


Abstract
Human cytochrome P450 3A4 (CYP3A4) is the most abundant hepatic and intestinal phase I drug-metabolizing enzyme, and participates in the oxidative metabolism of approximately 50% of drugs on the market. In the present study, a transgenic-CYP3A4(Tg-CYP3A4) mouse model that expresses CYP3A4 in the intestine and is phenotypically normal was generated, which was genotyped by both polymerase chain reaction and Southern blotting. Intestinal microsomes prepared from Tg-CYP3A4 mice metabolized midazolam (MDZ) to 1′-hydroxymidazolam about 2 times, and to 4-hydroxymidazolam around 3 times faster than that from wild-type (WT) mice. These increased MDZ hydroxylation activities were completely inhibited by an anti-CYP3A4 monoclonal antibody. The time course of plasma MDZ and its metabolite concentrations was measured after intravenous (0.25 mg/kg) and oral (2.5 mg/kg) administration of MDZ, and pharmacokinetic parameters were estimated by fitting to a noncompartmental model. Pretreatment with ketoconazole increased orally dosed MDZ maximum plasma concentration (Cmax), time of the maximum concentration, area under the plasma concentration-time curve from zero to infinity (AUC0-∞), and elimination half-life (t1/2) to 3.2-, 1.7-, 7.7-, 2-fold, and decreased MDZ apparent oral clearance about 8-fold in Tg-CYP3A4 mice. The ratios of MDZCmax, AUC0-∞,t1/2 and bioavailability between Tg-CYP3A4 and WT mice after the oral dose of MDZ were 0.3, 0.6, 0.5, and 0.5, respectively. These results suggest that this Tg-CYP3A4 mouse would be an appropriate in vivo animal model for the evaluation of human intestine CYP3A4 metabolism of drug candidates and potential food-drug and drug-drug interactions in preclinical drug development.
Cytochrome P450 (P450)4 represents a superfamily of heme-containing monooxygenases, many of which metabolize therapeutic drugs. They also play an important role in the oxidation of toxic chemicals and carcinogens, as well as endogenous steroids, fatty acids, and prostaglandins (Gonzalez and Gelboin, 1994; Guengerich, 2000; Nebert and Russell, 2002). Although approximately 60 P450 genes are known to exist in human genome, only a limited number belonging toCYP1A, 2C, 2D, and 3Asubfamilies play important role in drug oxidation (Guengerich, 1999,2000; Nebert and Russell, 2002). Human CYP3A subfamily contains four members, CYP3A4, 3A5, the predominantly fetal form 3A7, and relatively less understood3A43 (Nebert and Russell, 2002).
CYP3A4 is the most abundant P450 isozyme present in human liver and small intestine, and contributes to the biotransformations of approximately 50% of drugs currently on the market (de Wildt et al., 1999; Guengerich, 1999). In addition, CYP3A4 is involved in the oxidation of a variety of endogenous substrates, such as steroids and bile acids (Nebert and Russell, 2002). Like humans, the mouse has at least four CYP3A genes. Two of them, CYP3A11 and3A13, are expressed in adult mouse liver. CYP3A11 protein is 5- to 10-fold more abundant in liver than CYP3A13. The latter is also expressed in the small intestine (Schellens et al., 2000).
In recent years, several CYP3A4 substrates have been proposed in the literature as metabolic probes to evaluate the catalytic activity of CYP3A4 in vivo and in vitro. They include testosterone, erythromycin, midazolam, triazolam, cortisol, nifedipine, dapsone, dextromethorphan, and 7-benyloxy-4-trifluoromethylcoumarin (Zaigler et al., 2000;Williams et al., 2002). Testosterone represents endogenous steroid substrate and is commonly used in vitro. Fluorescent probe 7-benyloxy-4-trifluoromethylcoumarin has broad application in high-throughput screening for the prediction of drug-drug interaction in drug discovery. Midazolam (MDZ) seems to be an ideal probe for CYP3A4 activity not only in vitro but also in vivo because it is completely excreted through metabolism and is not a substrate of P-glycoprotein. Therefore, MDZ is commonly used in vivo to monitor CYP3A activity (Thummel et al., 1994a,b; Lin et al., 2001).
MDZ is a short-acting 1,4-benzodiazepine widely used in clinical practice for preoperative sedation, induction, and maintenance of anesthesia, and sedation of patients in intensive care. MDZ undergoes extensive first-pass metabolism in the intestinal wall and in the liver. The oxidation of MDZ leads to the major metabolite 1′-hydroxymidazolam (1′-OH-MDZ), the minor metabolite 4-hydroxymidazolam (4-OH-MDZ), and the secondary metabolite 1′,4-dihydoxymidazolam (Dundee et al., 1984; Kronbach et al., 1989;Thummel et al., 1994a,b). The major pathway 1′-hydroxylation of MDZ is well established as the index reaction for CYP3A activity.
Metabolism in the small intestine, the first site of exposure to orally dosed drugs, can activate or diminish the bioavailability of a large number of compounds, which may cause significant pharmacological effects clinically (Doherty and Pang, 1997; Bailey et al., 1998;Schellens et al., 2000; Doherty and Charman, 2002). Coadministration of drugs and food/beverages containing certain inducers or inhibitors is generally recognized phenomena, leading to marked drug-drug and food-drug interactions (Bailey et al., 1994; Bailey et al., 1998;Wienkers, 2001). Due to the high levels of CYP3A4 in the gut and the high percentage of drugs metabolized by this enzyme, there is increased interest in finding an animal model that has similar drug metabolism as the human intestine. Although several animal models such as Gottingen minipig, dog, and monkey have been proposed to assess human intestinal first-pass metabolism, the metabolism and disposition of CYP3A4 substrates using these species were not comparable with humans (Zuber et al., 2002). Several P450 gene knockout and CYP3A7 andCYP2D6 transgenic mice lines have been established for studying their functions in metabolism, pharmacology, and toxicity (Li et al., 1996, 1997; Gonzalez, 1998; Gonzalez and Kimura, 1999; Corchero et al., 2001). In this study, a phenotypically normal and viable transgenic mouse model expressing the human intestinal CYP3A4 was generated. This transgenic mouse model has been characterized with molecular biological techniques and validated in vitro and in vivo using MDZ as the marker for enzyme activity.
Materials and Methods
Chemicals and Enzymes.
Midazolam, ketoconazole, troleandomycin, dexamethasone, rifampin, α-naphthoflavone, furafylline, 8-methoxypsoralem, orphenadrine, sulfaphenazole, S-mephenytoin, quinine, quinidine, diethyldithiocarbamate, trypsin inhibitor, leupeptin, aprotinin, bestatin, phenylmethylsulfonyl fluoride and NADPH were obtained from Sigma-Aldrich (St. Louis MO). 1′-hydroxymidazolam, 4-hydroxymidazolam, pooled human liver microsomes (coded H161), and recombinant CYP3A4 enzymes were purchased from BD Gentest (Woburn, MA). Pooled human gut microsomes were bought from In Vitro Technologies (Baltimore, MD). Restriction enzymes were purchased from Roche Diagnostics (Indianapolis, IN), Invitrogen (Carlsbad, CA), or New England Biolabs (Beverly, MA) and were used in buffer systems provided by the manufacturers. All other chemicals and high-performance liquid chromatography grade solvents were of the highest grade of purity commercially available. Inhibitory and immunoblot monoclonal antibody to human CYP3A4 (mAb 3-29-9 and 275-1-2, respectively), and immunoblot monoclonal antibody to rat CYP3A1 (mAb 2-13-1) were characterized previously (Gelboin et al., 1995, 1999).
