


Abstract
The pharmacokinetics and metabolism of the direct thrombin inhibitor dabigatran (BIBR 953 ZW, β-alanine, N-[[2-[[[4-(aminoiminomethyl)phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl]-N-2-pyridinyl) were studied in 10 healthy males, who received 200 mg of [14C]dabigatran etexilate (BIBR 1048 MS, the oral prodrug of dabigatran) or an i.v. infusion of 5 mg of [14C]dabigatran. Radioactivity was measured in plasma, urine, and feces over 1 week. The metabolite pattern was analyzed by high-performance liquid chromatography with on-line radioactivity detection, and metabolite structures were elucidated by mass spectrometry. Dabigatran etexilate was rapidly converted to dabigatran, with peak plasma dabigatran concentrations being attained after approximately 1.5 h; the bioavailability of dabigatran after p.o. administration of dabigatran etexilate was 7.2%. Dabigatran was predominantly excreted in the feces after p.o. treatment and in the urine after i.v. treatment. The mean terminal half-life of dabigatran was approximately 8 h. The predominant metabolic reaction was esterase-mediated hydrolysis of dabigatran etexilate to dabigatran. Phase I metabolites accounted for ≤0.6% of the dose in urine and 5.8% of the dose in feces following p.o. administration and ≤1.5 and 0.2%, respectively, following i.v. administration. Dabigatran acylglucuronides accounted for 0.4 and 4% of the dose in urine after p.o. and i.v. dosing, respectively. In vitro experiments confirmed that dabigatran etexilate is metabolized primarily by esterases and that cytochrome P450 plays no relevant role. These findings suggest that pharmacologically active concentrations of dabigatran are readily achieved after p.o. administration of dabigatran etexilate and that the potential for clinically relevant interactions between dabigatran and drugs metabolized by cytochrome P450 is low.
Antithrombotic therapy plays an important role in the prevention and treatment of thromboembolic disorders. However, currently available agents are subject to certain limitations. Oral vitamin K antagonists, such as warfarin, have unpredictable pharmacokinetics and show numerous drug and food interactions (Ansell et al., 2004), whereas unfractionated and low molecular weight heparins and fondaparinux require parenteral administration. An orally active direct thrombin inhibitor would offer a number of potential advantages over these agents (Weitz and Bates, 2005).
Dabigatran is a reversible, competitive, direct thrombin inhibitor that has been shown to be an effective antithrombotic agent in animal models (Stassen et al., 2001; Wienen et al., 2001a,b) and to be efficacious and safe in the prevention of deep vein thrombosis in patients undergoing elective total hip or knee replacement (Eriksson et al., 2005). Dabigatran etexilate is currently in Phase III development for primary prevention of venous thromboembolism (VTE) in patients undergoing major orthopedic surgery, acute VTE treatment, and VTE secondary prevention, as well as stroke prevention in patients with atrial fibrillation. Pharmacokinetic studies in healthy volunteers and orthopedic surgery patients showed that dose-dependent concentrations of dabigatran are achieved after p.o. administration of dabigatran etexilate, with peak concentrations reached after approximately 2 h and with a slight delay up to 6 h on the day of surgery (Eriksson et al., 2004, 2005; Stangier et al., 2005).
This article describes a series of in vivo and in vitro studies performed to investigate the pharmacokinetics and metabolism of dabigatran in humans.
Materials and Methods
Reference Compounds and Other Materials. Radiolabeled [14C]dabigatran and [14C]dabigatran etexilate, with specific radioactivities of 0.486 and 0.859 MBq/mg ([14C]dabigatran etexilate) and 1.251 MBq/mg ([14C]dabigatran), were synthesized by the isotope chemistry laboratory of Boehringer Ingelheim GmbH (Ingelheim, Germany). The 14C-label was located at position 2 of the benzimidazole moiety. Radiolabeled purity of [14C]dabigatran etexilate and [14C]dabigatran was >98.3 and 98.1%, respectively. The 14C-labeled drugs were blended with the corresponding nonradiolabeled drug substances using Good Manufacturing Practice batches released for human use.
Nonradioactive dabigatran (BIBR 953 ZW), dabigatran etexilate (BIBR 1048 MS), and the reference compounds BIBR 951 CL, BIBR 1087 SE, BIBR 1151 ZW, CD00000338, BI00003521, BI00003522, BI00003543, and BI00003563 were synthesized and analyzed at Boehringer Ingelheim GmbH and Co KG (Biberach, Germany).
Other chemicals were of analytical grade or higher purity and were obtained from commercial suppliers.
In Vivo Pharmacokinetics and Disposition.Subjects/radiation protection. The pharmacokinetics of dabigatran and dabigatran etexilate were studied in 10 healthy Caucasian male volunteers at Charles River Laboratories International, Inc. (Tranent, Edinburgh, UK). The mean age of the participants was 39.9 years; their mean weight was 80.2 kg; and their mean body mass index was 25.8 kg/m2. The whole body radioactivity dose (effective dose) was less than 1 mSV. The study was approved by the independent Ethics Committee of Charles River Laboratories and conducted according to the principles of Good Clinical Practice and the Declaration of Helsinki. Written informed consent was obtained from all the participants.
Drug administration. Participants were randomized in a 1:1 ratio to receive a single p.o. dose of 200 mg of [14C]dabigatran etexilate or a single i.v. infusion of 5 mg of [14C]dabigatran administered over 30 min after an overnight fast. The total dose of radioactivity per subject was 2.8 MBq (76 μCi) in both cases. [14C]BIBR 1048 MS was administered in aqueous solution with mannitol, acidified with tartaric acid; [14C]BIBR 953 ZW was given in aqueous solution with mannitol together with polypropylene glycol, acidified with hydrochloric acid.
Sample collection. Blood samples were obtained either via an indwelling cannula in a forearm vein or by venipuncture. Blood sampling time-points were as follows:
-
p.o. administration: predose, 15, 30, and 45 min, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, 96, 120, 144, and 168 h; and
-
i.v. administration: predose, 5, 10, 15, 20, 25, 29 (immediately before end of infusion), 31, 33, 36, 40, 45, and 55 min, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, 96, 120, 144, and 168 h.
Samples were immediately placed on ice, and after removal of a 1-ml portion for measurement of radioactivity in whole blood, plasma was separated by centrifugation. Urine was collected before dosing, from 0 to 4, 4 to 8, 8 to 12, and 12 to 24 h after dosing, and thereafter over 24-h periods to 7 days after dosing. Plasma and urine samples were diluted 1:1 with 0.2 N hydrochloric acid. Feces were collected before dosing and subsequently over 24-h periods for 7 days. Pooled 24-h feces samples were acidified with 0.2 N hydrochloric acid and homogenized.
Sample storage and shipping conditions. After sample collection, blood, plasma, urine, and feces samples were stored frozen at –20°C until analysis. Samples were shipped to other analytical laboratories under dry ice.
Analysis of dabigatran and dabigatran acylglucuronides. Concentrations of dabigatran in plasma and urine samples were measured by means of validated liquid chromatography/tandem mass spectrometry (LC/MS/MS) methods.
The internal standard used was 13C6-labeled BIBR 953 ZW. The linear calibration curves covered a range of 1 to 400 ng/ml BIBR 953 ZW; the lower limit of quantitation was 1.00 ng/ml.
