


Abstract
The effect of P-glycoprotein (Pgp) and/or CYP3A on the disposition of xenobiotics has been extensively investigated and is often of interest during drug discovery lead optimization. We have previously described a monkey pharmacokinetic screen to rapidly estimate absorption and first-pass extraction. In the present work, this monkey screen has been expanded to include an assessment of Pgp/CYP3A effects on absorption and first-pass extraction, using ketoconazole as a prototypic dual Pgp/CYP3A inhibitor. To generate a ketoconazole dosing regimen, the pharmacokinetics of ketoconazole were first determined in the monkey and were found to be consistent with that previously described in the rat, dog, and human. Dose-ranging experiments demonstrated that a single 10-mg/kg intraduodenal ketoconazole dose would provide an appropriate exposure; this dose was used throughout subsequent interaction experiments. Next, erythromycin and propranolol were explored as positive and negative control substrates for Pgp/CYP3A interactions, respectively. As anticipated, ketoconazole produced no change in the absorption or first-pass extraction of propranolol but resulted in a substantial increase in absorption and decrease in first-pass extraction of erythromycin. Finally, this ketoconazole-based monkey screen was deployed in a drug discovery setting, and examples of such use are presented. These experiments have allowed a more complete characterization of ketoconazole as a prototypic dual Pgp/CYP3A inhibitor and its use as a tool in a preclinical setting and further demonstrate the use of the monkey to investigate the role of Pgp/CYP3A in limiting the oral bioavailability of new drug candidates.
The role of pharmacokinetic screening in a comprehensive drug discovery paradigm has been well established over the past decade (Smith, 1998; Tarbit and Berman, 1998). In particular, in vivo preclinical pharmacokinetic lead optimization has been widely used to enhance the quality of drug candidates (Cox et al., 2002). Because most drugs are intended for oral delivery, dependable methods for ascertaining the mechanism(s) governing the oral performance of new chemical entities (i.e., absorption versus first-pass hepatic extraction) are critical for productive lead optimization efforts. Among the various factors that have been identified as being important regulators of oral absorption, P-glycoprotein (Pgp1) and cytochrome P450 enzymes, especially CYP3A, are particularly prominent (Benet et al., 1996; Wacher et al., 2001). Thus, for molecules or entire chemical series with apparent absorption liabilities, elucidation of the role of Pgp and/or CYP3A in a relevant preclinical model may be a useful component of the overall lead optimization process. Numerous in vitro models for assessing the role of transporters and drug-metabolizing enzymes in limiting oral bioavailability have been described (Clarke, 1998; Polli et al., 2001). However, there appear to be few widely accepted in vivo models for investigating the role of these important potential absorption barriers in the major preclinical species, particularly models that can be used in a relatively high-throughput setting.
Previously (Ward et al., 2001), we have described the development of an in vivo screening model for the rapid characterization of drug candidates across several species, with particular emphasis on the monkey. In our earlier work, we set forth the theoretical basis for this model and described its implementation in a screening paradigm. As part of our ongoing efforts to further characterize this model and increase its utility in our in vivo screening efforts, we have further investigated its use to characterize the role of Pgp and/or CYP3A in regulating the absorption of new chemical entities. This report describes a set of studies in our laboratory designed to evaluate the use of ketoconazole, a prototypic dual Pgp/CYP3A modulator, in this monkey absorption model. The objectives of the present study were 1) to evaluate the pharmacokinetics of ketoconazole in the male cynomolgus monkey and identify an appropriate dosing regimen of ketoconazole for use in preclinical screening; 2) to validate the proposed screening model using erythromycin (a known Pgp/CYP3A substrate) (Wacher et al., 1995) and propranolol (known to have minimal interaction with Pgp/CYP3A) (McGinnity et al., 2000; Eneroth et al., 2001; Stephens et al., 2002); and 3) to demonstrate the application of the proposed screening model in the monkey during lead optimization.
Materials and Methods
Materials. Ketoconazole, erythromycin, and (±)-propranolol were purchased from Sigma-Aldrich (St. Louis, MO). Test compounds used in this study were proprietary compounds synthesized during lead optimization for various research programs by the Department of Medicinal Chemistry at GlaxoSmithKline (King of Prussia, PA). All other reagents and materials were purchased from standard vendors and were of the highest available purity. Unless otherwise noted, all dosages were prepared as solutions in analytical-grade water with up to 20% hydroxypropyl-β-cyclodextrin (Cerestar USA, Inc., Hammond, IN) or 5% polyethylene glycol (PEG-300; Sigma-Aldrich) and ≤3% dimethyl sulfoxide.