Animals.
Adult male mice described in this work (2–4 months old, body weight ranging from 25 to 30 g) were maintained under the controlled temperature (23 ± 1°C) and lighting (lights on 6:00 AM–6:00 PM) with food and water provided ad libitum. All animal experiments were conducted under National Institutes of Health guidelines for the use and care of laboratory animals and approved by National Institutes of Health Animal Care and Use Committee. Tg-CYP3A4 and WT mice were treated with dexamethasone (50 mg/kg p.o. in corn oil for 4 days) or rifampin (10 mg/kg p.o. in saline for 4 days) for induction analysis. Control mice were administered with vehicle. Three mice were used in each group.
Generation of CYP3A4 Humanized Mouse.
The CYP3A4 gene in a bacterial artificial chromosome described in a previous report (Hashimoto et al., 1993; Sata et al., 2000) was microinjected into a fertilized FVB/N mouse egg to produce a transgenic mouse line (Core Transgenic Facility, National Cancer Institute, Frederick, MD). Incorporation of the CYP3A4 DNA within the mouse genome was determined by both PCR genotyping and Southern blot analysis. The transgenic founder was mated with nontransgenic FVB/N (WT) mouse, and animals from this cross were subsequently crossed to each other to produce homozygous mice. Crossing them with WT mice and testing the progeny for transgene transmission confirmed mice homozygous for the transgene. WT and homozygous littermates were bred and maintained by brother-sister mating.
PCR Genotyping.
For CYP3A4 PCR analysis, approximately 50 ng of tail DNA was amplified in a 25-μl reaction mixture containing 2.5 mM MgCl2, 0.2 mM dNTPs, 1.25 U of AmpliTaq (PerkinElmer, Foster City, CA), and 20 pmol of CYP3A4gene-specific primers CYP3A4-forward 5′-GTA GGT GTG GCT TGT TGG GAT G-3′, CYP3A4-reverse 5′-TGC TCT TTG CTG GGC TAT GTG-3′, and microsomal epoxide hydrolase (mEH) gene-specific primers mEH-forward 5′-AGG TGA GTT TGC ATG GCG CAG-3′ and mEH-reverse 5′-CCC TTT AGC CCC TTC CCT CTG-3′. Cycling conditions were 90°C for 5 min and then 33 cycles of 90°C for 30 s, 60°C for 30 s, and 70°C for 1 min, followed by a 5-min extension at 70°C. mEH primers served as a positive control for amplification, yielding a fragment of 341 base pairs in all samples (Miyata et al., 1999). An additional band of 406 base pairs was amplified exclusively in CYP3A4 humanized animals.
Southern Blot Analysis.
Tail genomic DNA (1 μg/lane) was digested with ApaI and subjected to electrophoresis on a 0.5% agarose gel containing 0.5× Tris borate-EDTA. The DNA was hydrolyzed in 0.2 M HCl and transferred on to a Gene Screen Plus nylon membrane (DuPont, Wilmington, DE) by capillary blotting in 0.4 M NaOH. Blots hybridized with random-primer32P-labeled CYP3A4 cDNA probe (Gonzalez et al., 1988) at 42°C overnight, washed twice in 2× SSC (1× SSC is 150 mM NaCl plus 15 mM sodium citrate) and 0.5% SDS at 65°C for 15 min, twice in 0.1× SSC and 0.5% SDS for 5 min, and exposed to a PhosphorImager screen (Amersham Biosciences, Sunnyvale, CA) for 2 to 4 h. Transgene copy number was determined by Southern blotting using the purified genomic clone DNA as a standard. Cloned DNA was diluted with mouse DNA to yield the equivalent of 1, 2, 5, 10, and 100 copies of the gene per haploid genome (based on 3 × 109 base pairs per haploid genome). The DNA was digested with ApaI and subjected to Southern blot with DNA isolated from WT and Tg-CYP3A4 mice. The signals were quantified by use of a PhosphorImager, and the copy number was determined from a standard curve of the purified standard genomic clone.
Preparation of Microsomes.
Pooled microsomes of mouse small intestines and livers from Tg-CYP3A4 and WT mice were prepared according to the reported method (Emoto et al., 2000). Briefly, tissues were homogenized in 3 volumes of ice-cold buffer A [50 mM Tris-HCl buffer, pH 7.4, containing 150 mM KCl, 1 mM phenylmethylsulfonyl fluoride, 1 mM EDTA, 1 mg/ml trypsin inhibitor, 10 mM leupeptin, 0.04 unit/ml aprotinin, 1 mM bestatin, and 20% (v/v) glycerol] by three strokes using a motor-driven Teflon-tipped pestle. The homogenates were centrifuged at 9000g for 20 min at 4°C, and the supernatant was centrifuged at 100,000g for 60 min at 4°C. The microsomal pellet was resuspended in the same buffer, aliquoted, and stored at −80°C for further use. Protein concentrations were determined using a BCA protein assay kit (Pierce Chemical, Rockford, IL), following the manufacturer's instructions.
Immunoblot Analysis.
Microsomal proteins were separated by SDS-polyacrylamide gel electrophoresis with a 4% stack and 10% separating gel. Liver microsomes were used at 1 μg of protein per well, and intestinal microsomes were used at 10 μg of protein per well. Proteins were transferred onto nitrocellulose membrane. After incubated in 5% nonfat dry milk in phosphate-buffered saline (PBS), pH 7.4, for 1 h at room temperature, the membrane was incubated with a primary antibody (anti-CYP3A4 mAb at 1/400 dilution) in PBS containing 3% milk for an additional hour, washed with PBS containing 0.05% Tween 20, and then incubated with a secondary antibody at 1/10,000 dilution containing 3% milk. The secondary antibody, a peroxidase-labeled goat anti-mouse IgM (for the detection of CYP3A4) was detected with an ECL kit (Pierce Chemical) following the manufacturer's instruction.
In Vitro Metabolism Studies.
