


Abstract
Clinically, cimetidine therapy impairs the clearance of various drugs metabolized by CYP2D6, such as desipramine and sparteine. Cimetidine is known to reversibly inhibit CYP2D6 in vitro; however, Ki values are greater than plasma concentrations observed in vivo. There is evidence suggesting that this drug may act as an inactivator of cytochrome P450 (P450) enzymes after metabolic activation. Therefore, the purpose of this study was to determine whether cimetidine acts as a mechanism-based inactivator of CYP2D6. Dextromethorphan O-demethylation was used as a probe of CYP2D6 activity. The Vmax and Km of this reaction were 0.82 ± 0.06 nmol/min/nmol of P450 and 4.1 ± 0.1 μM, respectively, in pooled human liver microsomes; and 15.9 ± 0.8 nmol/min/nmol P450 and 1.4 ± 0.6 μM, respectively, with recombinant CYP2D6. With human liver microsomes, cimetidine competitively inhibited CYP2D6 (Ki = 38 ± 5 μM) and was a mixed inhibitor of recombinant CYP2D6 (Ki = 103 ± 17 μM). Preincubation of human liver microsomes with cimetidine and NADPH did not increase the inhibitory potency of cimetidine; however, preincubation with recombinant CYP2D6 resulted in enzyme inactivation that could be attenuated by the CYP2D6 inhibitor quinidine. The KI and kinact were estimated to be 77 μM and 0.03 min-1, respectively, and the half-life of inactivation was 25 min. Therefore, cimetidine may represent a class of compounds capable of inactivating specific cytochromes P450 in vivo, but for which conditions may not be achievable in vitro using human liver microsomes.
Cimetidine has been associated with many drug-drug interactions, some of which are thought to involve inhibition of cytochrome P450 (P4501) enzymes (reviewed in Somogyi and Gugler, 1982). One of the P450 enzymes that appears to be affected is CYP2D6. The clearance of several drugs known to be metabolized by this enzyme, including imipramine (Miller and Macklin, 1983; Abernethy et al., 1984), sparteine (Schellens et al., 1989), nortriptyline (Miller et al., 1983), and desipramine (Amsterdam et al., 1984), has been shown to be reduced 30 to 50% by concurrent administration of cimetidine. Furthermore, the effect of cimetidine on clearance of desipramine is limited to individuals with the rapid metabolizer phenotype; in poor metabolizers, who express no functional CYP2D6 enzyme, no interaction is observed (Steiner and Spina, 1987). Cimetidine also appears to inhibit the metabolism of debrisoquine (Philip et al., 1989) and dextromethorphan (Arnold et al., 1997), but clearance values were not obtained in these studies. Inhibition of CYP2D6 has also been investigated in vitro, and studies have demonstrated that cimetidine competitively inhibits bufuralol hydroxylase activity with a Ki of 50 to 55 μM (Knodell et al., 1991; Furuta et al., 2001). However, this value is approximately 5-fold greater than the maximal plasma level of cimetidine of 10 μM achieved during normal therapy (Brogden et al., 1978). Competitive inhibition alone, therefore, does not adequately explain the clinically observed effects of cimetidine on impaired CYP2D6-mediated drug clearance.
There is evidence to suggest that cimetidine may be able to alter P450 activity by a mechanism other than reversible inhibition. The metabolism of dapsone by human liver microsomes was found to be impaired by cimetidine; however, inhibition was observed only after microsomes had been preincubated with cimetidine and NADPH for 30 min (Tingle et al., 1991). The requirement for NADPH in the preincubation reaction suggests that cimetidine is catalytically converted to a more potent inhibitory species. In vivo, this postulated mechanism is supported by a delay in the onset of cimetidine drug-drug interactions. For example, the clearance of propranolol was reduced by 33% after 1 day of treatment with cimetidine; after 5 days of treatment, clearance was reduced by 51%, significantly lower than the effect seen on day 1 (Reimann et al., 1981). This observation is consistent with the requirement for metabolic activation of cimetidine, which would require a finite period of time.
Cimetidine has been shown to act as a selective and potent inhibitor of rat CYP2C11 through the formation of a metabolite-intermediate complex. When administered to rats, cimetidine inhibited the microsomal testosterone 2α-hydroxylase activity of CYP2C11 at the relatively low plasma concentrations attained after in vivo administration of the inhibitor (Chang et al., 1992a). In contrast, in vitro administration of cimetidine was found to inhibit the activities of rat hepatic microsomal CYP2B and CYP3A in addition to CYP2C11 with IC50 values in the range of 1.0 to 7.4 mM, concentrations much higher than those seen after in vivo administration of cimetidine (Chang et al., 1992b). The in vivo selectivity and potency of inhibition by cimetidine could only be emulated in vitro when the microsomes were first preincubated with the inhibitor and NADPH (Chang et al., 1992b). Spectral studies have demonstrated that a metabolite-intermediate complex is formed when rat hepatic microsomes are preincubated with cimetidine and NADPH, the formation of which can be attenuated by coincubation with anti-rat CYP2C11 antibodies (Levine and Bellward, 1995). Thus, at high concentrations, cimetidine appears to act as a relatively nonpotent, general P450 inhibitor; after preincubation, however, both the potency and specificity of inhibition are increased, suggesting that cimetidine may also be capable of acting as a mechanism-based inactivator. In addition to CYP2C11, CYP2C6 also appears to be inactivated by cimetidine in vitro (Levine et al., 1998).
