than that by erythromycin or clarithromycin (unpublished observation). This finding indicates the validity of the above assumption that azithromycin does not cause an interaction in the small intestine. However, because the Fg values used were in vivo data ignoring the time course of intestinal metabolism, efforts are being made by our group to predict the interaction involving the small intestine from in vitro data.
Recently, Mayhew et al.
(2000) proposed a more simple
method of predicting in vivo drug interactions involving metabolic
intermediate complex formation. Based on the inhibitor-induced change in the
steady-state enzyme level ([E]ss), the degree of increase in the AUC after
oral administration can be estimated by the following equation:
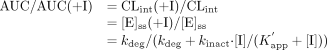 where (+I) represents the value in the presence of the inhibitor and [I]
represents the concentration of inhibitor. We have tried to predict the
above-described interactions involving macrolides according to eq. 17, using
the kdeg value for rat P450 as above and the
K′ app and kinact values for
α-hydroxylation of midazolam obtained in the present study
(Table 7). The maximum
concentration in blood (Imax), maximum unbound concentration in
blood (Imax,u), or maximum unbound concentration at the inlet to
the liver (Iin,u) was used as [I]. As shown in
Table 7, the predicted increase
in the midazolam AUC was comparable to the reported value when
Iin,u was used. Applying this methodology to other interactions
involving mechanism-based inhibitors should provide information for
determining what concentration of inhibitor (Imax, Iin,u
etc.) is appropriate for [I] in eq. 17.
where (+I) represents the value in the presence of the inhibitor and [I]
represents the concentration of inhibitor. We have tried to predict the
above-described interactions involving macrolides according to eq. 17, using
the kdeg value for rat P450 as above and the
K′ app and kinact values for
α-hydroxylation of midazolam obtained in the present study
(Table 7). The maximum
concentration in blood (Imax), maximum unbound concentration in
blood (Imax,u), or maximum unbound concentration at the inlet to
the liver (Iin,u) was used as [I]. As shown in
Table 7, the predicted increase
in the midazolam AUC was comparable to the reported value when
Iin,u was used. Applying this methodology to other interactions
involving mechanism-based inhibitors should provide information for
determining what concentration of inhibitor (Imax, Iin,u
etc.) is appropriate for [I] in eq. 17.
Prediction of the increase in midazolam AUC by macrolides using the steady-state enzyme level
The degree of interaction with CYP3A substrates varies among the macrolides. In the present study, some of these interactions have been successfully predicted from in vitro data. If an interaction involving mechanism-based inhibition of the enzyme is analyzed assuming a competitive inhibition, the in vivo interaction should be greatly underestimated. Quantitative predictions from in vitro data taking the inhibition type into consideration are essential for avoiding toxic interactions in clinical practice.
Acknowledgments
We express our appreciation to Dr. Yuichi Sugiyama (Graduate School of Pharmaceutical Sciences, University of Tokyo) for helpful discussions and reviewing the manuscript. We are grateful to Dr. Noriaki Shimada (Daiichi Pure Chemicals Co., Ltd.) for kindly providing human liver microsomes from BD Gentest.
Footnotes
-
↵1 Abbreviations used are: AUC, area under the concentration-time curve; o.d., once daily; P450, cytochrome P450; HPLC, high-performance liquid chromatography.
-
This work was supported in part by a Kitasato University Research Grant for Young Researchers.
- Received September 3, 2002.
- Accepted March 28, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}