The incubation was performed in 100 mM sodium phosphate buffer (pH 7.4) containing microsomes with 25 to 50 μg of protein, and MDZ in a final volume of 200 μl. The mixture was preincubated at 37°C in a shaking water bath for 5 min, and the reaction was initiated by the addition of 20 μl of 10 mM NADPH. The formation of metabolites was found to be linear up to 20 min using mouse and human microsomes. Incubation was stopped after 5-min reaction by adding 50 μl of 100 mM NaOH solution, and subsequently cooled on ice for 15 min. MDZ concentration ranged from 0.25 to 250 μM for kinetic analysis. All reactions were performed in duplicate, and each data point represented the average of duplicate determinations. The effects of the following P450-specific chemical inhibitors on the formation of 1′-OH-MDZ and 4-OH-MDZ were studied: furafylline (10 μM, CYP1A2), α-naphthoflavone (5 μM, CYP1A2), 8-methoxypsoralen (10 μM, CYP2A6/1A2), orphenadrine (200 μM, CYP2B6/2C/3A), sulfaphenazole (20 μM, CYP2C9), (200 μMS-mephenytoin, CYP2C19), quinine (5 μM, CYP2D6), quinidine (5 μM, CYP2D6), (100 μM diethyldithiocarbamate, CYP2A6/2B6/2E1), ketoconazole (0.010–10 μM CYP3A4), and (10 μM troleandomycin, CYP3A4). Mouse and human small intestinal and liver microsomes containing 25 μg of protein were incubated with 2.5 μM MDZ for 10 min for the inhibition analysis. Mechanism-based inhibitors were preincubated with microsomes and NADPH (1 mM, final concentration) for 10 min (8-methoxypsoralen), 15 min (furafylline or orphenadrine), or 20 min (troleandomycin) before the addition of MDZ.
Immunoinhibition was carried out using 25 μg of mouse and human small intestine and liver microsomes proteins and various amounts of monoclonal antibody against CYP3A4 in 100 mM sodium phosphate buffer (pH 7.4). The mixture was preincubated for 5 min at 37°C, and reaction was initiated by the addition of MDZ (2.5 μM) and NADPH (1 mM) to a final volume of 200 μl. Anti-lysozyme mAb (HyHEL) was used as a control for nonspecific binding. The reactions were incubated for 10 min and terminated with 50 μl of ice-cold 100 mM NaOH solution.
Pharmacokinetics Analysis.
Three mice were used in each group. Tg-CYP3A4 and WT mice were administered with MDZ intravenously (0.25 mg/kg) or orally through gavage (2.5 mg/kg). Blood samples were collected from suborbital veins using heparinized tubes at 0, 2.5, 5, 10, 20, 30, 60, 90, 120, and 180 min after oral administration and at 2.5, 5, 10, 20, 30, 60, 90, 120, and 180 min after i.v. administration of MDZ. To examine the effect of ketoconazole on MDZ pharmacokinetics, ketoconazole was dosed orally (40 mg/kg) to Tg-CYP3A4 and WT control mice at 45 min before the administration of MDZ. The ketoconazole treatment regimen was based on the preliminary pharmacokinetic study and existing published study in the literature (Fahey et al., 1998; Yamano et al., 1999). Blood samples were collected at different time intervals from suborbital veins as described above. Plasma was separated by centrifugation at 13,000g for 10 min and stored at −80°C until analysis.
Measurement of the Concentrations of MDZ and Its Metabolites.
Concentrations of MDZ and its metabolites 1′-OH-MDZ and 4-OH-MDZ were determined using a liquid chromatographic tandem mass spectrometric (LC-MS/MS) method based on previous method (Scott and Sobol, 1999) with minor modification. Briefly, 20 μl of internal standard solution (phenacetin, 5 μg/ml in methanol), 50 μl of aqueous sodium hydroxide (100 mM), and 3 ml of methyl-t-butyl ether were added to 50 μl of plasma or in vitro incubations. After vortexed for 1 min, aqueous and organic phases of the mixture were separated by centrifugation at 3000g at 4°C for 10 min. The aqueous layer was frozen on dry ice, and the organic phase was transferred to a new borosilicate glass tube and evaporated to dryness under a gentle stream of air at 30°C using a heater bloc (Pierce Chemical). The residue was reconstituted in 100 μl of acetonitrile/water (20:80, v/v) solution and transferred to polypropylene autosampler vial. Twenty-five microliters of each sample was injected for the LC-MS/MS analysis.
LC-MS/MS analysis was carried out using a high-performance liquid chromatography system consisting of a PerkinElmer Series 200 quaternary pump, vacuum degasser, and autosampler with a 100-μl loop interfaced to an API2000 SCIEX triple-quadrupole tandem mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA). MDZ, 1′-OH-MDZ, 4-OH-MDZ, and the internal standard were separated on a Phenomenex (Torrance, CA) Synergi Polar-RP 4-μm C18 column (2.0 × 50 mm). The mobile phase was consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). A linear gradient of solvent B from 5 to 95% over 4 min was applied on the column and then returned to the original condition and equilibrated for 1 min before the next injection. The sample was delivered with a flow rate of 0.200 ml/min, and each analysis lasted for 5.0 min.
The mass spectrometer was operated in the turbo ion spray mode with positive ion detection. The turbo ion spray temperature was maintained at 300°C, and a voltage of 4.8 kV was applied to the sprayer needle. Nitrogen was used as the turbo ion spray and nebulizing gas. The detection and quantification of analysts were performed using the multiple reactions monitoring mode. Multiple reactions monitoring analysis was performed with the transition m/z 326→291 for MDZ, m/z 342→203 for 1′-OH-MDZ, m/z 342→234 for 4-OH-MDZ, and m/z 180→110 for the phenacetin. All raw data were processed with PerkinElmer SCIEX Analyst Software, version 1.2. Calibration curves were linear from 2 to 2500 nM for MDZ concentrations and from 2 to 3000 nM for 1′-OH-MDZ and 4-OH-MDZ. The lower limit of quantitation was 2 nM for MDZ, 1′-OH-MDZ, and 4-OH-MDZ, where the coefficient variation was less 20%. The recoveries for MDZ, 1′-OH-MDZ, and 4-OH-MDZ at concentrations 50 and 1000 nM were 80 and 102%, respectively. Intraday and interday coefficients of variation at concentrations 50 and 1000 nM were less than 10%.
Estimation of Enzyme Kinetic and Pharmacokinetic Parameters.
The formation of 1′-OH-MDZ and 4-OH-MDZ in microsomes was calculated and expressed as picomoles per minute per milligram of protein. Kinetic parameters (Km,Vmax, Clint, andVmax/Km) were determined by nonlinear regression using GraphPad Prism, version 3.02 (GraphPad Software Inc., San Diego, CA). All analysis was performed using the mean values obtained from duplicate incubations. Pharmacokinetics parameters for MDZ and its metabolite 1′-OH-MDZ were estimated from the plasma concentration-time data by a noncompartmental approach using the software WinNonlin (Pharsight, Mountain View, CA). The peak concentration in serum (Cmax) and the corresponding time of maximum concentration (Tmax) were obtained from the original data. The area under the serum concentration-time curve from time 0 to 180 min (AUC0–180min) was calculated by the trapezoidal rule and the AUC0-∞ with extrapolation to infinity by dividing the last measured concentration by λ. The elimination rate constant (λ) was determined as the slope of linear regression for the terminal log-linear portion of the concentration versus time curve, and the elimination half-life (t1/2) was calculated from 0.693/λ. The mean residence time (MRT) value was determined as the ratio of the area under the first moment curve over AUC. The apparent oral clearance (CLoral) was calculated from Doral/AUC0–180min. The systemic clearance (CLiv) of MDZ was calculated as the i.v. dose divided by the AUC0-∞(Div/AUC0-∞). The oral bioavailability (Foral) of MDZ was determined form the ratio of the dose-normalized AUC values obtained from oral and i.v. administration [Foral = 100 × (Div × AUCoral)/(Doral × AUCiv)].