The purpose of the present study was to investigate the effect of cimetidine on CYP2D6 activity of human liver microsomes and recombinant CYP2D6 by determining whether characteristics of mechanism-based inactivation could be observed following preincubation of the enzyme with the inhibitor. To measure CYP2D6 activity, the metabolism of dextromethorphan to dextrorphan was used as the probe reaction. The formation of dextrorphan can be monitored using an HPLC assay with fluorescence detection (Lam and Rodriguez, 1993), and the conditions under which this reaction demonstrates greatest specificity for CYP2D6 have been well documented (von Moltke et al., 1998; Yu and Haining, 2001; Granvil et al., 2002).
Materials and Methods
Chemicals and Reagents. Dextromethorphan hydrobromide (DEX), cimetidine, quinidine, 1-aminobenzotriazole, β-nicotinamide adenine dinucleotide phosphate (NAPDH), catalase (CAT), superoxide dismutase (SOD), Sigmacote chlorinated organopolysiloxane in heptane, and ethylenediaminetetraacetic acid (EDTA) were purchased from Sigma-Aldrich (St. Louis, MO). Dextrorphan and 3-methoxymorphinan were obtained from BD Gentest (Woburn, MA), and 3-hydroxymorphinan was obtained from Sigma/RBI (Oakville, ON, Canada). HPLC-grade potassium phosphate and methanol were obtained from Fisher Scientific (Toronto, ON, Canada). Pooled human liver microsomes and recombinant CYP2D6 Supersomes were both purchased from BD Gentest, as was the NADPH regenerating system. The protein and total P450 content of the microsome samples were provided by the supplier.
Enzyme Incubations. Incubation mixtures contained pooled human liver microsomes (0.5 mg of protein/ml) or recombinant CYP2D6 (2.5 pmol of P450/ml) and various concentrations of dextromethorphan in incubation buffer, which consisted of 50 mM potassium phosphate (pH 7.4) and 1 mM magnesium chloride or 100 mM potassium phosphate with 3.3 mM magnesium chloride, for the microsomal or recombinant enzyme, respectively. Incubations were initiated by the addition of NADPH (1 mM) and were carried out in a final volume of 200 μl in a 37°C shaking water bath. Reactions were terminated with 20 μl of ice-cold perchloric acid after 20 or 15 min, for incubations with human liver microsomes or recombinant CYP2D6, respectively. All samples were then spun at 3000g for 10 min in a Beckman model J-6B centrifuge (Beckman Coulter, Fullerton, CA). The supernatant was passed through a 0.22-μm polyvinylidene fluoride membrane syringe-driven filtration unit (Millipore Corporation, Bedford, MA) prior to HPLC analysis.
Determination of Kinetic Constants. The apparent Vmax and Km of dextromethorphan O-demethylation were estimated by incubating pooled human liver microsomes and recombinant CYP2D6 with a range of substrate concentrations (1.25–60 μM for microsomal protein or 0.25–80 μM DEX for recombinant CYP2D6). The data were modeled with the Michaelis-Menten equation using nonlinear regression with the Sigma Plot enzyme kinetics module, version 1.1 (SPSS Inc., Chicago IL, 2001).
Inhibition Studies. The inhibition constants and mode of inhibition were determined for both quinidine and cimetidine. Pooled human liver microsomes were incubated with DEX (2.5, 4.5, and 7.5 μM; approximately 0.5, 1, and 2 times the Km) and cimetidine (25–250 μM) or quinidine (0.025–0.2 μM). With the recombinant enzyme, incubations were carried out in the presence of 0.5, 1, or 2 μM DEX and quinidine (0.5–7.5 nM) or cimetidine (25–100 μM). Ki values and mode of inhibition for each inhibitor were estimated with the Sigma Plot enzyme kinetics module using the following equations: 
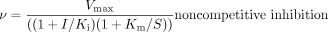
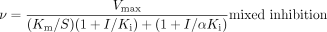 where S and I represent the concentration of substrate and inhibitor, respectively, and α is the factor change to the Km when the inhibitor is bound to the enzyme-substrate complex. The mode of inhibition that best described the data was determined by comparing r2 values for the fit of the various models.
where S and I represent the concentration of substrate and inhibitor, respectively, and α is the factor change to the Km when the inhibitor is bound to the enzyme-substrate complex. The mode of inhibition that best described the data was determined by comparing r2 values for the fit of the various models.
Inactivation Studies. The method used for the inactivation experiments involved two steps: a preincubation reaction with the inhibitor and an incubation reaction with the substrate, with these two steps conducted in separate reaction tubes. The preincubation mixture consisted of human liver microsomes (2 mg of protein/ml) or recombinant CYP2D6 (10 pmol/ml) and inhibitor in incubation buffer. After prewarming the mixture for 2 min in a 37°C shaking water bath, the preincubation reaction was initiated by the addition of 1 mM NADPH or an NADPH regenerating system consisting of 1.3 mM NADP+, 3.3 mM glucose 6-phosphate, and 0.4 U/ml glucose-6-phosphate dehydrogenase. In an attempt to maintain a high level of enzyme activity in the preincubation step, some reactions were carried out in the presence of CAT (2400 U/ml), SOD (2400 U/ml), and EDTA (1 mM), in siliconized glass test tubes prepared by pretreatment with Sigmacote. Scavengers of reactive oxygen species have been shown to have a stabilizing effect on CYP2D6 activity (Foti and Fisher, 2002). At appropriate time points, 50-μl aliquots of the preincubation mixture were removed and added to reaction tubes containing DEX and NADPH (1 mM) in incubation buffer in a volume of 150 μl. The incubation reaction, which was carried out in a final volume of 200 μl, was allowed to proceed for 20 or 15 min, for human liver microsomes or recombinant enzyme, respectively, before termination with 20 μl of perchloric acid. Samples were then processed as described above and analyzed by HPLC.