For the determination of free (nonconjugated) dabigatran, 40 μl of internal standard solution ([13C6]BIBR 953 ZW, 1000 ng/ml) was added to a 100-μl sample aliquot, then mixed, centrifuged, and 10 μl of the supernatant injected onto the LC/MS/MS system. For the determination of total (i.e., free plus conjugated) dabigatran, 40 μl of internal standard solution ([13C6]BIBR 953 ZW, 1000 ng/ml) was added to a 100-μl sample aliquot, then mixed, alkalized with 55 μl of 0.2 N NaOH, and incubated at 37°C for 2 h. The incubation of samples with NaOH was determined to be sufficient to cleave all the acylglucuronides. After centrifugation, 10 μl of the supernatant was injected onto the LC/MS/MS system. Analytes were extracted by on-line solid-phase extraction (column-switching) on a Bondapak C18 Porasil B (20 × 2 mm, 37–75 μm) enrichment column (Waters, Milford, MA). Chromatography was performed on a Purospher RP-18 E analytical column (60 × 2 mm, 5 μm) with a 20 × 2-mm guard column of the same material (Merck, Darmstadt, Germany). Mobile phases were 0.01 M ammonium formate buffer, pH 4.5 (A) and acetonitrile (B) using a gradient from 15 to 50% B at a flow rate of 0.3 ml/min. Analytes were quantified on a Sciex API 365 triple quadrupole mass spectrometer (Applied Biosystems, Foster City, CA) operated in positive electrospray mode. Transitions from m/z = 472.0 to m/z = 288.9 and from m/z = 478.0 to m/z = 294.9 were recorded for dabigatran and the internal standard, respectively. In addition, the transition from m/z = 648.2 to m/z = 288.9 was monitored for the presence of conjugates.
The fraction of acylglucuronide conjugates of dabigatran in plasma was assessed by determination of the concentration of free, nonconjugated, dabigatran in plasma and measurement of dabigatran concentrations after complete alkaline cleavage of conjugates (total dabigatran in plasma). The difference between total and free dabigatran concentrations in plasma was considered to represent dabigatran conjugates.
Protein binding. The binding of [14C]dabigatran to plasma proteins was measured by ultrafiltration. Plasma samples were spiked with unlabeled dabigatran to produce concentrations of 50, 500, and 5000 ng/ml and incubated for 10 min at 37°C. The radioactive concentration in the plasma was measured as described below, and duplicate portions of plasma were transferred to Centrifree tubes (Amicon Inc., Beverly, MA) and centrifuged at less than 2000g for 20 min. The radioactive concentration in the ultrafiltrate was measured, and protein binding was determined as a percentage of radioactivity in the ultrafiltrate relative to that in unfiltered plasma.
Pharmacokinetic evaluation. Pharmacokinetic parameters of dabigatran in plasma and urine were calculated by means of WinNonlin (version 3.1) software (Pharsight Corp., Mountain View, CA). The following pharmacokinetic parameters were determined: maximum plasma concentrations, Cmax; time to peak concentrations, Tmax; area under the plasma concentration-time curve extrapolated to infinity, AUC0-∞ (calculated as AUC0-t + Ct/λz, where Ct is concentration at time t and λz is the slope of the terminal elimination phase); mean residence time, MRT0-∞; CLtot/F and Vz/F, and total clearance, Cltot, and steady-state volume of distribution, Vss, after i.v. infusion. In addition, the absolute bioavailability of dabigatran was assessed.
Measurement of radioactivity. Duplicate samples of urine were mixed with 10 ml of Quickszint 1 (Zinsser Analytic GmbH, Frankfurt, Germany). Duplicate samples of whole blood and fecal homogenates were combusted in a Packard Tri-Carb 307 automatic sample oxidizer (PerkinElmer, Boston, MA). The [14C]carbon dioxide released was trapped in 3-methoxypropylamine (Carbosorb, PerkinElmer), and scintillation fluid (Permafluor E+, PerkinElmer) was added. Combustion of standards showed that recovery of [14C] was quantitative (>97%). Radioactivity was determined by liquid scintillation counting using a Packard TR 2100 (Canberra Industries, Meriden, CT) with automatic external quench correction. The standard counting time was 5 min. The resulting data were recorded in Debra data management software, version 5.2 (LabLogic, Sheffield, UK).
Sample preparation for metabolite investigation. Metabolite pattern analysis was performed in plasma, urine, and feces of individual subjects by high-performance liquid chromatography (HPLC) coupled to on-line radioactivity detection. Plasma samples obtained at 2, 4, and 6 h after p.o. administration of [14C]dabigatran etexilate and 40 min, 2 h, and 4 h after i.v. administration of [14C]dabigatran, and urine samples obtained over 0 to 4, 4 to 8, 8 to 12, 12 to 24, and 24 to 48 h after dosing were processed by solid-phase extraction on OASIS HLB columns (Waters) preconditioned with 0.1% formic acid in acetonitrile and equilibrated with 0.1% aqueous formic acid. After loading with sample, the columns were rinsed with 0.1% aqueous formic acid. Absorbed material was eluted with 0.1% formic acid in acetonitrile/water (1:1). Feces homogenates were exhaustively extracted by 0.1% formic acid in acetonitrile/water (1:1). Average extraction yields of plasma, urine, and feces samples were 102, 98, and 91%, respectively. Extracts and combined extracts of sample material were lyophilized, and the residues were dissolved in aqueous formic acid (0.1%) containing 3% methanol for HPLC and liquid chromatography/mass spectrometry (LC/MS) analysis.
Metabolite pattern analysis (HPLC coupled to on-line radioactivity detection). The HPLC systems used for metabolite pattern analysis consisted of autosamplers (LC PAL, CTC Analytics, Zwingen, Switzerland), HPLC pumps (G1312A, Agilent Technologies, Waldbron, Germany), degassers (G1379A, Agilent Technologies), and radioactivity detectors (LB 507B or LB 509, Berthold, Bad Wildbad, Germany) equipped with solid-phase scintillation cells (YG 150 S4D). The separation was additionally monitored by diode array detectors (G1315A, Agilent Technologies). The software used was Chromelion, version 6.2 (Dionex, Idstein, Germany). Samples were analyzed on 70 × 4-mm Nucleosil C100–5 C18 HD columns protected by 8 × 4-mm Nucleosil 100 C18 HD guard columns (both 5-μm particle size; Marcherey-Nagel, Düren, Germany). Metabolites were separated with a gradient of aqueous ammonium formate (0.02 M, pH 8, adjusted with ammonium hydroxide: mobile phase A) versus ammonium formate in methanol (0.02 M, pH 8, adjusted with ammonium hydroxide: mobile phase B) at a flow rate of 0.8 ml/min (gradient: 3% B at 0 min, linear to 20% B at 20 min, linear to 85% B at 31.5 min). With a signal to noise ratio = 2, the HPLC systems were linear (r2 ≥ 0.99) over the range of 425 to 88,935 dpm (absolute amount injected on column), respectively, as assessed by triplicate injections of [14C]dabigatran at various concentrations. The inaccuracy and imprecision of the method were ≤7.9 and ≤15%, respectively.
Metabolites were quantified based on the relative amount of radioactivity that was assigned to a given metabolite fraction in relation to the total amount of radioactivity present in the analyzed sample. Parent drug and metabolites were expressed as percentage of sample radioactivity in plasma or as percentage of the dose in excreta.
With a limit of detection at 425 dpm (absolute amount injected on column), the lower limits of detection for metabolites after i.v. administration of [14C]dabigatran were estimated to be 1 to 8 ng-Eq/ml in plasma, 1 to 19 ng-Eq/ml in urine, and 11 to 29 ng-Eq/ml in feces. The corresponding figures after p.o. administration of [14C]dabigatran etexilate were 61 to 109, 16 to 374, and 0.1 to 14.9 ng-Eq/ml, respectively.
Elucidation of Metabolite Structures by LC/MS. Aliquots of plasma, urine, and feces samples were analyzed by electrospray ionization mass spectrometry in positive ion mode using a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Micromass/Waters, Manchester, UK) equipped with a combined Z-spray/lockspray interface. The instrument was coupled to the postcolumn flow of an HPLC/radioactivity detection system and gradient as described above (see under Metabolite pattern analysis). Samples were separated on 70 × 2-mm Nucleosil C100–5 C18 HD columns protected by 8 × 3-mm Nucleosil 100 C18 HD guard columns (both 5-μm particle size; Marcherey-Nagel) at a flow rate of 0.4 ml/min.