Animals. Male cynomolgus monkeys (Macaca fascicularis; Charles River Primate Laboratories, Houston, TX) with indwelling femoral, hepatic portal, and duodenal access ports were used in these studies. The monkeys were housed according to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals in individual cages in unidirectional airflow rooms with controlled temperature (22 ± 2°C) and relative humidity (50 ± 10%) and 12-h light/dark cycles. Animals were fed a standard animal diet (Purina, St. Louis, MO); food was available ad libitum except for overnight periods before dosing. Whenever overnight fasting was used, food was provided after the 240-min blood sample was obtained unless otherwise stated. All animal use was conducted according to protocols approved by the Institutional Animal Care and Use Committee before the study, and all surgical procedures were conducted using aseptic techniques in special-purpose operating suites. In addition, a standard complete blood chemistry panel was performed on each animal before each study leg. Studies were not conducted unless the blood chemistry values were within normal ranges. Monkeys also were conditioned for the restraint system used to collect blood samples before the initiation of the study. All study days were separated by a minimum interval of 1 week.
Ketoconazole Pharmacokinetics. Studies were conducted to evaluate the intravenous pharmacokinetic parameters of ketoconazole in the monkey, as well as to determine the portal and systemic plasma ketoconazole concentration profiles obtained after intraduodenal administration. The dose solution of ketoconazole was prepared in analytical-grade water (pH 3.5) with 5% PEG-300 and 3% dimethyl sulfoxide. Monkeys (n = 3) received ketoconazole at a dose of 1 mg/kg as a 60-min intravenous infusion or 10 mg/kg as a 2-min intraduodenal infusion (4 ml/kg dose volume for both routes of administration). For all studies, blood was collected from the femoral vein (during and after intravenous administration) or simultaneously from the femoral and hepatic portal veins (during and after intraduodenal administration) at predetermined time points and centrifuged to obtain plasma.
P-Glycoprotein/CYP3A Modulation Validation. To validate the proposed screening model, erythromycin was used as a prototypic dual P-glycoprotein/CYP3A substrate, and propranolol was used as a negative control. Erythromycin was administered as an intraduodenal bolus at a dosage of 5 mg/kg (formulated in PEG-300, 0.5 ml/kg dose volume), and propranolol was administered as an intraduodenal bolus at a dosage of 1 mg/kg (formulated in water, 0.5 ml/kg dose volume). Three hours after the initial administration of erythromycin or propranolol, ketoconazole (10 mg/kg) was administered as a 10-min intraduodenal infusion, as described above. A second bolus of erythromycin or propranolol was then administered 10 min after the completion of the ketoconazole infusion. Blood samples were collected simultaneously from femoral and hepatic portal vein catheters starting 5 min after the first bolus of test compound and continuing until 3 h after the second bolus of test compound, with plasma isolated from each blood sample by centrifugation. The animals were fasted for the duration of this experiment.
Deployment of the Interaction Screen. During the course of lead optimization, various test compounds were screened for evidence of favorable absorption properties in the monkey as described previously (Ward et al., 2001). Based on these screens, selected compounds with limited bioavailability were identified for investigation in the interaction screen to determine the role of Pgp/CYP3A in restricting their oral bioavailability in the monkey. Studies were conducted as described for erythromycin and propranolol above, with the exception that the dosing interval between the initial compound dose and the repeat dose with ketoconazole was shortened from 3 to 2 h. This study design change was invoked to minimize stress on the animals and presumably would have minimal effect on the experimental data.
Analytical Procedures. Plasma samples were subjected to protein precipitation by acetonitrile and concentrations of test compounds quantified using liquid chromatography/tandem mass spectrometry using an atmospheric pressure chemical ionization interface to a Sciex API 4000 mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA). Positive-ion multiple reaction monitoring was used for the tandem mass spectrometric detection of erythromycin, ketoconazole, and propranolol. The selected [M+H]+ precursor ions were m/z 734.5 for erythromycin, m/z 531.2 for ketoconazole, and m/z 260.1 for propranolol, and the product ions monitored were at m/z 158.3, 82.3, and 116.3 for erythromycin, ketoconazole, and propranolol, respectively. The lower limit of quantification was 2.0 ng/ml for erythromycin and propranolol and 5.0 ng/ml for ketoconazole. Using a (1/x) weighted linear regression analysis of the calibration curves, linear responses were observed for erythromycin and propranolol concentrations ranging from 2 to 5000 ng/ml and for ketoconazole concentrations ranging from 5 to 5000 ng/ml. For the proprietary compounds evaluated in the screening studies, assay conditions were dependent upon the structural characteristics of the compounds under analysis. In general, positive-ion Turbo IonSpray ionization was used for mass spectrometric detection. Using 50 μl of plasma, the lower limit of quantification for each test compound ranged from 2.0 to 10.0 ng/ml. Each of the analytical methods used demonstrated adequate reproducibility, with <5% day-to-day variability in response.