Statistical analyses were performed using GraphPad Prism, version 3.02. Kinetic parameters for each group were compared using Student'st test for paired data. Difference was considered significant if the probability (P value) was less than 5%.
Results
Generation of Transgenic-CYP3A4 Mouse Line.
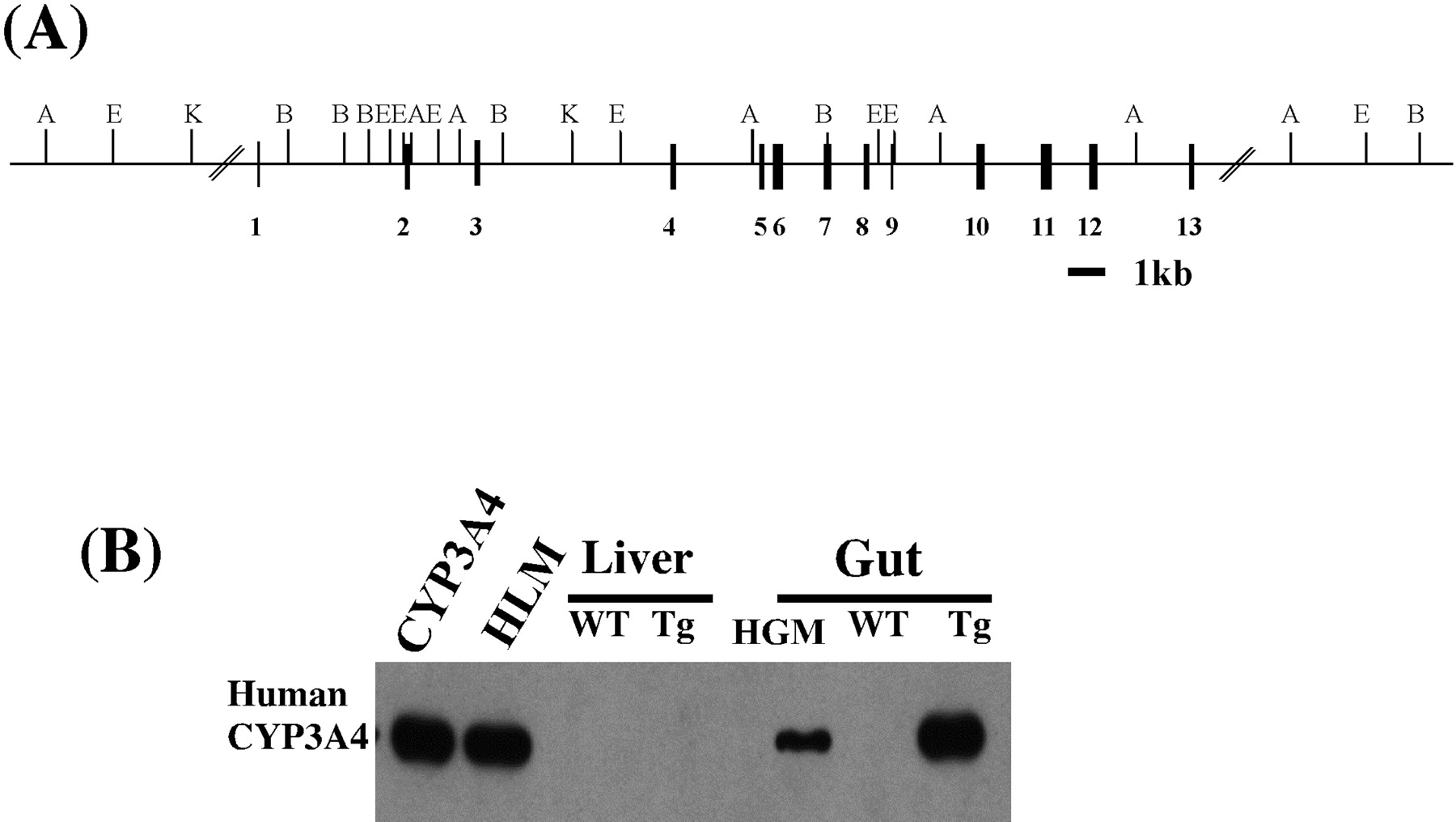
The CYP3A4 gene was isolated from a human BAC genomic library (d7-90 clone; Genome Systems, St. Louis, MO). The screening and the restriction map of the BAC d7-90 genomic clone were the same with those of the previously published CYP3A4 gene (Sata et al., 2000). This d7-90 BAC clone containing CYP3A4 gene (Fig.1A) was microinjected into a fertilized FVB/N mouse egg to produce a transgenic mouse line. The transgenic founders with incorporation of CYP3A4 DNA into the germ line were identified by Southern blot analysis. Genomic DNA from WT and Tg-CYP3A4 mice was digested with ApaI and probed with the CYP3A4 cDNA. Hybridization signals were found only in the lanes with DNA from Tg-CYP3A4, but not from WT FVB/M mice. The size of the band corresponded exactly with the predicted sizes calculated from the sequence of the CYP3A4 gene. The copy number of the transgene in these animals was approximately 5, as estimated by comparing the intensity of the hybridization signal to that obtained with known amounts of human DNA. The transgenic mice were indistinguishable from their wild-type controls by gross pathological examination. The mice were fertile and produced normal-sized litters. Western blotting was performed to assess the expression of the CYP3A4 protein in microsomal preparations from WT and Tg-CYP3A4animals using a CYP3A4-specific monoclonal antibody that does not react with mouse CYP3A (Gelboin et al., 1995). Human CYP3A4 expression was only found in the intestine, not in the liver of the transgenic mice (Fig. 1B) or other tissues such as brain, spleen, kidney, heart, lung, and muscles. To determine the expression level of CYP3A4 along the length of the gut, it was segmented in to the duodenum, jejunum, ileum, and colon. Western blotting of microsomal proteins revealed the highest CYP3A4 expression in the duodenum and jejunum and about one-third in the ileum (Fig. 2).

Generation and analysis of the CYP3A4 transgenic mouse.
A, schematic diagram of the wild-type CYP3A4 gene used for microinjection. Restriction sites for EcoRI (E),ApaI (A), KpnI (K), andBamHI (B) are depicted. Black boxes representCYP3A4 exons. The bar represents 1 kb. B, Western blot analysis of CYP3A4 protein expression in WT and transgenic-CYP3A4 (Tg-CYP3A4) mice. Liver, duodenum microsomal proteins (1 μg each) and expressed cDNA-CYP3A4 (0.4 pmol) were separated by SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. A CYP3A4-specific monoclonal antibody was used to assess CYP3A4 protein expression. The antibody only reacts with CYP3A4 and does not recognize mouse CYP3A proteins. Human liver microsomes (HLM), pooled human gut microsomes (HGM), and recombinant CYP3A4 were used as controls.