Mean (± S.D.) concentrations of free and total dabigatran in plasma following p.o. administration of 200 mg of [14C]dabigatran etexilate (2.8 MBq) (A) or i.v. infusion of 5 mg of [14C]dabigatran (2.8 MBq) (B) in healthy male volunteers (n = 5 per treatment).
The electrospray interface was operated with nitrogen as nebulizer and desolvation gas at flow rates of 20 and 300 l/h, respectively, and a desolvation temperature of 250°C. The capillary and cone voltage was set to 3.0 kV and 60 V, respectively. The first quadrupole was operated at unit mass resolution. Collision-induced dissociation experiments were performed with collision energies of 10 to 30 eV with argon as collision gas at a pressure of 1.1 × 10–3 mbar. The time of flight analyzer was operated in V-mode optics at an average resolution of 11,000 (full-width at half-maximum definition) with a pusher frequency of 16 kHz. High-resolution mass spectra in “full scan” and “product ion scan” mode were acquired in the range of m/z 50 to 1000. Full scan mass spectra were calibrated using [(H3PO4)5 + H]+, m/z 490.8923 of phosphoric acid (0.01%); product ion mass spectra were internally calibrated by known pseudomolecular [M+H]+ or fragment ions. Metabolite structures were elucidated by the detailed analysis of the fragmentation pattern in comparison with synthetic reference compounds.
In Vitro Studies.Human liver microsomes. Human liver microsomes [pools of 15–29 donors; 0.354–0.422 nmol cytochrome P450 (P450)/mg protein] were obtained from Xenotech LLC (Kansas City, KS). Information on enzyme activities was provided by the supplier.
Microsomes from a human B-lymphoblastoid cell line expressing human CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C9-Arg, CYP2C19, CYP2D6-Val, CYP2E1, CYP3A4, CYP4A11 cDNAs and control microsomes from cells lacking the human P450 cDNA insert were from BD Biosciences (Woburn, MA) and delivered by NatuTec (Frankfurt, Germany).
Human plasma. Human plasma was prepared from two male and two female healthy volunteers. Blood was taken shortly before use and collected in heparin-containing vials as an anticoagulant. Plasma was prepared by centrifugation of blood (10,000g, 15 min), pooled, and stored on ice until used for experiments.
Incubation experiments. Reaction mixtures consisted of 0.1 M Tris buffer (pH 7.4 at 37°C), 5 mM magnesium chloride, up to 0.5 mg of microsomal protein/ml, 1 to 2 mM NADPH, the test compound, and, where necessary, the inhibitor in a total volume of 0.2 or 0.25 ml. Reactions were started by addition of [14C]dabigatran etexilate and carried out at 37°C for up to 60 min.
For incubations with human plasma, plasma was diluted with an equal volume of 0.2 M Tris buffer and preincubated at 37°C for 3 min. The reaction was started by addition of [14C]dabigatran etexilate and performed for up to 4 h at 37°C in a total volume of 0.25 ml.
Incubations containing esterase inhibitors [physostigmine, paraoxon, bis-(p-nitrophenyl) phosphate sodium (BNPP), p-chloromercuribenzoate (PCMB), phenylmethylsulfonyl fluoride (PMSF), tetraisopropyl pyrophosphoramide (iso-OMPA)] were preincubated for 20 min before reactions were initiated by the addition of [14C]dabigatran etexilate.

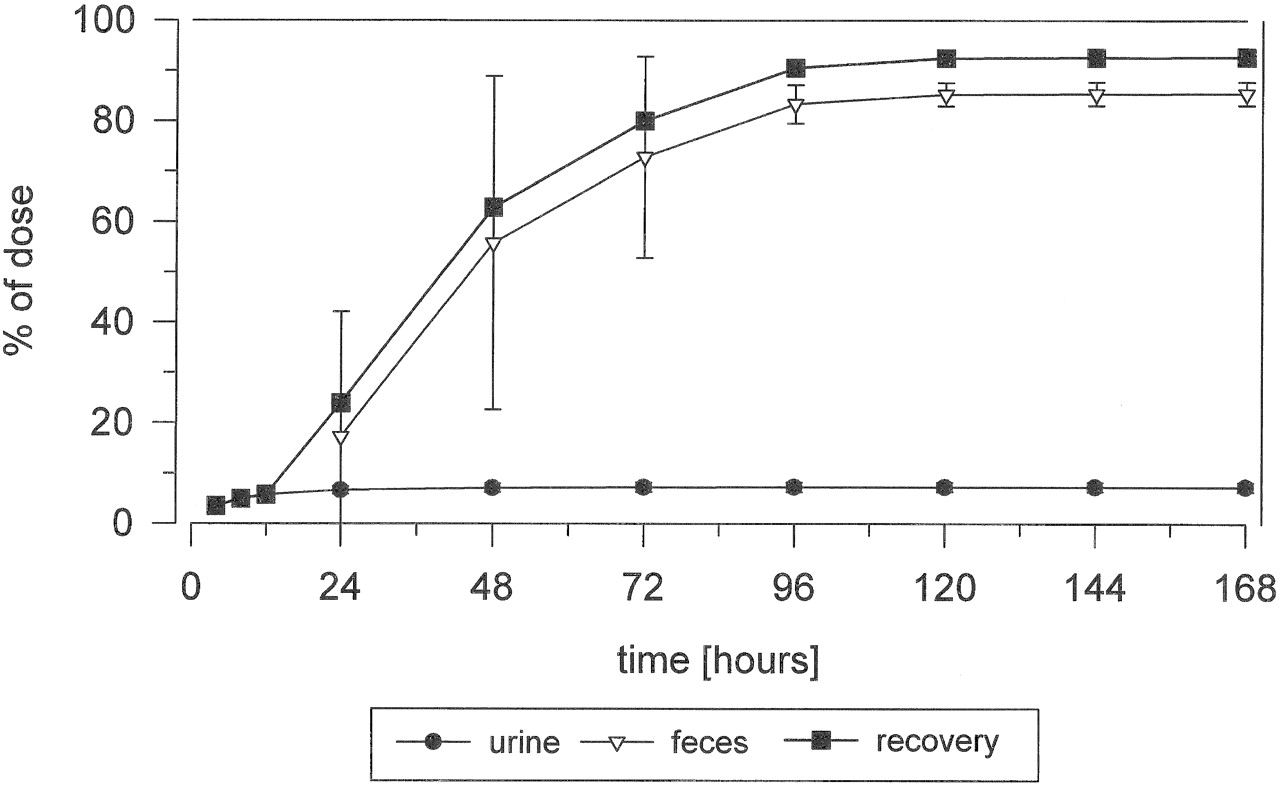
Mean cumulative excretion of total radioactivity following a single p.o. dose of 200 mg of [14C]dabigatran etexilate (2.8 MBq) in healthy male volunteers (n = 5).
Reactions were terminated by cooling to 4°C, adding an equal volume of 0.2 M hydrochloric acid, and vortex-mixing. All the incubations were performed at least in duplicate.
HPLC of dabigatran and metabolites in incubates. Incubates were centrifuged, and 100-μl samples were injected directly into the HPLC system, which consisted of an autosampler (HTC PAL, CTC Analytics AG), a PU-980 pump with ternary gradient unit, 3-line degasser (Jasco, Groβumstadt, Germany), a radioactivity detector (flow scintillation analyzer TR 525, PerkinElmer), a fluorescence detector (FP 920, Jasco) at an excitation and emission wavelength of 320 and 400 nm, respectively, and a UV-975 detector (Jasco) at a wavelength of 292 nm. Chromatographic separation of metabolites was carried out on a 5-μm LiChroCART Purospher RP 18-e 125–2 column (Merck) protected by a 5-μm LiChroCART Purospher RP 18-e 10–2 column (Merck). The mobile phase consisted of a linear gradient of 50 mM formic acid, adjusted to pH 4.0 with ammonia solution, versus acetonitrile at a flow rate of 0.4 ml/min.