Pharmacokinetic Data Analysis. Concentration versus time profiles were obtained for each analyte, and standard noncompartmental analysis was performed on the data using WinNonlin Professional Version 3.3 (Pharsight, Mountain View, CA) to recover area under the curve (AUC) and other noncompartmental parameters. Bioavailability was estimated by dividing the dose-normalized AUC (DNAUC) (0 - t) resulting from intraduodenal administration by the DNAUC (0 - t) resulting from intravenous administration (where t is the last time point with measurable drug concentrations in the study; i.e., AUC values were not extrapolated to infinity). Additionally, for the screening studies, bioavailability was estimated using the theoretical maximum DNAUC concept described previously (Ward et al., 2001). Where appropriate, data for each pharmacokinetic parameter were averaged and reported as mean ± standard deviation. A two-tailed Student's t test was used for statistical evaluation of the observed data as appropriate. In all cases, a probability level of p ≤ 0.05 was predetermined as the criterion of significance.
Results
Ketoconazole Pharmacokinetics. Ketoconazole demonstrated a systemic plasma clearance in the male cynomolgus monkey of 12.5 ± 1.2 ml/min/kg, with a volume of distribution of 0.661 ± 0.119 liter/kg and an apparent half-life of 50.6 ± 6.5 min (Table 1). To help design future drug interaction studies, the absorption and systemic exposure of ketoconazole were determined after intraduodenal administration. A preliminary dose-ranging exposure study was first conducted at 0.3, 1.0, and 3.0 mg/kg (n = 1 monkey per dose), followed by a more definitive exposure study at 1.0 and 10.0 mg/kg (n = 3 additional monkeys per dose). The results of these experiments are summarized in Fig. 