Inactivation constants were determined by measuring the half-life of inactivation at various concentrations of cimetidine. The y- and x-intercepts of a Kitz-Wilson plot of half-life versus inverse inactivator concentration were used to provide estimates of the rate constant of inactivation (kinact) and the concentration of inactivator required for half-maximal inactivation (KI).
HPLC Assay. The chromatographic system consisted of a model 1525 binary pump, a model 717plus autosampler, and a model 474 scanning fluorescence detector (all from Waters, Milford, MA). Operation of all equipment, as well as data acquisition and peak integration, was managed with Waters' Breeze software. Separation was achieved on a Waters SymmetryShield RP18 column (4.6 mm × 150 mm, 5-μm particle size) equipped with a SymmetryShield RP18 guard column (3.9 mm × 20 mm, 5-μm particle size). A mobile phase of 60% 50 mM potassium phosphate (pH 4.75) and 40% methanol was pumped through the column at 1 ml/min at ambient temperature. An injection volume of 25 μl was used and the fluorescence excitation and emission wavelengths were set at 280 and 310 nm, respectively.
Assay Validation. Calibration curves for dextrorphan, 3-hydroxymorphinan, and 3-methoxymorphinan were linear over the range of 0.1 to 5 μM. Interassay and intra-assay variability was assessed by analyzing quality control samples (N = 5) on the same day or three separate days, respectively. The percentage coefficient of variation and percentage bias were less than 7% at the high- (5 μM) and mid- (2.5 μM) range of the calibration curve, and less than 15% at the limit of quantitation (0.1 μM).
Data Analysis. Statistical analysis of the preincubation experiments was carried out by two-way analysis of variance followed by the Student Newman-Keuls test. Significant interactions between preincubation time and the presence of inhibitor were used to demonstrate that the loss in enzyme activity following preincubation was greater than would be expected due to either of these factors alone. The level of significance was set, a priori, at p < 0.05. All statistical analyses were performed using SigmaStat for Windows, version 1.0 (Jandel Inc., San Rafael, CA, 1994).
Results
The Vmax and Km of dextromethorphan O-demethylation were 0.82 ± 0.06 nmol/min/nmol of P450 and 4.1 ± 0.1 μM (n = 3, mean ± S.D.), respectively, with pooled human liver microsomes, whereas with recombinant CYP2D6, these values were 15.9 ± 0.8 nmol/min/nmol of P450 and 1.4 ± 0.6 μM (n = 3, mean ± S.D.), respectively.
Cimetidine inhibited dextrorphan formation with a potency three to four orders of magnitude lower than that of quinidine (Fig. 1), a known competitive inhibitor of CYP2D6 (Newton et al., 1995). However, the mode of inhibition by cimetidine was different for the two model systems used (Table 1), with the recombinant enzyme exhibiting mixed inhibition and human liver microsomes demonstrating competitive inhibition. The estimated Ki values for cimetidine are greater than the maximal plasma levels seen after in vivo administration of the drug, which are in the low micromolar range (Brogden et al., 1978); this is consistent with the idea that competitive inhibition alone does not explain the effect of cimetidine on CYP2D6.
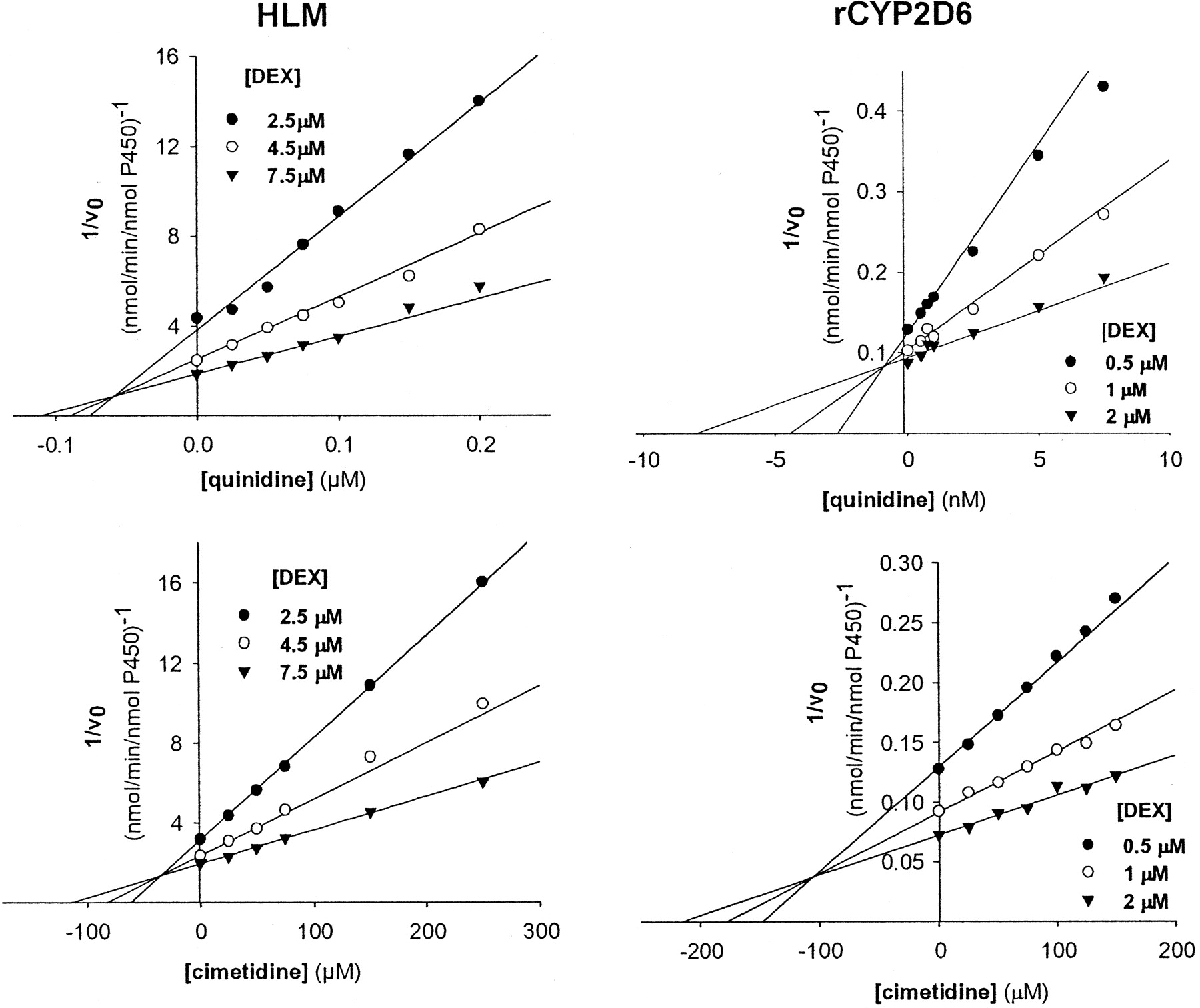
Inhibition of dextromethorphan O-demethylase activity by cimetidine and quinidine.