P450-dependent reactions. Standard assays were used to measure the activities of P450 isoenzymes in human liver microsomes and to investigate the effect of dabigatran etexilate, dabigatran, BIBR 951 CL, and BIBR 1987 SE on the various test reactions. CYP1A1 and CYP1A2 activity was measured by phenacetin O-deethylation (Tassaneeyakul et al., 1993), CYP2C19 and CYP2B6 by S-mephenytoin 4′-hydroxylation (Wedlund and Wilkinson, 1996) and N-dealkylation (Heyn et al., 1996), respectively, CYP2C9 by tolbutamide hydroxylation (Ludwig et al., 1998), CYP2D6 by bufuralol 1′-hydroxylation (Yamazaki et al., 1994), CYP2E1 and CYP4A11 by lauric acid 11- and 12-hydroxlation, respectively (Clarke et al., 1994; Amet et al., 1995), and CYP3A by nifedipine oxidation (Guengerich et al., 1986) and testosterone 6β-hydroxylation (modified from the technique of Newton et al., 1995). All the incubations were performed in duplicate.

Mean cumulative excretion of total radioactivity following a single i.v. dose of 5 mg of [14C]dabigatran (2.8 MBq) in healthy male volunteers (n = 5).
Results
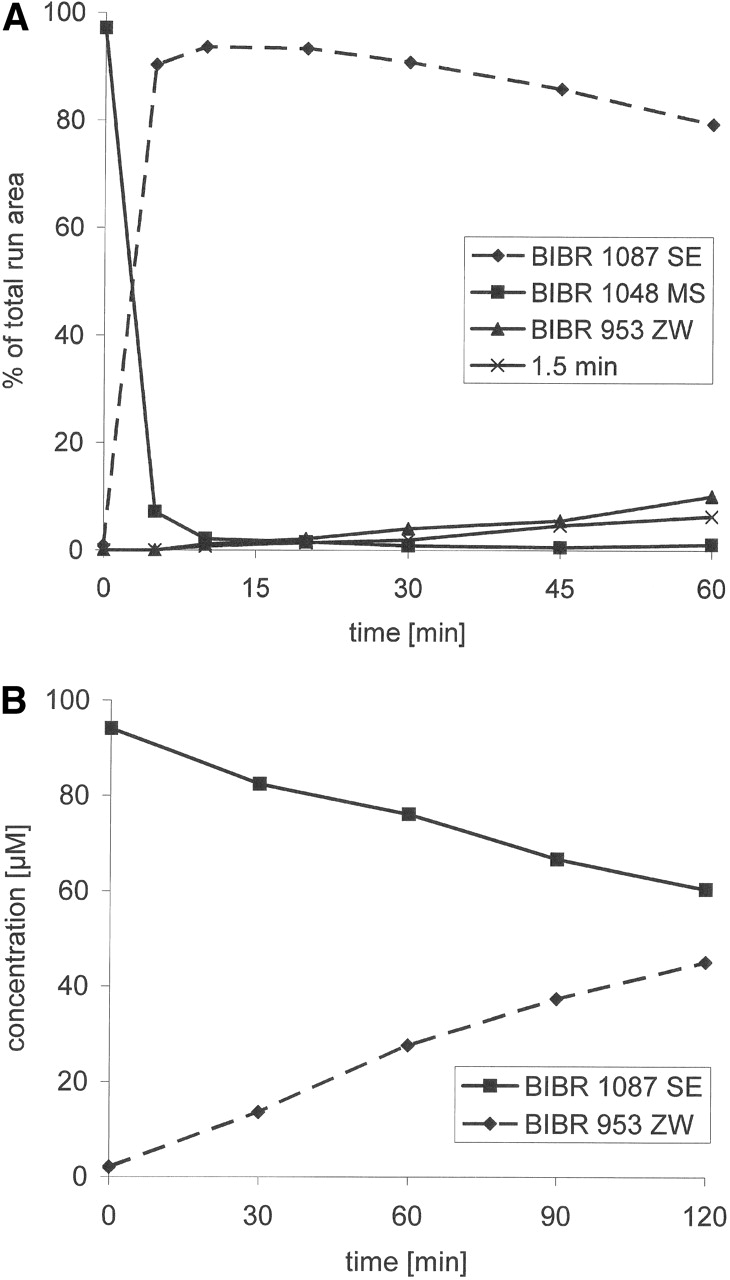
Pharmacokinetics and Disposition of Dabigatran. Total radioactivity in plasma following p.o. administration of [14C]dabigatran etexilate or i.v. administration of [14C]dabigatran is shown in Fig. 1, A and B, respectively. The p.o. administration of 200 mg of [14C]dabigatran etexilate resulted in a mean peak concentration of [14C]dabigatran etexilaterelated radioactivity of 244.4 ± 56.8 (S.D.) ng-Eq/ml, which was attained 1.5 h after dosing. Total radioactivity declined thereafter to a mean of 35.8 ng-Eq/ml at 12 h; all the subsequent samples were below the limit of reliable measurement (i.e., less than 30 dpm above background). The peak plasma concentration of [14C]dabigatran following i.v. administration of 5 mg of [14C]dabigatran was 258.2 ± 43.1 ng-Eq/ml, which was observed at the end of infusion (Fig. 1B). By 36 h after dosing, concentrations had decreased to 2.5 ng-Eq/ml, and all the subsequent samples were below the limit of reliable measurement. The total radioactivity in whole blood resembled that in plasma but at lower levels.
Following p.o. administration of [14C]dabigatran etexilate, the mean (S.D.) erythrocyte/plasma concentration ratio was between 0.1 ± 0.1 at 30 min after dosing and 0.4 ± 0.2 at 4 h after dosing. Similarly, after i.v. administration of [14C]dabigatran, the mean erythrocyte/plasma concentration ratio at the end of infusion and 8 h postdose was between 0.1 ± 0.2 and 0.3 ± 0.2.
The percentage of dabigatran bound to plasma proteins was approximately 35% and remained constant over the concentration range 50 to 5000 ng/ml dabigatran, indicating that the extent of protein binding does not depend on dabigatran plasma concentrations.
Pharmacokinetic parameters of dabigatran following p.o. administration of [14C]dabigatran etexilate or i.v. infusion of [14C]dabigatran are summarized in Tables 1 and 2. The mean plasma concentration-time profiles of dabigatran after p.o. and i.v. administration are shown in Fig. 1, A and B. After p.o. administration of dabigatran etexilate, dabigatran was formed rapidly by complete conversion of the prodrug. Dabigatran was the predominant compound in plasma. The prodrug and its intermediates BIBR 1087 and BIBR 951 were hardly detectable in plasma. Maximal plasma concentrations of dabigatran were attained at about 1.5 h after p.o. administration and followed by a biexponential decrease in plasma concentrations, with the terminal phase apparent from approximately 8 to 12 h after dosing. The mean terminal half-life of dabigatran after p.o. administration was 8.8 h.
Pharmacokinetic parameters of dabigatran, calculated from free dabigatran concentration/time data, following p.o. administration of 200 mg of [14C]dabigatran etexilate (2.8 MBq) or i.v. infusion of 5 mg of [14C]dabigatran (2.8 MBq) in healthy male volunteers (n = 5 per treatment)
Results are presented as mean ± S.D.
Pharmacokinetic parameters calculated from total dabigatran radioactivity data
Results are presented as mean ± S.D.