1, and the average hepatic portal and systemic ketoconazole plasma concentration versus time profiles for the 10 mg/kg intraduodenal dose are displayed in Fig. 2. Ketoconazole was reasonably well absorbed (55.2 ± 11.2%) and underwent substantial apparent hepatic extraction (61.1 ± 6.9%) in the monkey, consistent with its plasma clearance (∼60% hepatic plasma flow) (Table 1). Portal and systemic Cmax of ketoconazole after the 10-mg/kg intraduodenal dose were approximately 14,000 and 1300 ng/ml (26 and 2.4 μM), respectively. Similar and dose-proportional observations were made with the other doses of ketoconazole (Fig. 1).
Pharmacokinetic parameters obtained for ketoconazole in the monkey after either intravenous or intraduodenal administration All data are expressed as mean ± standard deviation for n = 3 monkeys per dose.
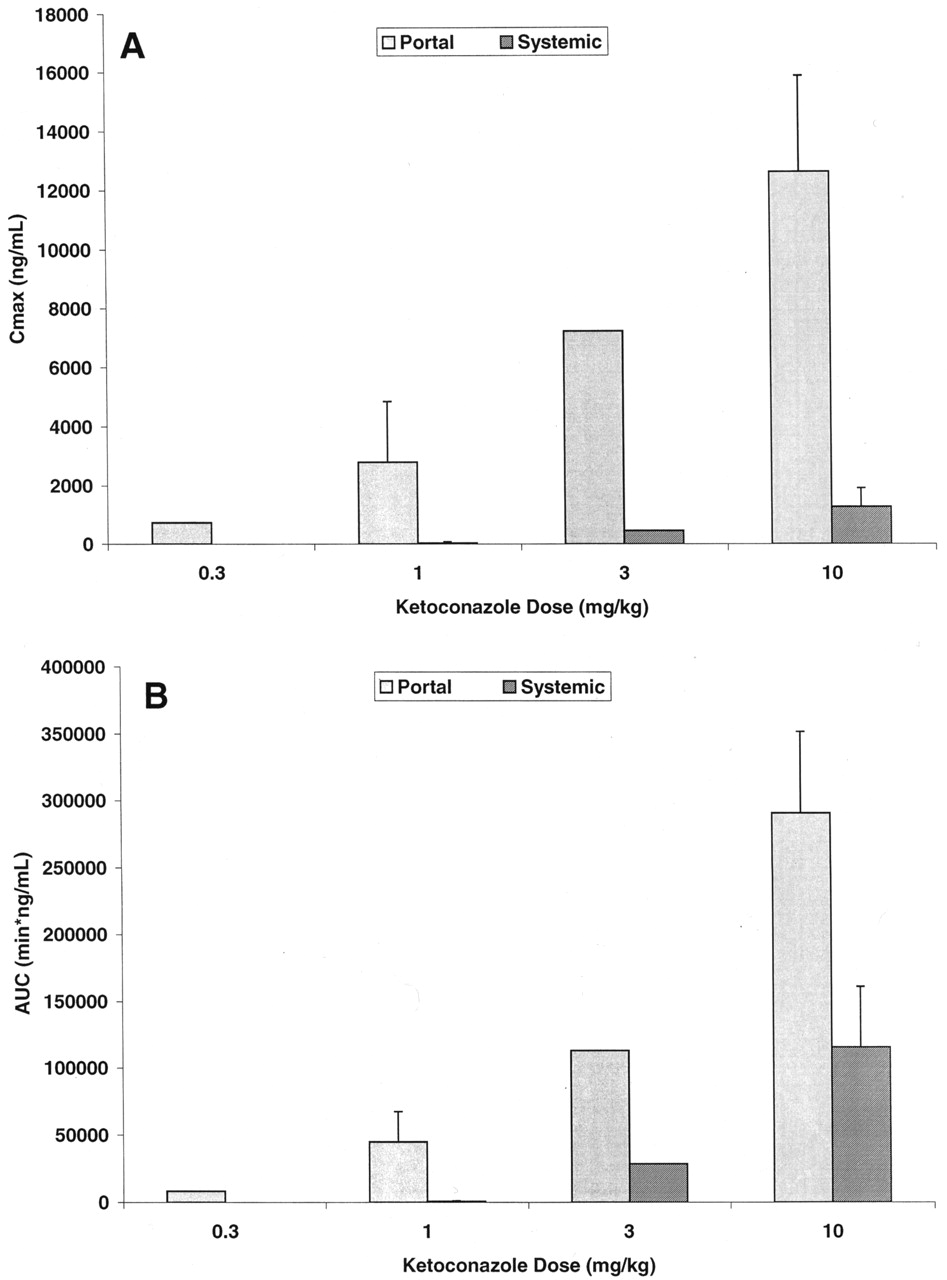
Portal and systemic Cmax(A) and AUC (B) of ketoconazole in the monkey after intraduodenal administration.
Solid bars represent data from the hepatic portal vein; hashed bars represent data from the femoral vein.

Portal (open symbols) and systemic (closed symbols) exposure of ketoconazole after a 10-mg/kg intraduodenal administration to the monkey.
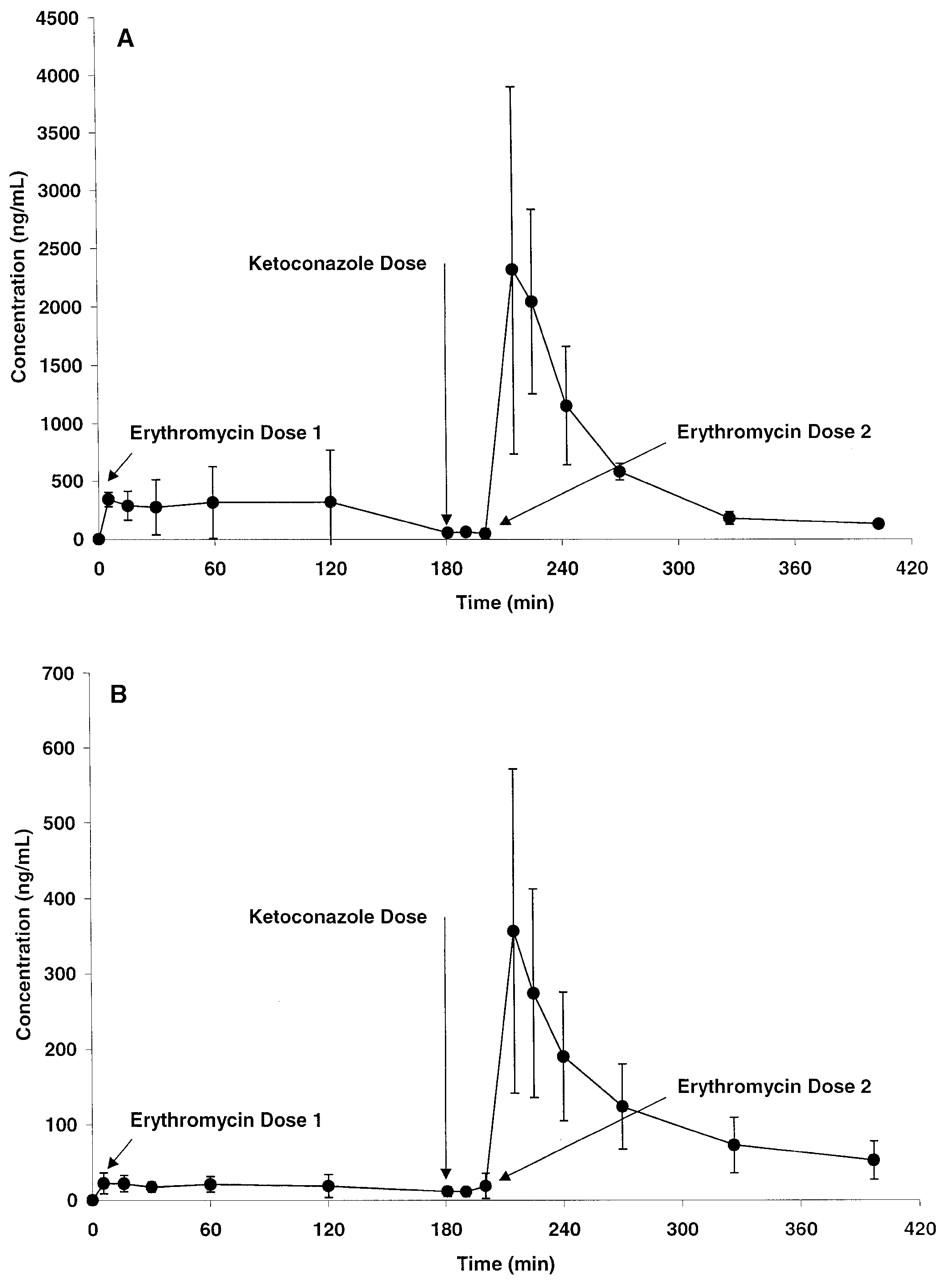
Validation of Ketoconazole Modulation of Pgp/CYP3A. To confirm that the ketoconazole dosing regimen selected for the interaction studies was adequate to modulate the activity of Pgp/CYP3A, the effect of ketoconazole on the disposition of erythromycin in the monkey was evaluated. The portal and systemic plasma concentrations of erythromycin before and after a concomitant administration of ketoconazole are shown in Fig. 3. In the absence of ketoconazole, the absorption and systemic exposure of erythromycin in the monkey were