Pooled human liver microsomes (HLM; left panels) and recombinant CYP2D6 (rCYP2D6; right panels) were incubated with cimetidine and quinidine as described in the legend for Table 1. Data shown are representative Dixon plots. Each point represents the mean of duplicate measurements. v0 indicates initial reaction velocity.
Inhibition of dextromethorphan O-demethylation by cimetidine and quinidine Pooled human liver microsomes (HLM; 0.5 mg of protein/ml) or recombinant CYP2D6 (rCYP2D6; 2.5 pmol/ml) were incubated with NADPH (1 mM) and various concentrations of DEX and cimetidine or quinidine, and Ki values were determined as described under Materials and Methods. Data are expressed as the mean ± S.D. of three independent determinations.
To determine whether metabolism of cimetidine can result in mechanism-based inactivation of CYP2D6, the drug was preincubated with NADPH and the enzyme before the addition of the substrate (dextromethorphan). Preincubation of pooled human liver microsomes with NADPH alone resulted in a 45% decrease in enzyme activity over a 120-min period (Fig. 2A). Beyond 120 min, metabolite levels could not be reliably measured due to loss of enzyme activity. Inclusion of cimetidine in the preincubation reaction at a concentration of 25 or 100 μM did not result in further loss of enzyme activity, as would be expected in the case of mechanism-based inactivation. The loss in activity due to NADPH alone was attenuated by the presence of SOD, CAT, and EDTA. Under these conditions, enzyme activity was maintained at a measurable level for up to 24 h. However, addition of NADPH or the regenerating system during the prolonged incubation further accelerated the loss of CYP2D6 activity (data not shown). Even with the longer preincubation times, however, cimetidine did not result in inactivation of CYP2D6 (Fig. 2B). In fact, conversely, cimetidine appeared to have a protective effect that may be explained by the drug's antioxidant properties, which have been documented elsewhere (Lambat et al., 2002; Liu et al., 2002).

Effect of preincubation of human liver microsomes with cimetidine.
A, preincubation of pooled human liver microsomes with cimetidine for up to 120 min. Pooled human liver microsomes (2 mg of protein/ml) were preincubated with cimetidine (25 μM or 100 μM) or incubation buffer (control) and NADPH (1 mM). At 30-min intervals, 50 μl of the preincubation mixture was removed and added to a reaction tube containing DEX and NADPH, in a volume of 150 μl, and incubated for another 20 min. In the final incubation, the concentration of DEX was 5 μM and the concentration of microsomal protein was 0.5 mg/ml. Each point represents the mean of triplicate measurements ± S.D. ⋆, significantly less (p < 0.05) than the control group preincubated for the same period of time. B, effect of scavengers of reactive oxygen species on preincubation of human liver microsomes with cimetidine. Pooled human liver microsomes were preincubated with cimetidine (100 μM) as described for Fig. 1B, except that CAT (2400 U/ml), SOD (2400 U/ml), and EDTA (1 mM) were included. The preincubation reaction was initiated with either 1 mM NADPH (top panel) or an NADPH regenerating system (bottom panel). At the indicated times, 50 μl of the preincubation mixture was removed and incubated with DEX and NADPH, as described previously. Each point represents the mean of four measurements ± S.D. ⋆, significantly greater (p < 0.05) than the control group preincubated for the same period of time.