There was evidence of a multiexponential decrease in dabigatran concentrations following i.v. administration of dabigatran, with the terminal phase apparent from approximately 6 h after dosing. The Vss of 69 to 90 l indicated that dabigatran is not confined to the body water volume. Plasma clearance of dabigatran after i.v. administration was 149 ml/min. The mean terminal half-life of dabigatran was 8.3 h.
In terms of the dabigatran area under the plasma concentration-time curve calculated from zero to the time of last quantifiable drug concentration (AUC0-tz), the amount of dabigatran liberated by cleavage of dabigatran glucuronides was 19.5 and 3.4% of the total exposure to dabigatran following p.o. and i.v. dosing, respectively. Conversely, free, nonconjugated dabigatran accounted for 80.5 and 96.6% of the systemic exposure to total (free plus conjugated) dabigatran in plasma.
The absolute bioavailability of dabigatran following p.o. administration of dabigatran etexilate was approximately 6.0%. With the pharmacologically active glucuronide conjugates of dabigatran in plasma included, the absolute bioavailability of total dabigatran amounted to 7.2%.
Excretion and Mass Balance in Urine and Feces. The mean cumulative excretion of radioactivity following p.o. administration of [14C]dabigatran etexilate is shown in Fig. 2. A mean of 85.5% (range, 82.6–88.6%) of the dose was excreted by 168 h, predominantly in the feces. Within the first 24 h after dosing, a mean of 7.3% (range, 6.3–8.4%) and 17.3% (range, 0.04–52.6%) of the dose was excreted in the urine and the feces, respectively. The mean total recovery of the administered dose during the collection period was 92.7% (range, 88.9–95.7%).
The mean cumulative excretion of radioactivity following i.v. infusion of [14C]dabigatran is shown in Fig. 3. Radioactivity was predominantly excreted via the urine, with a mean of 85.4% (range, 83.3–87.2%) of the dose being excreted via this route within 168 h after dosing; a mean of 78% was excreted within the first 24 h after dosing. The mean total recovery by the end of the collection period was 91.3% (range, 87.9–93.9%).
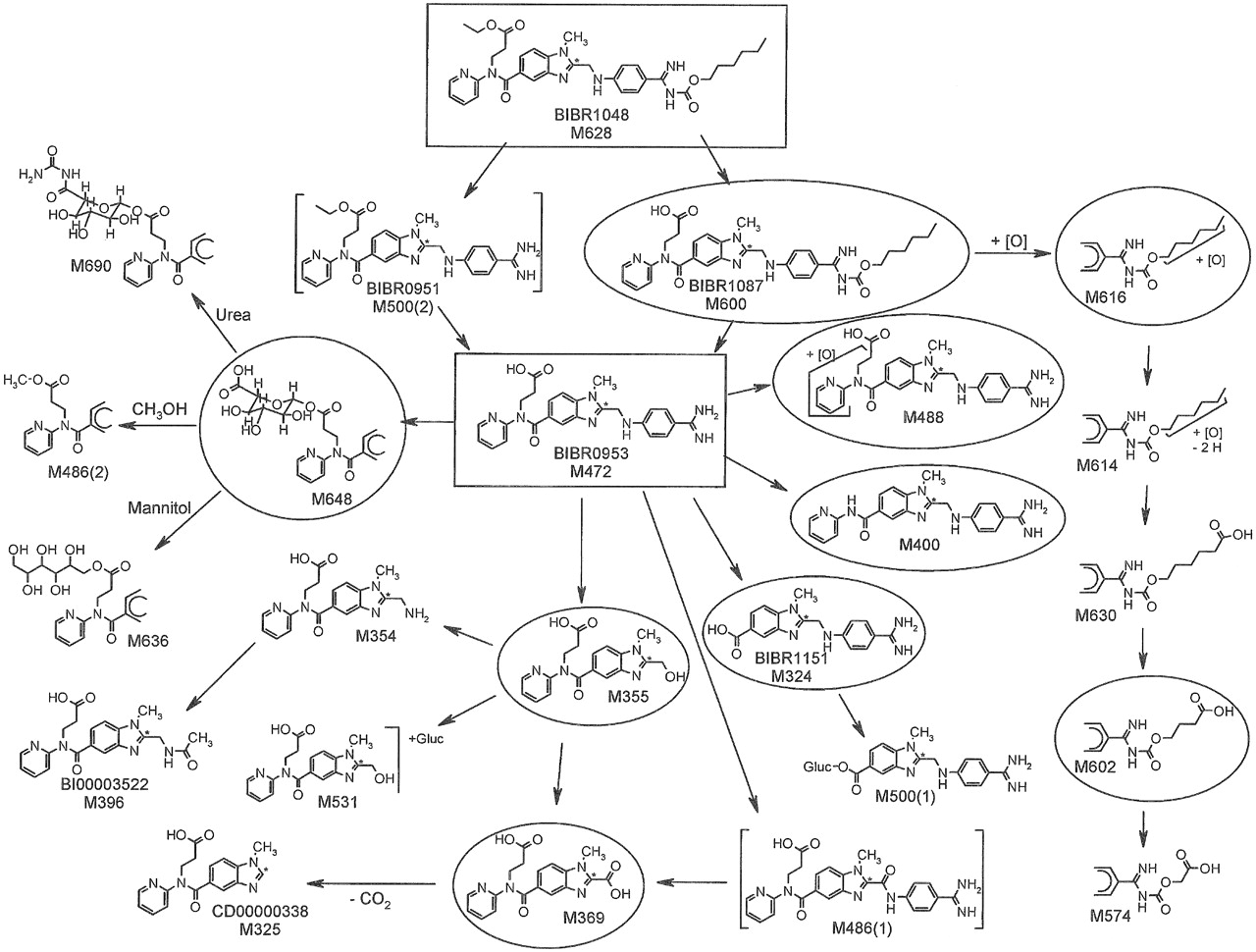
Metabolite Characterization by LC/MS. Metabolite structures were elucidated by LC/MS (Table 3), with detailed analysis of fragmentation pathways of pseudomolecular [M+H]+-metabolite ions in comparison with product ion spectra and retention times of authentic reference compounds (BIBR 951 CL, BIBR 1087 SE, BIBR 1151 ZW, CD00000338, BI00003521, BI00003522, BI00003543, and BI00003563). The proposed structures are given in Fig. 4.
LC/MS data of dabigatran etexilate and dabigatran and metabolites in plasma, urine, and feces following p.o. administration of 200 mg of [14C]dabigatran etexilate or i.v. infusion of 5 mg of [14C]dabigatran to healthy male volunteers
Compounds are listed in order of nominal [M + H]+ masses. Proposed chemical structures are shown in Fig. 4.
The proposed fragmentation pathway of the pseudomolecular ion [M+H]+ of dabigatran, m/z 472, is shown in Fig. 5. The spectrum was dominated by m/z 289, which was formed via m/z 324 by an intramolecular six-ring cyclic rearrangement reaction with loss of ketene (CH2CO) and 2-(methylenimino)pyridine (C6H6N2), and subsequently via m/z 307 and m/z 306 by loss of water and ammonia, respectively. Other fragments were formed by combinations of the loss of acrylic acid (C3H4O2), 4-aminobenzamidine (C7H9N3), 2-pyridinylisocyanate (C6H4N2O), 2-aminopyridine (C6H5N2), water, ammonia, carbon dioxide, and carbon monoxide.
The metabolites were subject to similar fragmentation reactions to dabigatran and dabigatran etexilate, which allowed the localization and elucidation of the metabolic changes in substructures of the parent drugs.