poor; coadministration of 10 mg/kg ketoconazole produced a 4.0 ± 2.1-fold increase in absorption and a 7.5 ± 0.4-fold increase in systemic exposure. Also, ketoconazole increased the apparent oral systemic half-life of erythromycin in the monkey from 80.7 ± 33.5 min to 131 ± 44 min (p = 0.07, two-tailed paired Student's t test), indicating an effect of ketoconazole on both the absorption and elimination of erythromycin. In a separate experiment using the same monkeys 2 weeks later, the effect of ketoconazole on the disposition of propranolol in the monkey was evaluated. Ketoconazole had no significant effect on either the absorption (portal AUC = 42.0 versus 32.9 μg · min/ml in the absence and presence of ketoconazole, respectively) or systemic exposure (systemic AUC = 0.883 versus 1.39 μg · min/ml in the absence and presence of ketoconazole, respectively) of propranolol in the monkey.

Portal (A) and systemic (B) exposure of erythromycin before and after a concomitant 10-mg/kg dose of ketoconazole in the monkey.
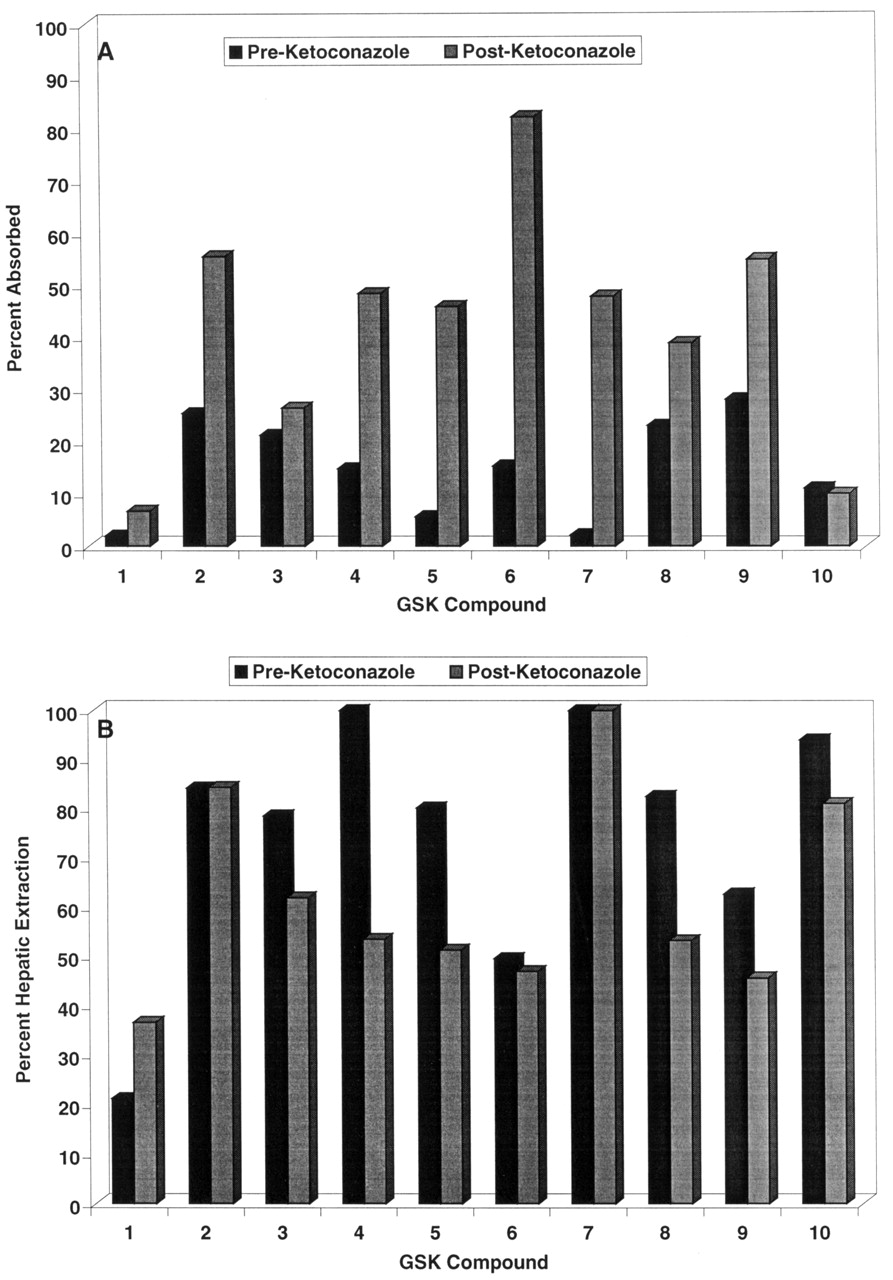
Deployment of the Ketoconazole Interaction Screen. During the course of lead optimization for several different efforts, a monkey oral bioavailability issue was identified, and a monkey screen was initiated in the fashion described previously (Ward et al., 2001). A total of 10 compounds were identified from these efforts to be evaluated in the ketoconazole screen. The 10 compounds selected for the ketoconazole interaction studies demonstrated poor systemic exposure after intraduodenal administration (<25% estimated bioavailability) when dosed in cassettes of up to four components each in the initial monkey screen. These compounds represent six different structural classes directed at four different therapeutic targets