Immunoblot analysis of CYP3A4 expression in duodenum, jejunum, ileum, and colon in wild-type and Tg-CYP3A4 mice.
Aliquots of intestinal microsomal protein (10 μg) and recombinant CYP3A4 (0.4 pmol) were subjected to SDS-polyacrylamide gel electrophoresis, and Western blotting was performed using monoclonal anti-CYP3A4 and anti-CYP3A1. HGM, human gut microsomes; rCYP3A4, recombinant CYP3A4.
In Vitro Midazolam Metabolism.
Midazolam metabolism in intestinal and hepatic microsomes
To determine whether the CYP3A4 present in the Tg-CYP3A4 and wild-type mice is active, in vitro metabolism was conducted by monitoring the formation of 1′-OH–MDZ and 4-OH-MDZ from MDZ, a common probe for CYP3A4 activity. The rate of 1′-OH-MDZ and 4-OH-MDZ formation in gut microsomes from Tg-CYP3A4 mice was significantly higher than those from WT mice and humans (Fig.3). Similar to previous reports (Perloff et al., 2000; Schrag and Wienkers, 2001; Williams et al., 2002), substrate inhibition was observed when MDZ concentrations were higher than 10 μM. We attempted to estimate the substrate inhibition constant (Ki), but the fit of the data by visual inspection, random distribution of residuals, and the standard error of the estimate parameters were unsatisfactory. Michaelis-Menten kinetic parameters were estimated using the truncated data (MDZ concentrations ranging from 0 to 10 μM for 1′-OH-MDZ, from 0 to 50 μM for 4-OH-MDZ), from which substrate inhibition was not observed. The calculated intrinsic clearance (Vmax/Km) in Tg-CYP3A4 mice intestinal microsomes was 2-fold higher for 1′-OH-MDZ and 3-fold higher for 4-OH-MDZ than that of WT mice intestinal microsomes (Table 1). Kinetics parameters were similar among the Tg-CYP3A4, WT mouse, and human liver microsomes (Table 1). These results are consistent with immunoblot results, indicating that CYP3A4 enzyme was not expressed in Tg-CYP3A4 mouse livers.

Velocity plots (enzyme activity versus concentration) of 1′-hydroxymidazolam (A) and 4-hydroxymidazolam (B) in Tg-CYP3A4 and wild-type mice, and human small intestinal microsomes.
Incubations were performed in 100 mM potassium phosphate buffer, pH 7.4, at 37°C for 5 min. values represent the mean, and the vertical lines represent the S.E.M. obtained from duplicate experiments.
Apparent kinetic parameters of midazolam metabolism in pooled intestinal and hepatic microsomes from wild-type, Tg-CYP3A4 mice and human
Inhibitory effect of ketoconazole on the metabolism of MDZ in wild-type and Tg-CYP3A4 mouse intestinal microsomes.
The effect of various chemical inhibitors on the MDZ oxidation was investigated using Tg-CYP3A4 and WT mouse and human intestinal microsomes. α-Naphthoflavone (5 μM), furafylline (10 μM), 8-methoxypsoralem (10 μM), orphenadrine (200 μM), sulfaphenazole (20 μM), S-mephenytoin (200 μM), quinidine (5 μM), quinine (5 μM), and diethyldithiocarbamate (100 μM) showed minor or no effect on 1′-OH-MDZ and 4-OH-MDZ formation (Table 2). As expected, potent CYP3A inhibitors ketoconazole (2.5 μM) and troleandomycin (10 μM) inhibited 1′-OH-MDZ and 4-OH-MDZ formation by more than 90% in mouse and human intestinal microsomes. The effect of ketoconazole on MDZ hydroxylation in Tg-CYP3A4 and WT mouse intestinal microsomes was further examined (Fig. 4, A and B). The IC50 values (micromolar concentration) for ketoconazole-inhibited 1′-OH-MDZ and 4-OH-MDZ production were 0.014 and 0.121 in Tg-CYP3A4 and 0.011 and 0.036 in WT mouse gut microsomes, respectively. Ketoconazole also showed strong inhibition to both 1′-OH-MDZ and 4-OH-MDZ formation in Tg-CYP3A4 and WT mouse liver microsomes (Fig. 4, C and D). In pooled human liver microsomes, ketoconazole inhibited the formation of 1′-OH-MDZ and 4-OH-MDZ, with IC50 values of 0.112 and 0.124 μM, respectively (data not shown). These data suggested that the oxidation of midazolam is dependent on CYP3A isoform in the intestinal microsomes.
Effects of P450-selective inhibitors on the formation of 1′-and 4-hydroxymidazolam in wild-type and Tg-CYP3A4 mouse, and human intestinal microsomes

Inhibition of 1′-hydroxymidazolam (A) and 4-hydroxymidazolam (B) formation by ketoconazole in Tg-CYP3A4 (○) and wild-type (●) mouse small intestinal microsomes, and the effects of ketoconazole on 1′-hydroxymidazolam (C) and 4-hydroxymidazolam (D) formation in Tg-CYP3A4 (○) and wild-type (●) mouse liver microsomes.
Values represent the mean ± S.E.M. from duplicate experiments.
Inhibition of midazolam biotransformation by anti-CYP3A4 monoclonal antibody.