The metabolites M324, M325, M355, M396, M400, and M600 were identified by comparison of LC/MS data of synthetic reference compounds. The amine M354 showed the fragmentation pattern of the alcohol M355. The carboxylic acid M369 showed a weak [M+H]+ because of a rapid decarboxylation. The site of oxidation of the two isomers M488(1) and M488(2) could be assigned to the pyridine moiety because of the shift of the fragment m/z 149 to m/z 165. M488(1) showed the loss of oxygen of the fragment m/z 416 to m/z 400, suggesting a pyridine N-oxide structure. M500(1) was identified as acylglucuronide of the aromatic carboxylic acid M324. The fragment m/z 365 suggested the carboxylic acid of the benzimidazole moiety as site of glucuronidation. The metabolite fraction M531 consisted of isomeric acylglucuronides of the aliphatic carboxylic acid M355. The extracted ion chromatogram of m/z 531 of urine samples showed a broad peak of unresolved isomers at a retention time of 12.2 min. The metabolites M574, M602, M614, M616, and M630 were formed by oxidation of the hexyl side chain. Four isomeric acylglucuronides of dabigatran, M648(1) through M648(4), were identified that were formed by glucuronidation of the aliphatic carboxylic acid and subsequent acyl migration. Two secondary metabolites were elucidated: M636 was identified as the mannitol ester of dabigatran. It was formed via the acylglucuronides of dabigatran by transesterification with mannitol, which was a constituent of the i.v. and p.o. formulation. The isomers M690(1) through M690(4) were identified as condensation products of isomeric dabigatran acylglucuronides with urea. The site of condensation was tentatively localized at the carboxylic acid of the glucuronide moiety. The formation of M636 and the isomers of M690 was confirmed in vitro by incubation experiments of mannitol and urea with isolated dabigatran acylglucuronides.

Metabolic fate of dabigatran etexilate and dabigatran (both in rectangle) following p.o. administration of 200 mg of [14C]dabigatran etexilate or i.v. infusion of 5 mg of [14C]dabigatran to healthy male volunteers. Structures of metabolites were partially or completely characterized by LC/MS and/or by comparison with authentic reference compounds. Metabolites in excreta with ≥0.5% of the administered dose are indicated by a circle.
Metabolism of Dabigatran in Vivo. Representative radio chromatograms of plasma, urine, and feces following p.o. administration of [14C]dabigatran etexilate and i.v. administration of [14C]dabigatran are shown in Figs. 6 and 7, respectively. The metabolic pathway of dabigatran is presented in Fig. 4. Mass spectrometry data of individual metabolites are summarized in Table 3. The quantitative recovery of metabolites in plasma, urine, and feces following i.v. administration of dabigatran and p.o. administration of dabigatran etexilate is summarized in Tables 4 and 5, respectively.
Metabolite pattern in plasma, urine, and feces after i.v. infusion of 5 mg of [14C]dabigatran in healthy volunteers
Data are mean percentage recovery from five individuals.
Metabolite pattern in plasma, urine, and feces after p.o. administration of 200 mg of [14C]dabigatran etexilate in healthy volunteers
Data are mean percentage recovery from five individuals.
Dabigatran was the predominant compound in plasma, urine, and feces following both p.o. administration of dabigatran etexilate and i.v. administration of dabigatran. The principal metabolic reaction following p.o. administration of the etexilate was hydrolysis by esterases of the ethyl ester and the hexyloxycarbonyl moiety, yielding dabigatran via the two intermediates BIBR 1087 SE and BIBR 951 CL. The parent compound, dabigatran etexilate, was not detectable in feces. Only trace amounts were identified by LC/MS analysis in urine. The intermediate metabolite BIBR 951 CL was completely hydrolyzed to dabigatran, whereas a small amount (equivalent to 0.2% of the total dose) of BIBR 1087 SE was excreted in the feces. Oxidation of the hexyl side chain of BIBR 1087 SE resulted in the formation of M616, which accounted for 2.0% of the dose in feces; subsequent oxidation of this metabolite produced M614, M630, and M602, each of which was present in amounts ≤0.5% of the dose in the feces. The isomeric metabolites M488 accounted for 1.2% of the dose in feces and 0.1% of the dose in urine. Other metabolites that were only detectable in plasma by high-sensitivity LC/MS were M324 (BIBR 1151 ZW), its glucuronide M500(1), M355, the acylglucuronide M648, the oxidation product M488, and M396 (BI00003522), the N-acetylated derivative of M354. The acylglucuronides, M648, and their derivatives M690 (generated by condensation with urea) and M636 (a transesterification product with mannitol) accounted for approximately 0.4% of the dose in urine; other metabolites were present in low concentrations, each accounting for less than 0.6% of the dose. In feces, M324 and M400, which were formed by oxidative dealkylation of dabigatran, accounted for 2.3 and 5.8%, respectively, of the dose.
Following i.v. administration of dabigatran, the acylglucuronides accounted for 2.0% of the dose in plasma at 2 h and 4.3% at 4 h; M369 accounted for 0.7% of the dose at 40 min. Unchanged dabigatran accounted for 76.8% of the dose in urine and 1.9% of the dose in feces. Acylglucuronides and their secondary products M690 and M636 accounted for 4% of the dose in urine. M324 and M355 accounted for 1.5 and 1.4%, respectively, of the dose in urine. Other metabolites were present in urine in trace amounts only. The only dabigatran metabolite detectable in feces was M324, which accounted for 0.2% of the dose.
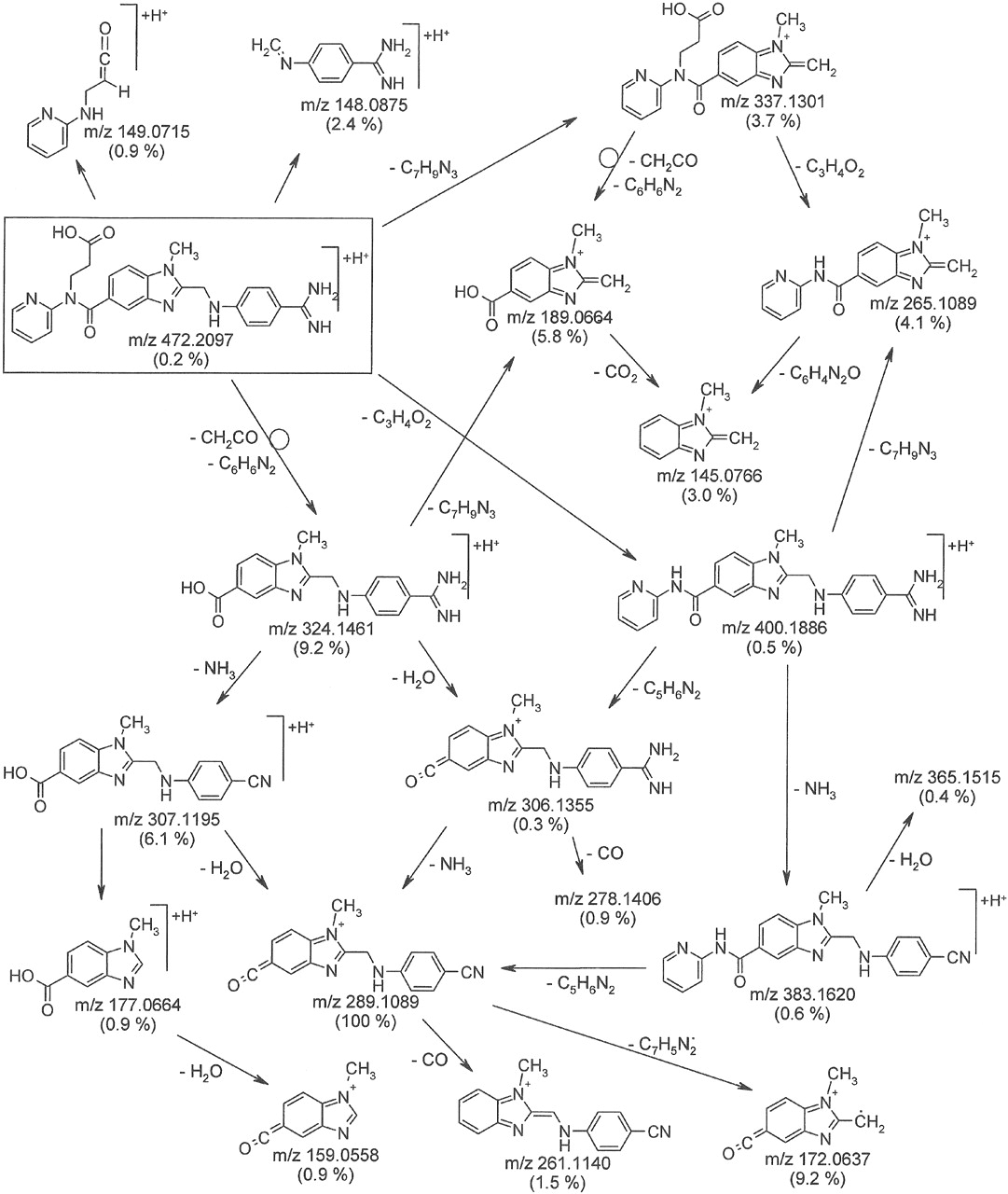
Proposed fragmentation pathway of [M+H]+, m/z 472 of dabigatran (rectangle): collision energy 30 eV, exact masses of fragment ions, relative abundance in brackets, differences between measured and calculated masses: –2.3 to 2.6 mmu.