and thus demonstrate some structural diversity. The estimated absorption and hepatic extraction in the presence and absence of ketoconazole for these 10 compounds are displayed in Fig. 4, and the overall plasma concentration versus time profile for a representative compound (compound 8) is shown in Fig. 5. Ketoconazole substantially enhanced the absorption of all compounds except compounds 1, 3, and 10, indicating that factors other than Pgp/CYP3A limit the absorption of these analogs. Furthermore, ketoconazole substantially suppressed the first-pass hepatic extractions of compounds 4, 5, 8, and 9, indicating that Pgp/CYP3A limits not only absorption but also hepatic extraction of these molecules.
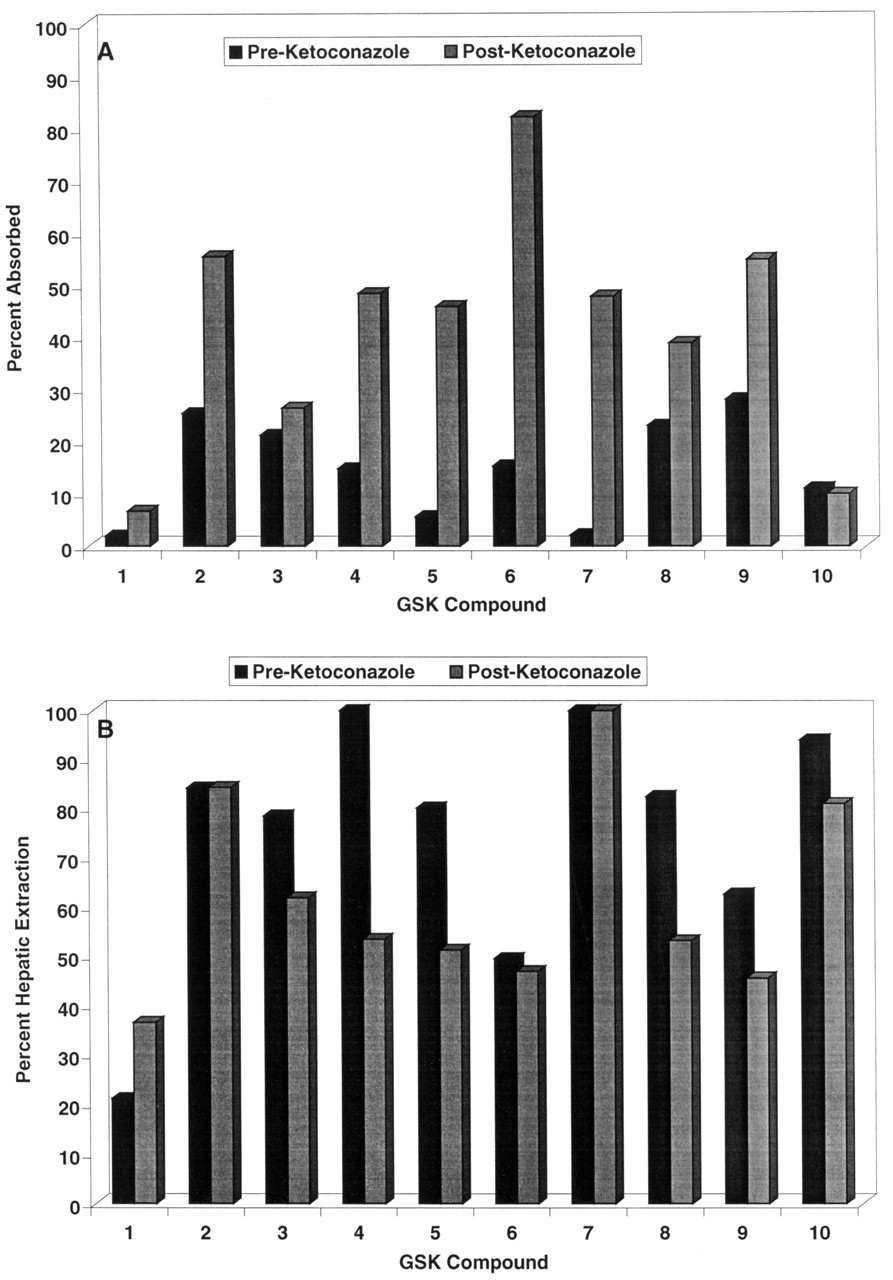
Absorption (A) and hepatic extraction (B) for 10 compounds in the absence (closed bars) or presence (hashed bars) of ketoconazole in the monkey.

Portal (closed symbols) and systemic (open symbols) exposure of compound 8 after intraduodenal administration in the presence or absence of a concomitant dose of ketoconazole in the monkey.
Discussion
The important role of Pgp/CYP3A in limiting the oral bioavailability of certain classes of molecules has been well established in recent years (Wacher et al., 2001). At the same time, the number of tools available to study the role of transporters and drug-metabolizing enzymes has increased, both in vitro (isolated enzyme preparations, specific overexpressing cell lines) and in vivo (specific Pgp inhibitors, mdr1a/b(-/-) mice) (Adachi et al., 2001; Polli et al., 2001; Yamazaki et al., 2001). However, many of the available research tools are ill suited for application in a drug discovery setting. For example, often in drug discovery a species-specific pharmacokinetic issue is identified, such