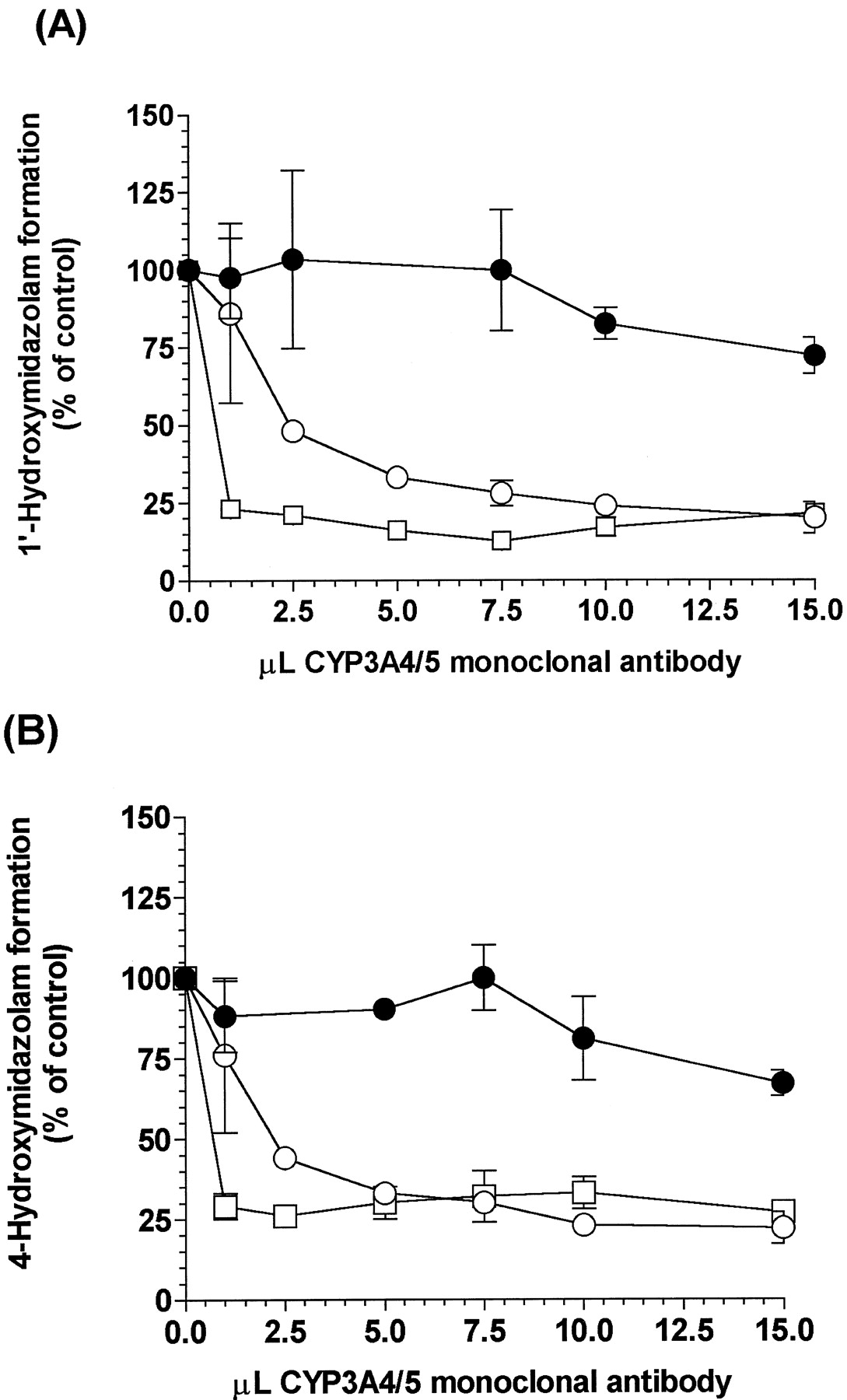
The effect of inhibitory monoclonal antibody against CYP3A4 on MDZ hydroxylation in Tg-CYP3A4 and WT mouse, and human intestinal microsomes was examined (Fig.5). CYP3A4 monoclonal antibody completely blocked the increased MDZ oxidation activities in Tg-CYP3A4intestinal microsomes. As shown in Fig. 5, the formation of 1′-OH-MDZ and 4-OH-MDZ in Tg-CYP3A4 and human gut microsomes was inhibited more than 80% by the antibody. As expected, it did not show strong inhibition to 1′-OH-MDZ and 4-OH-MDZ formations in WT mouse intestinal microsomes.

Inhibition of 1′-hydroxymidazolam (A) and 4-hydroxymidazolam (B) formation in Tg-CYP3A4 (○) and wild-type (●) mice, and human (▪) small intestinal microsomes by an anti-CYP3A4 monoclonal antibody.
Final MDZ concentration in each reaction was 2.5 μM, which lasted at 37°C for 10 min. Anti-lysozyme mAb (HyHEL) was used as a control for nonspecific binding. Values represent the mean ± S.E.M. from duplicate experiments.
Effects of rifampin and dexamethasone on MDZ metabolism in Tg-CYP3A4 and wild-type mice.
Pretreatment of WT mice with dexamethasone (50 mg/kg) resulted in about 3-fold increase of 1′-OH-MDZ formation and about 3-fold increase of 4-OH-MDZ formation in intestine (Fig. 6). As expected, no induction was observed in WT mice treated with rifampin (10 mg/kg) because it is not a ligand for the mouse pregnane X receptor (Willson and Kliewer, 2002). Dexamethasone resulted in about 30% increase of 1′-OH-MDZ formation (P = 0.032), but no increase of 4-OH-MDZ in Tg-CYP3A4 mouse intestine, which may be due to its high CYP3A4 expression background. In Tg-CYP3A4 mice pretreated with rifampin, a significant decrease of MDZ hydroxylation was observed (Fig. 6), and the reason is unknown at this point. Dexamethasone increased the formation of 1′-OH-MDZ and 4-OH-MDZ production by approximately 9- and 6-fold, respectively, in WT mouse livers (data not shown), suggesting a profound hepatic enzyme induction. Same trend was observed in Tg-CYP3A4 mouse liver pretreated with dexamethasone, with 5- and 7-fold increase of 1′-OH-MDZ and 4-OH-MDZ formation, respectively. As expected, no significant difference was observed in MDZ oxidation in WT mice treated with rifampin versus the control.

Formation of 1′-hydroxymidazolam (A) and 4-hydroxymidazolam (B) in intestinal microsomes from Tg-CYP3A4 and wild-type mice treated with dexamethasone (50 mg/kg p.o.) and rifampin (10 mg/kg p.o.) for 4 days. Control mice were treated with vehicle.
Values are the mean ± S.E.M. from duplicate experiments.
Pharmacokinetics of Midazolam in Tg-CYP3A4 Mice and the Effect of Ketoconazole.
Pharmacokinetic analysis of midazolam metabolism
The mean (± S.E.M) plasma concentration-time curves of MDZ (Fig.7A), 1′-OH-MDZ (Fig. 7B), and the ratio of 1′-OH-MDZ over MDZ (Fig. 7C) in WT and Tg-CYP3A4 mice after single oral administration of midazolam (2.5 mg/kg) were determined. Corresponding pharmacokinetic parameters were estimated by noncompartmental analysis and are listed in Table3. MDZ maximum plasma concentrations were 1190 ± 957 and 348 ± 93.7 nM in WT and Tg-CYP3A4mice, respectively. Elimination half-life in Tg-CYP3A4 mice (18.7 ± 0.6 min) was about one-half of that in WT mice (34.3 ± 4.2 min) and is statistically significant. Mean residence time was 26.0 ± 2.7 and 23.0 ± 15.3 min for WT and Tg-CYP3A4 mice, respectively. MDZ disposition was fast with a clearance of 0.9 l/h in WT mice, which was approximately equal to that (0.93 l/min) in Tg-CYP3A4 mouse. TheCmax values of MDZ major metabolite, 1′-OH MDZ, were 707 ± 199 and 2,290 ± 69.6 nM for WT and Tg-CYP3A4 mice, respectively. The AUC of 1′-OH-MDZ was 3-fold higher in Tg-CYP3A4 than in WT mice (102,000 ± 6,800 versus 35,500 ± 8,560 nmol min/l), which is statistically significant. These results suggested that the human CYP3A4 enzyme expressed in Tg-CYP3A4 mouse intestine contributed to extrahepatic first-pass metabolism of orally dosed MDZ.