In control experiments, 1-aminobenzotriazole was used as a mechanism-based inactivator of both human liver microsomal and recombinant CYP2D6 (Balani et al., 2002). After preincubation of pooled human liver microsomes with 20 μM ABT and NADPH, a 34% loss in activity was observed, whereas preincubation of a 4 μM concentration of the inactivator with the recombinant enzyme resulted in a 52% decrease in activity (Fig. 3). As expected, in both cases, no inhibition was observed without preincubation.
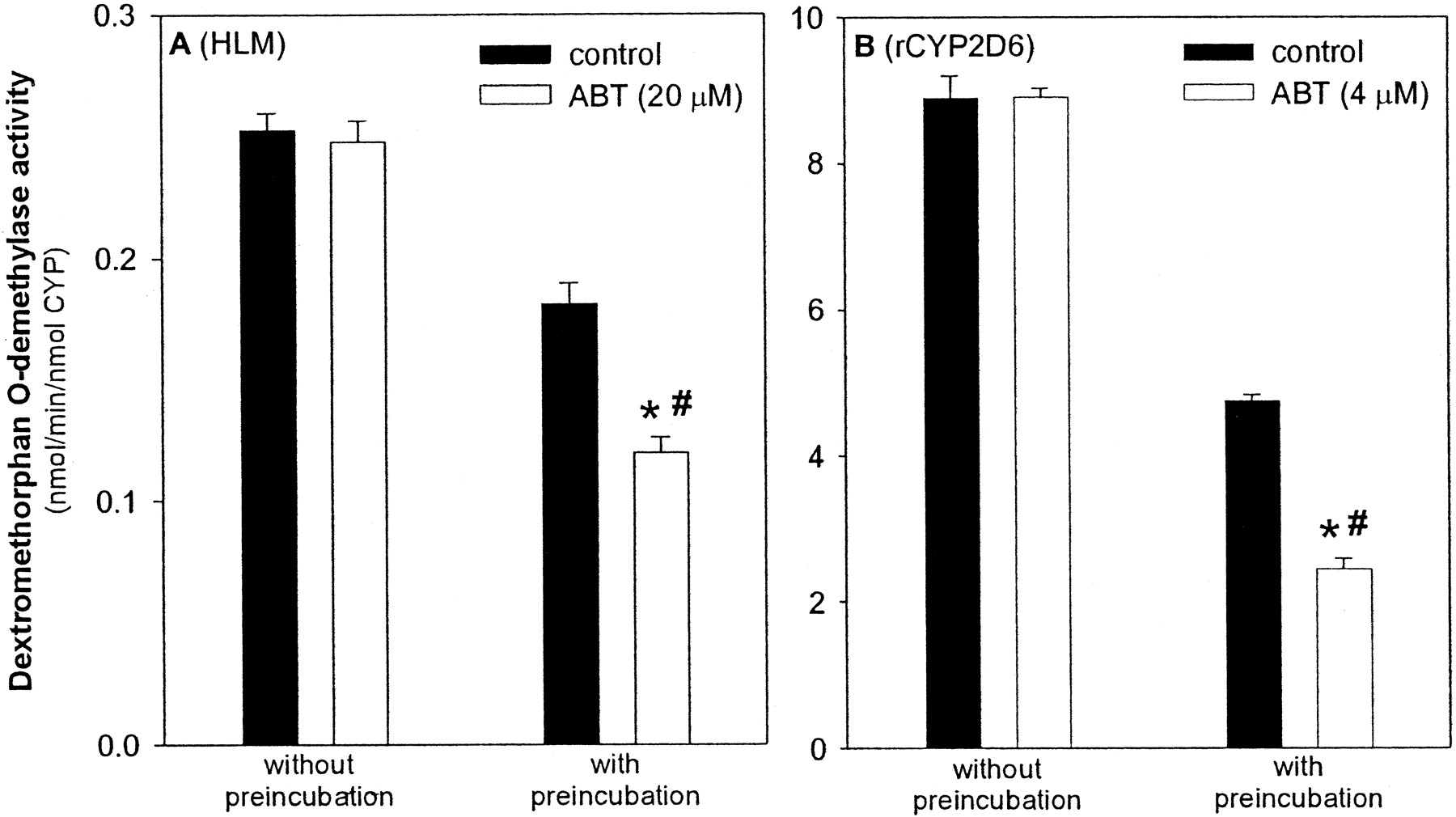
Preincubation of pooled human liver microsomes and recombinant CYP2D6 with ABT.
A, pooled human liver microsomes (2 mg of protein/ml) were preincubated with ABT (20 μM) or an equal volume of buffer (control), and NADPH (1 mM) for 30 min. At this time, a 50-μl aliquot was removed and added to a reaction tube containing DEX and NADPH and incubated for another 20 min. The final concentration of DEX in the incubation reaction was 5 μM. B, recombinant CYP2D6 (10 pmol/ml) was preincubated with ABT (4 μM) or an equal volume of buffer (control), and NADPH (1 mM) for 20 min. A 50-μl aliquot was then removed and added to a reaction tube containing DEX and NADPH, and incubated for another 15 min. The final concentration of DEX in the incubation reaction was 20 μM. All data shown are the mean of four measurements ± S.D. ⋆, significantly different (p < 0.05) from control group preincubated for the same period of time. #, there was a significant interaction (p < 0.05) between preincubation time and ABT treatment.