as poor monkey oral bioavailability. In those instances, in vivo knockout mouse models may be instructive, but do not directly address the problem at hand. Similarly, many readily available in vitro systems are aimed mostly at human or rodent tissues, and a species-appropriate in vitro model may not be available. Consequently, there is a need for characterized research tools to study Pgp/CYP3A interactions that can be readily adapted to a drug discovery setting.
Although ketoconazole is a potentially useful tool for inhibiting Pgp/CYP3A, before this investigation the pharmacokinetic parameters for ketoconazole in the monkey have not been described. In the rat, after a 5 mg/kg dose, ketoconazole displays a plasma clearance of 14.4 ml/min/kg, a volume of distribution that approximates total body water, and a half-life of 0.58 h (Remmel et al., 1987). After a 20 mg/kg dose in the dog, clearance is 2.7 ml/min/kg with a volume of distribution that approximates total body water and a half-life of 2.7 h (Baxter et al., 1986). Our data for the disposition of ketoconazole in the cynomolgus monkey are consistent with these observations, although the monkey demonstrates somewhat higher plasma clearance (as a fraction of hepatic plasma flow) than that reported in the rat or dog. This difference may be due in part to the nonlinear elimination of ketoconazole that has been demonstrated in the rat (Matthew et al., 1993). In the present study, there was no direct evidence of nonlinearity of ketoconazole exposure in the monkey over the dose range explored, although a rigorous exploration of linearity was not conducted. Together, these data demonstrate that substantial portal, hepatic, and systemic exposure of ketoconazole is achievable in the monkey with a reasonable dosing regimen.