Time course of plasma concentrations of midazolam (A), 1′-hydroxymidazolam (B), and their ratios after single oral administration of midazolam (2.5 mg/kg) to Tg-CYP3A4 and wild-type mice without treatment (Cont), or pretreated with ketoconazole (Ket, 45 mg/kg).
Values represent the mean, and the vertical lines represent the S.D. of midazolam and 1′-hydroxymidazolam concentrations from three mice.
Pharmacokinetic parameters estimated for midazolam and 1′-hydroxymidazolam in WT and Tg-CYP3A4 mice
After intravenous administration of MDZ (0.25 mg/kg), plasma concentrations of 1′-OH-MDZ in Tg-CYP3A4 mice were slightly higher compared with those of the WT mice (Fig.8). Estimated pharmacokinetics parameters are shown in Table 4. The terminal half-life (14.3 versus 20.8 min) and the mean residence time (17.0 and 19.2 min) of midazolam were not significantly different between WT and Tg-CYP3A4 mice. The elimination was fast with a clearance of 0.10 l/h in Tg-CYP3A4 mice, which was approximately equal as that (0.12 l/h) in WT mice. The Cmaxvalues of 1′-OH-MDZ were 135.0 ± 28.9 and 185.0 ± 49.6 nM in WT and Tg-CYP3A4 mice, respectively. These results were consistent with the findings from in vitro studies that the humanCYP3A4 gene is not expressed in Tg-CYP3A4 mice livers. Surprisingly, the plasma 1′-OH-MDZ AUC values (nanomoles per minute per liter) in Tg-CYP3A4 mice (9140 ± 1630) were different from those in WT mice (4750 ± 367). However, the basis for this difference is not clear.
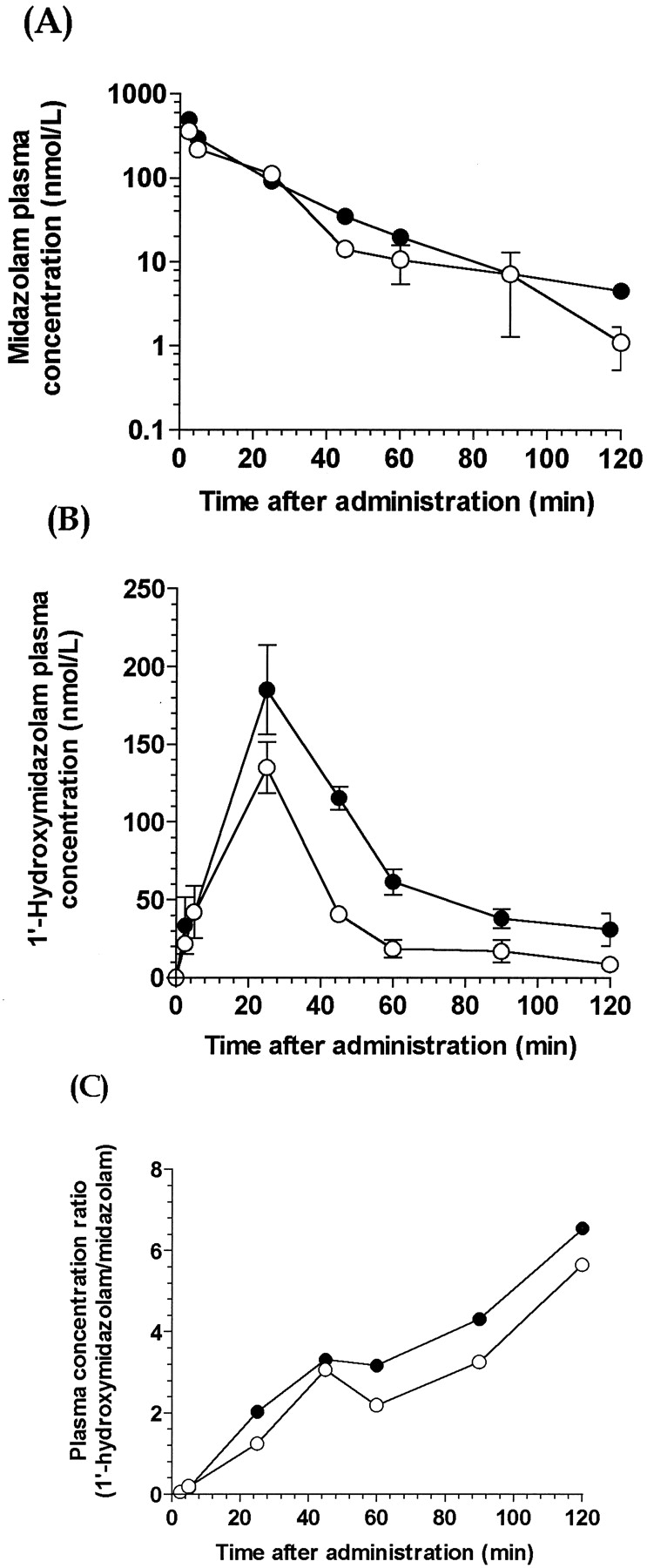
Time course of plasma midazolam (A) and 1′-hydroxymidazolam (B) concentrations and their ratios after single intravenous administration of midazolam (0.25 mg/kg) to Tg-CYP3A4 (●) and wild-type (○) mice.
Values represent the mean and the vertical lines represent S.D. of midazolam and 1′-hydroxymidazolam concentrations from three mice.
Pharmacokinetic parameters for midazolam and 1′-hydroxymidazolam after single intravenous administration of midazolam (0.25 mg/kg) in wild-type and Tg-CYP3A4 mice
The F value of orally administrated midazolam in Tg-CYP3A4mice (10.1%) was decreased about 50% compared with that of WT mice (21.5%), which is statistically significant.
Effect of ketoconazole on pharmacokinetics of midazolam metabolism.
Pretreatment with ketoconazole (40 mg/kg, oral) significantly increasedCmax of MDZ, prolonged MRT of MDZ andTmax of its metabolite, increased AUC of MDZ, and decreased MDZ CLoral (Fig. 7; Table3). Ketoconazole showed stronger inhibition of MDZ metabolism in the Tg-CYP3A4 mice than in the WT mice. As shown in Table 3,Cmax andt1/2 values of MDZ,Tmax andCmax values of 1′-OH-MDZ revealed no significant change in WT mice with and without ketoconazole treatment. However, these values became significantly different in Tg-CYP3A4 mice after the treatment of ketoconazole. Interestingly, MDZ Cmax (1130 ± 180 nM) of Tg-CYP3A4 mice treated with ketoconazole was similar as that (1190 ± 957 nM) of WT mice without treatment, suggesting that human CYP3A4 enzyme activity presenting in the Tg-CYP3A4 mouse intestine was almost totally inhibited.