To further characterize the effect of cimetidine on CYP2D6, experiments were carried out with the recombinant enzyme. A similar decline in activity was observed when recombinant CYP2D6 was preincubated with NADPH alone. However, cimetidine appeared to have an inhibitory effect, which was apparent even in the presence of a saturating concentration of dextromethorphan (20 μM) in the incubation. After 20 min of preincubation with NADPH, 100 μM cimetidine resulted in a modest 18% (95% confidence interval, 13–22%) decrease in activity, whereas in the absence of preincubation, no inhibition was observed (Fig. 4A). Furthermore, as determined by the two-way analysis of variance, there was a significant interaction between treatment with cimetidine and preincubation time, indicating that the observed effect was greater than would be expected due to either of these factors alone. The addition of CAT, SOD, and EDTA to the preincubation reaction had a stabilizing effect on the recombinant enzyme, but not to the same extent that was seen with human liver microsomes. After 20 min of preincubation under these conditions, the loss in control activity was decreased from 48% to 36%. Longer preincubation times resulted in continued loss in enzyme activity. In the presence of CAT, SOD, and EDTA, preincubation with 100 μM cimetidine resulted in a 31% (95% confidence interval, 25–36%) inhibition of enzyme activity (Fig. 4B), again with a significant interaction found between treatment with cimetidine and preincubation time.
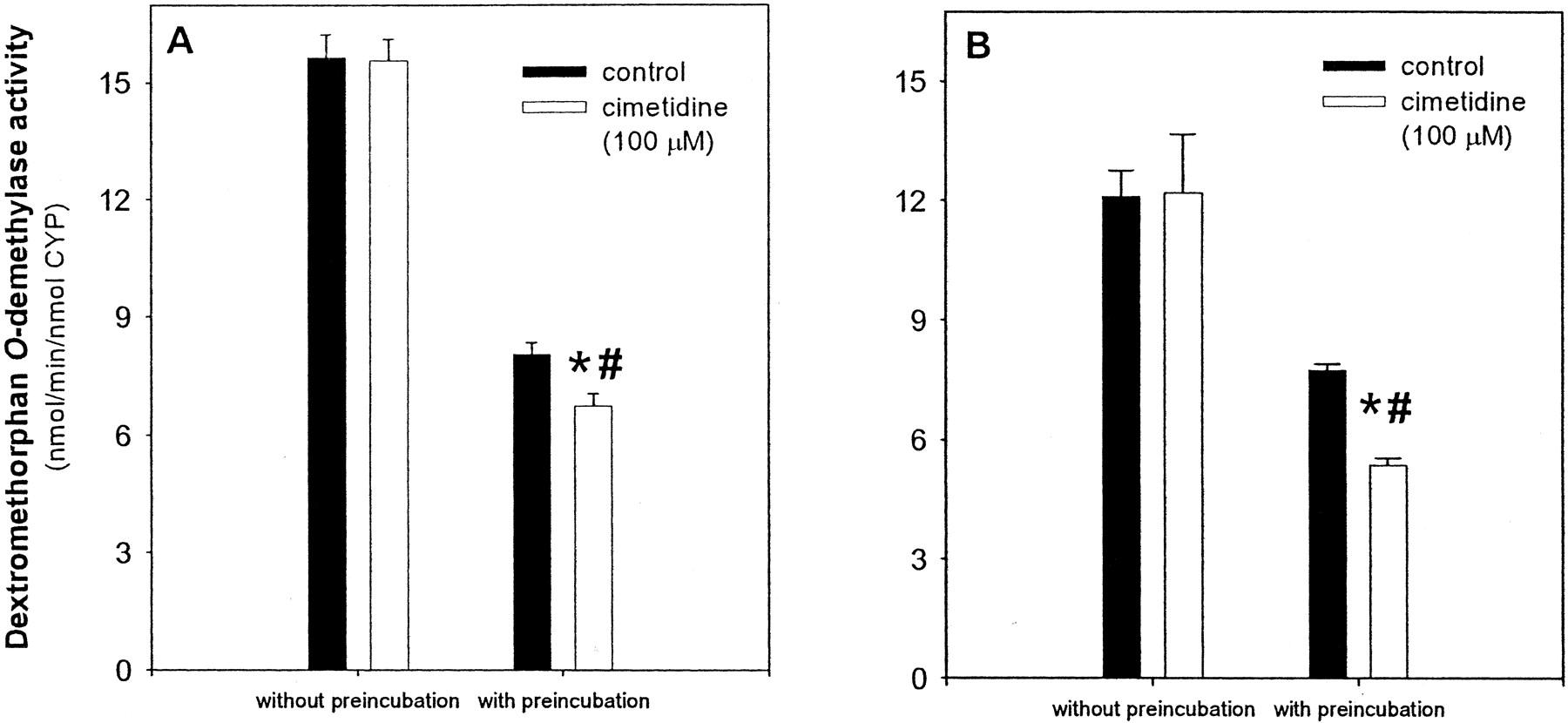
Preincubation of recombinant CYP2D6 with cimetidine.