After establishing the pharmacokinetics of ketoconazole in the monkey, we wanted to ensure that an appropriate dose was administered to inhibit Pgp/CYP3A. However, as no directly applicable literature data were available, the monkey concentration-effect relationship was inferred from in vitro data and from in vivo data in humans, dogs, and rats. In vitro, ketoconazole inhibits human CYP3A metabolism with a Ki of approximately 0.02 to 5 μM (10-2700 ng/ml) and reverses P-glycoprotein-mediated cellular accumulation in vitro with a Ki ranging from 3 to 25 μM (Yasuda et al., 2002). In clinical interaction studies with the prototypic CYP3A substrate midazolam (Tsunoda et al., 1999; Lee et al., 2002), systemic ketoconazole concentrations were maintained above 300 ng/ml (resulting from multiple 3 mg/kg dosages) and produced significant changes in midazolam pharmacokinetics. No estimate of ketoconazole Cmax was provided; however, previous data suggest that a 3 mg/kg oral dose should result in a Cmax >5000 ng/ml in humans (Baxter et al., 1986). In dogs, a single 16 mg/kg oral dose of ketoconazole (which should correspond to a Cmax of ∼12,000 ng/ml) (Baxter et al., 1986) resulted in significant changes in midazolam pharmacokinetics (Kuroha et al., 2002). Several ketoconazole interaction studies have been performed in the rat, with estimated ketoconazole concentrations of 1500 to >5000 ng/ml producing significant effects on the pharmacokinetics of antipyrine (Ervine et al., 1996), midazolam (Yamano et al., 1999), K11002 (Zhang et al., 1998), and O6-benzylguanine (Ewesuedo and Dolan, 2000). Finally, it has been demonstrated that total (rather than unbound) ketoconazole concentrations should be used when planning drug-drug interaction studies (Venkatakrishnan et al., 2000); therefore, monkey plasma protein binding of ketoconazole was not considered. Altogether, these data suggested that the concentrations achieved using a 10 mg/kg intraduodenal dose in the monkey would be sufficient for both intestinal and hepatic Pgp/CYP3A inhibition. This suggestion was confirmed in our experiments using erythromycin as a Pgp/CYP3A substrate, in which a 10 mg/kg ketoconazole coadministration produced significant enhancement in both absorption of and total systemic exposure to erythromycin.
Based on the promising results from our validation experiments, we have deployed this monkey Pgp/CYP3A interaction screen in several drug discovery efforts, as described under Results and in Fig. 4. Our experience to date indicates that use of this interaction screen as part of a comprehensive preclinical pharmacokinetic lead optimization program can effectively direct the efforts of research teams toward solving mechanism-based bioavailability issues. For example, compound 1 in this study was an early lead molecule for a respiratory disease program that had demonstrated poor monkey oral bioavailability. As shown in Fig. 4, compound 1 demonstrated poor absorption in the monkey, with no marked enhancement by ketoconazole. These data implicated a mechanism other than Pgp/CYP3A in the poor bioavailability of compound 1; subsequent in vitro experiments in fact demonstrated compound 1 to have poor passive permeability (data not shown). Extensive lead optimization with the use of in vitro permeability assays and in vivo absorption screening was able to overcome the permeability issue, resulting in molecules such as compound 2, which had improved absorption but were subject to a substantial ketoconazole effect. To minimize potential clinical drug-drug interactions, additional monkey absorption screening was conducted, and compound 3 was identified, with absorption that is not markedly enhanced by ketoconazole. Compound 3 subsequently was selected for further development. In a different research program, compounds 4, 5, 6, and 7 were early lead molecules that suffered from poor monkey oral bioavailability; the data from the present study indicate an important role of Pgp/CYP3A in limiting the absorption of these four compounds. After substantial lead optimization using in vitro permeability and cytochrome P450 inhibition assays, compounds 8 and 9 have been identified, which have substantially less ketoconazole interaction and are being further pursued in development. Finally, compound 10 demonstrated poor oral bioavailability in the monkey that was clearly mediated by poor absorption and high first-pass hepatic extraction; however, these phenomena did not appear to be mediated by Pgp/CYP3A. Additional investigative work is ongoing into the mechanism(s) underlying the poor solution bioavailability of compound 10 in the monkey, and the present screening data have directed experiments away from Pgp/CYP3A as areas of investigation for this research.
In summary, we have described the further development of a monkey screening model using ketoconazole to probe Pgp/CYP3A interactions with compounds of interest in a discovery setting. The development and deployment of this screen are ongoing, and future work will focus on novel methods for the dissection of intestinal and hepatic drug interactions, the use of alternate probe inhibitors to evaluate the role of additional transporters/drug-metabolizing enzymes in limiting oral bioavailability, and the application of these areas of research to other major preclinical species.
Acknowledgments
We acknowledge the expert technical assistance of members of the Cardiovascular and Urogenital Center of Excellence in Drug Discovery Drug Metabolism and Pharmacokinetics group and the Laboratory Animal Sciences group, including Earl Jenkins and Guy Vaden, in conducting the in vivo experiments described here. We also acknowledge the valuable assistance of members of CVU CEDD DMPK (including Ryan McGee, Bonnie Orr, and Amanda Wright) and Worldwide Bioanalysis (including Yanwen Qian, Pamela Souder, Renee Person, and Lorrie Day) in the quantitative analysis of numerous molecules investigated in the monkey screening protocol.
Footnotes
-
↵1 Abbreviations used are: Pgp, P-glycoprotein; PEG-300, polyethylene glycol 300; AUC, area under the concentration-time curve; DNAUC, dose-normalized AUC.
-
These data were presented in part at the 33rd Annual Gordon Research Conference on Drug Metabolism, Plymouth, NH, July 2003.
- Received June 30, 2003.
- Accepted September 26, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}