In Vitro Studies.Metabolism of dabigatran etexilate in vitro. Incubation of [14C]dabigatran etexilate (100 μM) with human liver microsomes resulted in the rapid formation of BIBR 1087 SE, which was subsequently hydrolyzed to form dabigatran and trace amounts of a metabolite with a retention time of 1.5 min that was not further characterized (Fig. 8A). When BIBR 1087 (100 μM) was incubated with liver microsomes for up to 2 h, concentrations of BIBR 1087 SE decreased by approximately 50%, resulting in the formation of an equivalent amount of dabigatran (Fig. 8B).
The cleavage of [14C]dabigatran etexilate to BIBR 1087 SE was completely inhibited by the esterase inhibitors BNPP, PMSF, and paraoxon (100 μM). Iso-OMPA reduced cleavage to 16% of control levels, whereas EDTA (1 and 10 mM), physostigmine, and PCMB (100 μM) had no effect (Fig. 9A). Similarly, iso-OMPA, BNPP, PMSF, and paraoxon reduced the cleavage of BIBR 1087 SE to dabigatran to 10 to 15% of control levels, whereas physostigmine produced a 40% inhibition (Fig. 9B).

Representative radio chromatograms of plasma (A), urine (B), and feces (C) following p.o. administration of 200 mg of [14C]dabigatran etexilate in healthy volunteers.
In other experiments, [14C]dabigatran etexilate (100 μM) was incubated with human plasma for 60 min in the presence and absence of esterase inhibitors. Only EDTA at a concentration of 10 mM inhibited the cleavage of dabigatran etexilate.
Incubation of [14C]dabigatran etexilate with microsomes containing recombinant human P450s showed that the P450 system does not contribute to the metabolism of dabigatran etexilate to a significant extent. The only P450 isoenzyme shown to metabolize dabigatran etexilate to any extent was CYP3A4: this resulted in the production of small amounts of a metabolite that accounted for approximately 2% of total radioactivity and was not further investigated. In all the experiments, a basal level of BIBR 1087 SE formation (less than 4%/60 min) was observed.
Effect of dabigatran metabolites on P450-dependent reactions. Dabigatran etexilate, dabigatran, BIBR 1087 SE, and BIBR 951 CL had no relevant effect on any of the P450-dependent reactions studied in human liver microsomes at concentrations of up to 10 μM. At a concentration of 100 μM, dabigatran etexilate produced approximately 50% inhibition of CYP2E1-dependent lauric acid 11-hydroxylation and CYP3A4-dependent testosterone 6β-hydroxylation.

Representative radio chromatograms of plasma (A), urine (B), and feces (C) following i.v. administration of 5 mg of [14C]dabigatran etexilate in healthy volunteers.
Discussion
The pharmacokinetic profile of the direct thrombin inhibitor dabigatran, the active form of the prodrug dabigatran etexilate, was investigated in healthy male subjects with emphasis on the conversion of the prodrug, the absolute bioavailability of dabigatran, and its metabolism.
The direct inhibition of thrombin is most effectively achieved by an ionic interaction between Asp 189, which is located in the substrate binding pocket of thrombin, and strongly basic functional groups of potential small-molecule thrombin inhibitors (Hauel et al., 2002). This approach, however, results in strongly basic compounds that are permanently charged at physiological pH and are therefore very hydrophilic. As a result, such compounds show very poor intestinal absorption after p.o. dosing. The use of less basic and hence less hydrophilic prodrugs is a suitable approach to overcoming this obstacle to the development of orally available anticoagulants and offers a means of producing more permeable drugs with favorable pharmacokinetic properties. Such strategies have been used for thrombin inhibitors and the factor Xa inhibitors (Schipper et al., 2001; Ries and Wienen, 2003).
In the case of dabigatran, the strongly basic benzamidine group (pKa ∼12.4) was converted to a lipophilic n-hexyl carbamate ester. Because of this modification and the ethyl ester conversion of the carboxylate functional group, the polarity was reduced from an lgD (pH 7.4) of –0.6 for dabigatran to an lgD of 3.7 for the double prodrug dabigatran etexilate. The transformation of the double prodrug to the polar active principle is accomplished by ester cleavage and is catalyzed by serine esterases. Unlike thrombin inhibitors that are hydroxylamine-structured prodrugs, such as ximelagatran (Clement and Lopian, 2003), P450 enzymes or other oxidoreductases are not involved in the proteolytic reactions that convert the double prodrug dabigatran etexilate into the active drug dabigatran. Esterase-catalyzed prodrug activation has the advantage that esterases are enzymes with high catalytic capacity and low substrate specificity and are therefore less prone to be affected by drug-drug interactions and interindividual differences in enzyme expression than P450 enzymes.
Dabigatran was formed by the rapid esterase-catalyzed prodrug conversion of dabigatran etexilate, via the two intermediate prodrugs BIBR 1087 SE and BIBR 951 CL. Following the p.o. administration of dabigatran etexilate in healthy volunteers in this study, peak plasma concentrations of dabigatran were achieved within 1 to 2 h, concentrations subsequently declined in a biexponential manner, with a terminal half-life of approximately 9 h after single dose. The values of Cmax, Tmax, half-life, and AUC0-∞ recorded in this study are comparable with those obtained previously (Stangier et al., 2005) in healthy male volunteers receiving a single p.o. dose of 150 mg of dabigatran etexilate in a capsule formulation.
The total radioactivity, expressed as concentration equivalents of dabigatran, was higher than the sum of unconjugated and conjugated dabigatran in plasma (Fig. 1, A and B). This could be explained by the presence of metabolites in plasma. Dabigatran undergoes conjugation with activated glucuronic acid to yield pharmacologically active, but unstable, glucuronide conjugates. After p.o. administration, glucuronides represented approximately 20% of the total drug exposure in plasma, which at least in part explained the difference between parent drug and total radioactivity in plasma.
Renal excretion was the predominant elimination pathway of dabigatran. In urine, unchanged dabigatran accounted for 77% of the i.v. dose of dabigatran, with a further 4% being accounted for as glucuronide conjugates. Overall, more than 80% of systemically available dabigatran is eliminated by renal excretion. Because renal function declines with increasing age, it is anticipated that the elimination of dabigatran in elderly patients will be prolonged. In a pharmacokinetic study in elderly subjects aged >65 years, a mean terminal half-life of 12.7 h was determined after 7 days of twice-daily treatment with 150 mg of dabigatran etexilate (Stangier et al., 2007b).