Recombinant CYP2D6 (10 pmol/ml) was preincubated with 100 μM cimetidine and NADPH (1 mM), or NADPH alone (control), for 20 min (panel A). In panel B, CAT (2400 U/ml), SOD (2400 U/ml), and EDTA (1 mM) were added to the preincubation mixture containing cimetidine and NADPH. A 50-μl aliquot was then removed and added to a reaction tube containing DEX and NADPH (1 mM), and samples were incubated for another 15 min. In the final incubation step, the concentration of recombinant CYP2D6 was 2.5 pmol/ml and the concentration of DEX was 20 μM. Data shown are the mean of four (A) or three (B) separate experiments ± S.D. ⋆, significantly different from preincubated control (p < 0.05). #, significant interaction (p < 0.05) between preincubation time and treatment with cimetidine.
To further characterize the inactivation effects of cimetidine, a number of other properties of mechanism-based inactivation were demonstrated. Inactivation was found to be dependent on both the presence of NADPH and preincubation time, suggesting that the inactivating species required catalytic activation (Fig. 5). If the inactivator species is indeed a product of CYP2D6-mediated catalysis, inhibition of this enzyme should block the inactivation of the enzyme. By including quinidine, a potent inhibitor of CYP2D6 (Newton et al., 1995), in the preincubation reaction, the degree of inactivation by cimetidine could be attenuated. At a concentration equivalent to half the Ki for competitive inhibition of dextromethorphan O-demethylase (0.6 nM), quinidine reduced the magnitude of inactivation by cimetidine from 17% to 10%, whereas at higher concentrations (1.3 and 2.6 nM), the effect of preincubation was completely blocked by quinidine (Fig. 6).

Time- and NADPH-dependent inactivation of CYP2D6 by cimetidine.
Recombinant CYP2D6 (10 pmol/ml) was preincubated with cimetidine (100 μM) and NADPH (1 mM), or NADPH (1 mM) alone. Aliquots (50 μl) were removed at 5, 10, and 20 min following initiation of the preincubation reaction and were added to reaction tubes containing DEX (20 μM) and NADPH (1 mM), and reactions (200 μl total volume) were incubated for another 15 min. A second set of samples was preincubated with or without cimetidine in the absence of NADPH. Each point represents the mean of three measurements ± S.D. ⋆, significantly different from the groups preincubated with NADPH alone for the same period of time. #, significantly different from the groups preincubated with NADPH and cimetidine for 0, 5, and 10 min.

Effect of quinidine on inactivation of recombinant CYP2D6 by cimetidine.
Recombinant CYP2D6 (10 pmol/ml) was preincubated with cimetidine (100 μM) and varying concentrations of quinidine (0–2.6 nM) and NADPH (1 mM) for 0 or 20 min. A 50-μl aliquot was removed and added to a reaction tube containing DEX and NADPH in a volume of 150 μl and was incubated for another 15 min. The concentration of DEX in the final incubation was 20 μM. Activity in each group was normalized to the activity of the control group, which was preincubated with NADPH (1 mM) for 0 or 20 min in the absence of both cimetidine and quinidine. Data are expressed as the mean of three experiments ± S.D. ⋆, significantly less (p < 0.05) when compared with the group containing the same concentration of quinidine that was not preincubated. There was no difference in the activity of the groups that were not preincubated.
The kinact and KI were estimated from a Kitz-Wilson plot to be 0.03 min-1 and 77 μM, respectively (Fig. 7). Based on this kinact value, the time required to inactivate 50% of the recombinant enzyme molecules under these experimental conditions was approximately 25 min.

Estimation of inactivation constants of cimetidine.
Recombinant CYP2D6 (10 pmol/ml) was preincubated with buffer (control) or various concentrations of cimetidine, and NADPH (1 mM) for 0, 5, 10, and 20 min. Aliquots (50 μl) of the preincubation mixture were then added to reaction tubes containing DEX (20 μM) and NADPH (1 mM) and were incubated for another 15 min. A, plot of log percentage of control activity versus preincubation time. Each point represents the mean of two separate experiments in which measurements were made in triplicate. The concentration of cimetidine reported is that in the preincubation reaction. B, Kitz-Wilson plot of half-life of inactivation versus inverse concentration of cimetidine.
Discussion
The present study has shown that cimetidine is a weak, reversible inhibitor of CYP2D6 in vitro, a finding consistent with reports from other laboratories (Martinez et al., 1999; Furuta et al., 2001). Additionally, evidence presented here suggests that cimetidine acts as a mechanism-based inactivator of CYP2D6. A 31% decrease in recombinant CYP2D6 activity was observed following preincubation with cimetidine, NADPH, and antioxidants. This loss in activity was time-dependent and was consistent with the catalysis-dependent formation of a reactive metabolite or metabolite-intermediate species, as previously demonstrated in animal studies with CYP2C11 and CYP2C6 (Chang et al., 1992b; Levine and Bellward, 1995; Levine et al., 1998). Furthermore, inactivation of recombinant CYP2D6 was attenuated by quinidine. As a competitive inhibitor of CYP2D6, quinidine would be able to reduce the formation of the putative inactivating species in a concentration-dependent manner if CYP2D6-mediated metabolism is involved in the inactivation process.
The rate of turnover of cimetidine to the putative inactivating species by recombinant CYP2D6 was slow (t1/2 inact = 25 min) relative to the loss in activity with preincubation time. This apparent enzyme instability may have been caused by NADPH-induced lipid peroxidation, which has been shown to result in P450 dysfunction (Levin et al., 1973; Renton et al., 1976).