Discussion
CYP3A4 is a major P450 enzyme expressed in the liver and gastrointestinal tract and plays an important role in the metabolism of a majority of therapeutic drugs (Guengerich, 1999; Wrighton et al., 2000). Expression of CYP3A5 in the human small intestine has been debated due to discrepancies in the specific detection of its mRNA and protein (Kolars et al., 1992, 1994; Lown et al., 1994; Paine et al., 1997). However, a recent comprehensive study demonstrated and supported the notion that CYP3A4 is the major P450 isoform expressed in this tissue (Zhang et al., 1999). In particular, the intestinal contribution of CYP3A4 to food-drug, drug-drug interactions and the determination of the bioavailability of a number of CYP3A substrates are of major medical importance (Venkatakrishnan et al., 2001; Nebert and Russell, 2002). However, an appropriate model to study the human small intestinal CYP3A4 is lacking. In the present study, a new transgenic mouse that expresses the human CYP3A4 (Tg-CYP3A4) in the small intestine was generated. CYP3A4 expression in the small intestine was higher than that found in pooled human gut microsomes, and its mobility was similar to the recombinant CYP3A4 as judged by immunoblot with a monoclonal antibody that is specific to CYP3A4 and does not cross-react with CYP3A5 or other mouse CYP3A proteins (Gelboin et al., 1995). Surprisingly, CYP3A4 is not expressed in the livers of transgenic mice. The lack of CYP3A4 expression in liver, a major site of expression in humans, could be due to the lack ofcis-acting binding sites for liver-enriched transcription factors in the BAC transgene. Alternatively, the lack of expression could be due to the effect of the integration site on tissue-specific expression of the transgene, which is present in approximately five copies per genome. In any case, the high level of expression of CYP3A4 in the small intestine of the transgenic mouse yields a potential model to study the expression of human CYP3A4 in the small intestine in an intact animal model.
In humans, the hydroxylation of MDZ to its 1′-OH-MDZ (major metabolite) and 4-OH-MDZ metabolites is well established to be dependent on CYP3A enzymes (Kronbach et al., 1989; Thummel et al., 1994a,b). The results of chemical inhibition and immunoinhibition studies indicate that CYP3A4 is a major P450 isoform mediating hydroxylation of MDZ in intestinal microsomes. Thus, hydroxylation of MDZ or 1′-OH-MDZ/MDZ ratio can be used as an in vitro and in vivo approach to evaluate the expression of the human CYP3A4. In vitro studies were carried out to determine the CYP3A4 activities present in the intestinal and hepatic metabolism in Tg-CYP3A4 and WT mice. TheVmax for the formation of 1′-OH-MDZ and 4-OH-MDZ was much higher in Tg-CYP3A4 than in the WT mouse, and pooled human gut microsomes. The apparentKm was similar between Tg-CYP3A4 and WT mice, suggesting that the difference in the CLint is due to CYP3A4 expression in the small intestine of the transgenic mouse. These kinetic parameters are in accordance with those reported previously (Emoto et al., 2000). Our data demonstrate that ketoconazole is a potent inhibitor to both 1′-OH-MDZ and 4-OH-MDZ formation from MDZ in Tg-CYP3A4 as well as WT mice, and human intestinal microsomes.
MDZ metabolism was used to determine the activity of CYP3A4 expression in the small intestine of the Tg-CYP3A4 mice in vivo as well. After a single oral dose of MDZ, the calculated pharmacokinetic parameters, peak plasma concentration, half-life, and bioavailability were significantly decreased in Tg-CYP3A4 mice. However, the clearance, systemic exposure (AUC), and mean residence time were not significantly different between WT and Tg-CYP3A4 mice. The half-life, clearance, and bioavailability were in the range of those previously reported for MDZ in the rat (Cleton et al., 1999;Higashikawa et al., 1999). It is shown that hepatic CYP2C also catalyzes the production of 1′-OH-MDZ from MDZ in addition to CYP3A in mice liver microsomes (Perloff et al., 2000). It is likely that this enzyme has some contribution in the oxidative metabolism of MDZ in vivo to hydroxylated metabolites that are rapidly conjugated and eliminated in mice.
In addition, the successful expression of the human CYP3A4 in the small intestine of the transgenic mouse led to a significant 3-fold higher plasma concentration of 1′-OH-MDZ (an index of the CYP3A4 activity) in Tg-CYP3A4 compared with WT mice. The maximum plasma concentration and systemic exposure (AUC) ratio of 1′-OH-MDZ/MDZ was also statistically different after oral administration of MDZ between Tg-CYP3A4 than WT mice. These data are in agreement with human studies measuring the ratio of 1′-OH-MDZ/MDZ as an indicator of hepatic CYP3A4 activity (Streetman et al., 2001).
CYP3A enzyme is constitutively expressed in the intestine and liver of different species such as mice, rats, dogs, and humans, and regulated by many classes of compounds (Guengerich, 1999; Wrighton et al., 2000). To evaluate the validity of our mouse model, the effect of ketoconazole, a potent inhibitor of CYP3A4, on the pharmacokinetics of MDZ in Tg-CYP3A4 mice was investigated. Ketoconazole significantly changed the half-life, clearance, and systemic exposure in both Tg-CYP3A4 and WT mice. The inhibitory potency of ketoconazole toward MDZ and 1′-OH-MDZ was more significant in Tg-CYP3A4 than in WT mice. Our observations are consistent with previous studies in which coadministration of ketoconazole (400 mg daily) with 7.5 mg of MDZ orally increased the AUC about 16-fold (Olkkola et al., 1994). However, the maximum plasma concentration and the AUC of 1′-OH-MDZ showed little or no inhibitory effect by ketoconazole in WT mice, whereas these parameters were decreased in Tg-CYP3A4 mice. More significant change of MDZ pharmacokinetics is due to the expression of CYP3A4 in this transgenic mouse. This observation suggests that this Tg-CYP3A4 mouse could be used to study food-drug, drug-drug interactions of CYP3A4 substrates in vivo.
Acknowledgment
We thank John Buckley for technical assistance and Jeffrey Idle for valuable discussions.
Footnotes
-
↵1 Current address: CombinatoRx Inc., 650 Albany St., Boston, MA 02118.
-
↵2 Current address: Departamento De Farmacologia Y Toxicologia, Seccion De Toxicologia Ambiental, Cinvestav-IPN, AP 14-740, México D.F. 07000.
-
↵3 Current address: Merck Research Laboratories, 126 East Lincoln Ave., Rahway, NJ 07065.
- Abbreviations used are::
- P450
- cytochrome P450
- MDZ
- midazolam
- 1′-OH-MDZ
- 1′-hydroxymidazolam
- 4-OH-MDZ
- 4-hydroxymidazolam
- PBS
- phosphate-buffered saline
- mAb
- monoclonal antibody
- Tg-CYP3A4
- transgenic-CYP3A4
- WT
- wild-type
- PCR
- polymerase chain reaction
- mEH
- microsomal epoxide hydrolase
- SSC
- standard saline citrate
- LC-MS/MS
- liquid chromatography tandem mass chromatography
- AUC
- area under the concentration-time curve
- MRT
- mean residence time
- Received December 3, 2002.
- Accepted January 27, 2003.
- U.S. Government




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}