The bioavailability of dabigatran after p.o. administration of dabigatran etexilate was low (6–7%). One factor contributing to this low bioavailability may be incomplete absorption of dabigatran etexilate. If absorption is incomplete, bioavailability may increase at higher doses. Dose escalation studies showed that the low oral bioavailability of dabigatran etexilate was not caused by a saturable first-pass process because dabigatran plasma concentrations were shown to increase in a dose-proportional manner (Stangier et al., 2007a).
A, time-dependent cleavage of dabigatran etexilate in human liver microsomes. [14C]Dabigatran etexilate (100 μM) was incubated with human liver microsomes (0.5 mg protein/ml) in the absence of NADPH for up to 60 min. B, time-dependent cleavage of BIBR 1087 SE in human liver microsomes. BIBR 1087 SE (100 μM) was incubated with human liver microsomes (0.5 mg of protein/ml) in the absence of NADPH for up to 120 min.
The in vivo metabolism study showed that dabigatran, being a polar and hydrophilic compound, does not undergo quantitatively relevant oxidative metabolism and is mainly excreted unchanged. It was the predominant compound in plasma, urine, and feces after both p.o. administration of dabigatran etexilate and i.v. administration of dabigatran. Although a considerable number of metabolites have been identified by high-resolution LC/MS, only a few may be formed by P450 enzymes, and these metabolites were observed only in small amounts. They accounted for up to 0.6% of the dose in urine and 5.8% of the dose in feces following p.o. administration of dabigatran etexilate, and up to 1.5 and 0.2%, respectively following i.v. administration of dabigatran.
The predominant metabolic reaction was, as intended by the prodrug design of dabigatran etexilate, esterase-mediated hydrolysis yielding dabigatran via BIBR 1087 SE and BIBR 951 CL. Consequently, dabigatran was the only compound detectable in plasma by radioactivity monitoring. Trace amounts of minor metabolites were detected by LC/MS analysis but escaped detection by HPLC and radioactivity detection because of the low specific activity of plasma samples.

A, effect of esterase inhibitors on the cleavage of [14C]dabigatran etexilate by human liver microsomes. [14C]Dabigatran etexilate (100 μM) was incubated with human liver microsomes (0.5 mg of protein/ml) and esterase inhibitors (100 μM) in the absence of NADPH for 20 min. B, effect of esterase inhibitors on the cleavage of BIBR 1087 SE by human liver microsomes. BIBR 1087 SE (100 μM) was incubated with human liver microsomes (0.5 mg of protein/ml) and esterase inhibitors (100 μM) in the absence of NADPH for 20 min.
In contrast to the negligible role of Phase I metabolic reactions, the formation of the 1-O-acylglucuronide was the predominant route of dabigatran metabolism in humans. The 1-O-acylglucuronide and its isomeric rearrangement products that were formed by nonenzymatic acyl migration (Fenselau, 1994) accounted for 4 and 0.4% of the dose in urine after i.v. and p.o. dosing, respectively. The glucuronides were present in plasma up to 4 h after i.v. treatment. The possibility cannot be excluded that the low concentrations of acylglucuronides present in plasma samples used for metabolic profiling were underestimated because of instability during prolonged sample storage. Measured by LC/MS/MS, a fraction of 19.5% of drug-related material in the plasma was assigned to the acylglucuronides of dabigatran by comparison of the AUC of free (nonconjugated) dabigatran and the AUC of total dabigatran after complete alkaline hydrolysis of the acylglucuronides. The acyl glucuronides of dabigatran may contribute to the overall clinical effect of the drug because they prolong the activated partial thromboplastin time to a similar extent to dabigatran (T. Ebner et al., Boehringer Ingelheim, data on file).
A secondary metabolite with a peculiar chemical structure, namely, the mannitol ester of dabigatran, M636, was identified in small amounts in urine after i.v. and p.o. administration of dabigatran and dabigatran etexilate, respectively. Mannitol was used as a constituent in the i.v. and p.o. formulations and is known be eliminated via the urine after i.v. administration. After p.o. administration approximately 20% of mannitol is absorbed (Better et al., 1997). In vitro incubations of dabigatran-acylglucuronide with mannitol in buffered aqueous solutions (S. Blech, unpublished observations) showed that M636 was formed by nonenzymatic transesterification. Therefore, such reactions may occur for the dabigatran-acylglucuronides with mannitol that was eliminated and enriched in the bladder.
The in vitro studies using human liver microsomes underlined the central role of esterases in the metabolism of dabigatran etexilate to dabigatran. The esterase inhibitors iso-OMPA, BNPP, PMSF, and paraoxon (Thomsen et al., 1991; Madhu et al., 1997; Kraut et al., 2000) inhibited the conversion of dabigatran etexilate to BIBR 1087 SE and of BIBR 1087 SE to dabigatran. By contrast, the esterase inhibitor PCMB had no effect on the hydrolysis of dabigatran etexilate. These findings suggest that the hydrolysis of dabigatran etexilate and BIBR 1087 SE is mediated by microsomal carboxylesterases, which are members of the family of serine esterases. Dabigatran is not metabolized by P450 isoenzymes to any significant extent and does not inhibit P450-dependent reactions at concentrations of up to 10 μM. Only in the presence of microsomes containing recombinant CYP3A4 (which do not contain esterases) was a small amount of a metabolite that accounted for about 2% of total radioactivity detected, and this was not further characterized. Together, these findings suggest that the P450 system plays no relevant role in the metabolism of dabigatran etexilate and dabigatran, and hence the potential for interactions with drugs metabolized by this system is likely to be low. This is in marked contrast to the situation with warfarin, which is subject to numerous food and drug interactions that necessitate careful monitoring of its anticoagulant effect (Ansell et al., 2004).
In conclusion, these studies have shown that pharmacologically active concentrations of dabigatran are rapidly formed after p.o. administration of dabigatran etexilate, predominantly via esterase-mediated hydrolysis. Complete recovery of radioactive substance (>90% within 168 h) was observed. The main elimination pathway of dabigatran is unchanged excretion via the kidney. The absolute bioavailability was low (6–7%). Low binding of dabigatran to plasma proteins (about 35%) was observed, and hence the risk of protein binding interactions is low. The P450 system plays little part in the formation or metabolism of dabigatran, and hence at therapeutic doses dabigatran is likely to have little potential for clinically relevant interactions with drugs metabolized by this system.
Acknowledgments
We thank Ralf Laux, Annamaria Grimminger, Helga Ott, and Melanie Schmitke (Boehringer-Ingelheim, DMPK, Drug Metabolism) for their technical work; Heinz Switek for the preparation of the radiolabeled drugs; and Dr. Norbert Hauel for the synthesis of reference compounds.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.019083.
-
ABBREVIATIONS: Dabigatran, β-alanine, N-[[2-[[[4-(aminoiminomethyl)phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl]-N-2-pyridinyl; Dabigatran etexilate, β-alanine, N-[[2-[[[4-[[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl]-N-2-pyridinyl-, ethyl ester, methanesulfonate; VTE, venous thromboembolism; LC/MS/MS, liquid chromatography/tandem mass spectrometry; AUC0-∞, area under the plasma concentration-time curve extrapolated to infinity; HPLC, high-performance liquid chromatography; LC/MS, liquid chromatography/mass spectrometry; P450, cytochrome P450; BNPP, bis-(p-nitrophenyl) phosphate sodium; PCMB, p-chloromercuribenzoate; PMSF, phenylmethylsulfonyl fluoride; iso-OMPA, tetraisopropyl pyrophosphoramide.
- Received October 5, 2007.
- Accepted November 14, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}