In the human liver microsomal studies, there was a significant decrease in enzyme activity in all groups due to NADPH. Even after 2 h of preincubation, no cimetidine-induced inactivation occurred. In a control experiment, mechanism-based inactivation of microsomal dextromethorphan O-demethylase activity was observed with ABT, indicating that the lack of inactivation of microsomal CYP2D6 by cimetidine was not due to the preincubation methods. If the rate of turnover of cimetidine in human liver microsomes is slow, then this, along with the decline in activity, may explain why no inactivation was observed in the microsomal system.
To allow more time for a possible metabolite-intermediate to be generated in human liver microsomes, we performed preincubation experiments in the presence of CAT and SOD, and EDTA. Under these conditions, either with the addition of NADPH or an NADPH regenerating system, we were able to maintain measurable enzyme activity for up to 24 h. However, cimetidine significantly attenuated the loss of enzyme activity with time, and thus, enzyme inactivation could not be demonstrated. This ability of cimetidine to attenuate the loss in activity is consistent with cimetidine acting as an antioxidant, which has been reported elsewhere (Lambat et al., 2002; Liu et al., 2002).
The present data indicate that in the recombinant system, cimetidine does generate mechanism-based inactivation of CYP2D6, but this could not be demonstrated in human liver microsomes. It is unlikely that the inactivation of recombinant CYP2D6 was an artifact, since there is considerable evidence from clinical studies supporting the role of cimetidine as an inactivator of CYP2D6 (Miller and Macklin, 1983; Miller et al., 1983; Abernethy et al., 1984; Amsterdam et al., 1984; Philip et al., 1989; Schellens et al., 1989; Arnold et al., 1997). The most direct evidence is provided by the effect of cimetidine on the metabolism of sparteine and desipramine in vivo.
Sparteine has been frequently used as a probe for CYP2D6, which is thought to be responsible for the major metabolic pathway in vivo. The urinary metabolic ratio of sparteine to its dehydroxy metabolites correlates well with the debrisoquine metabolic ratio (Inaba et al., 1983). Also, the main metabolite, 5-dehydroxysparteine, is undetectable in the plasma of poor metabolizers of sparteine (Eichelbaum et al., 1979). In a study designed to determine the effect of enzyme inhibition on the metabolism of several drugs, cimetidine reduced the formation clearance of 5-dehydroxysparteine in extensive metabolizers by 37% (Schellens et al., 1989), indicating that cimetidine had a large effect on the CYP2D6-mediated metabolism of sparteine. In poor metabolizers, plasma metabolite levels were below the level of detection, so that the effect of cimetidine on these individuals could not be assessed.
Like sparteine, the clearance of desipramine is dependent on CYP2D6 activity. In individuals phenotyped with debrisoquine, the formation clearance of 2-hydroxydesipramine was reduced by more than 98% in poor metabolizers compared with extensive metabolizers, indicating that CYP2D6 is the major contributor to the formation of this metabolite in humans (Spina et al., 1987). The effect of cimetidine on poor and extensive metabolizers was investigated by Steiner and Spina (1987). Cimetidine inhibited the formation clearance of 2-hydroxydesipramine by 47% in extensive metabolizers, an observation consistent with a large inhibitory effect of cimetidine on CYP2D6 activity. In poor metabolizers, desipramine's clearance was unaffected. Similarly, Abernethy et al. (1984) investigated the effect of cimetidine on imipramine and desipramine, its N-demethylated metabolite, and found that the clearance of both imipramine and desipramine was reduced. In this case, cimetidine appeared to be inhibiting CYP2D6 as well as other P450 enzymes. The formation of 2-hydroxydesipramine from desipramine is primarily mediated by CYP2D6, whereas CYP2C19 appears to be responsible for the conversion of imipramine to desipramine (Koyama et al., 1997).
It is important to note that in the cases described above, the administration of cimetidine began a number of days before that of the studied drug and was continued throughout the sampling period. As well, in the study by Reimann et al. (1981), the inhibition of propranolol clearance was significantly greater after 5 days of cimetidine treatment (51%) than after 1 day of treatment (33%). The long treatment period associated with inhibition in vivo is consistent with the gradual formation of a metabolite-intermediate species in patients treated with cimetidine.
In summary, initial evidence that cimetidine is a mechanism-based inactivator of recombinant CYP2D6 is presented. The results suggest that the rate of metabolic transformation of cimetidine to the putative inactivating species is slow. We were unable to achieve the appropriate in vitro conditions to observe this inactivation in human liver microsomes, despite strong evidence that cimetidine is an inhibitor in vivo. Cimetidine may represent a class of compounds capable of inactivating specific cytochromes P450 in vivo, but for which conditions may not be achievable in vitro using human liver microsomes. It may be possible in the future to use the humanized mouse model to address some of these questions (Corchero et al., 2001). However, during drug development, cytochrome P450 inhibition studies will still likely need to be performed in humans to avoid incorrect conclusions.
Footnotes
-
↵1 Abbreviations used are: P450, cytochrome P450; ABT, 1-aminobenzotriazole; CAT, catalase; DEX, dextromethorphan; HPLC, high-performance liquid chromatography; SOD, superoxide dismutase.
-
This research was supported by the Canadian Institutes of Health Research (CIHR) Grant MOP-37974 to G.D.B. M.M. received a University Graduate Fellowship from the University of British Columbia and a Merck Research Laboratory Traineeship in Drug Metabolism and Pharmacokinetics. T.K.H.C. received a Research Career Award in the Health Sciences from CIHR and the Rx&D Health Research Foundation.
- Received June 30, 2003.
- Accepted December 29, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}