


Abstract
A new glutathione adduct (M4) was tentatively identified, likely as 2′-hydroxy-3′-(glutathione-S-yl)-monoclofenac, using liquid chromatography-tandem mass spectrometry analysis of incubations of diclofenac with human liver microsomes. The same conjugate was not detected in incubations with either rat or monkey liver microsomes. Formation of M4 was mediated specifically by CYP2C9 in human liver microsomes, as evidenced by the following observations: 1) cDNA-expressed CYP2C9-catalyzing formation of M4; 2) inhibition of M4 formation by sulfaphenazole, a CYP2C9-selective inhibitor; and 3) strong correlation between the production of M4 and CYP2C9-mediated tolbutamide 4-hydroxylase activities in a panel of human liver microsome samples. Formation of M4 suggests the existence of a new reactive intermediate as diclofenac-2′,3′-oxide. A tentative pathway states that diclofenac is oxidized to diclofenac-2′,3′-oxide that reacts with glutathione (GSH) to form a thioether conjugate at the C-3′ position, followed by a concomitant loss of chlorine to give rise to M4. Furthermore, a likely mechanism leading to the formation of diclofenac oxides is rationalized: CYP2C9-catalyzed oxidation at the C-3′ position of the dichlorophenyl ring to form a cationic σ-complex that subsequently results in diclofenac-3′,4′-oxide and diclofenac-2′,3′-oxide; the former oxide is converted to 4′-hydroxy-diclofenac as a major metabolite and can be trapped by GSH to produce 4′-hydroxy-3′-glutathione-S-yl diclofenac (M2), whereas the latter oxide forms 3′-hydroxy-diclofenac and can be trapped by GSH to produce M4. This mechanism is consistent with the structural modeling of the CYP2C9-diclofenac complex, which reveals that both the C-3′ and C-4′ of the dichlorophenyl ring are proximate to the heme group.
Diclofenac is a nonsteroidal anti-inflammatory drug that is widely prescribed for the treatment of osteoarthritis, rheumatoid arthritis, ankylosing spondylitis, and acute muscle pain conditions (Small, 1989). Presumably, the pharmacological efficacy of diclofenac is produced from inhibition of cyclooxygenases. Treatment with diclofenac has been associated with a rare, but severe, incidence of hepatic injury (Banks et al., 1995; Boelsterli, 2003), which is often described as idiosyncratic toxicity. Although the exact mechanism of diclofenac hepatotoxicity is not clearly understood, it has been proposed that metabolic activation of the drug and subsequent covalent modifications of host proteins by reactive metabolites may play an important role, either by directly impairing cellular signal transduction cascades or indirectly eliciting an immune response, since modified proteins can be recognized as foreign antigens by host cells (Boelsterli, 2003).
As shown in Scheme 1, diclofenac contains a dichlorophenyl ring and a phenyl acetic acid moiety. In humans, metabolism of diclofenac is mediated by both glucuronidation and oxidative biotransformation (Tang, 2003). Glucuronidation of the carboxylic acid group is catalyzed by uridine diphosphate glucuronosyltransferase 2B7 (King et al., 2001); formed diclofenac-1-O-acyl glucuronide has been shown to be chemically reactive and covalently binds to cellular proteins via glycation and/or transacylation (Kretz-Rommel and Boelsterli, 1994a,b). Oxidation of the aromatic rings of diclofenac is mediated by cytochromes P450 (P450s). As seen in Scheme 1, hydroxylation of the dichlorophenyl ring is catalyzed specifically by CYP2C9 to produce 4′-hydroxydiclofenac as the major metabolite (Leemann et al., 1993). At higher drug concentrations, CYP3A4-mediated oxidation of the phenyl acetic acid moiety results in 5-hydroxydiclofenac (Shen et al., 1997b). In rat bile, after dosing with diclofenac, three glutathione adducts have been previously identified (Tang et al., 1999a) as 5-hydroxy-4-glutathione-S-yl diclofenac (M1), 4′-hydroxy-3′-glutathione-S-yl diclofenac (M2), and 5-hydroxy-6-glutathione-S-yl diclofenac (M3). Subsequent in vitro studies have shown that both 4′-hydroxydiclofenac and 5-hydroxydiclofenac could be further oxidized to benzoquinone imine intermediates that were trapped as GSH adducts M1 to M3, respectively (Tang et al., 1999a). These observations have led to a two-step bioactivation mechanism: 1) diclofenac is first converted to 4′-hydroxydiclofenac and 5-hydroxydiclofenac via direct hydroxylation; and 2) these two metabolites are further oxidized to form benzoquinone imine intermediates by CYP2C9 and CYP3A4, respectively (Scheme 1) (Tang et al., 1999a,b). These findings are significant as the first line of evidence to suggest that P450-mediated reactive metabolites may play an important role in toxicity of the drug.
Although several protein adducts have been detected in rats treated with diclofenac (Shen et al., 1997a; Jones et al., 2003), the relationship between covalent modifications and diclofenac hepatotoxicity has not been clearly established (Tang, 2003). Characterization of reactive metabolites would be very helpful for understanding biochemical mechanisms of idiosyncratic toxicity associated with diclofenac. In this study, we report a novel reactive metabolite in the form of a glutathione adduct in NADPH-fortified human liver microsomes incubated with diclofenac; in addition, it was found that formation of the reactive metabolite was specifically mediated by CYP2C9. These data are important for further understanding the relationship between metabolic activation of diclofenac and the hepatotoxicity of the drug observed in clinic.

Structure of diclofenac and proposed oxidative pathways.
Materials and Methods
Materials. Reagents and solvents used in the current study were of the highest possible grade available. The following chemicals were purchased from Sigma-Aldrich (St. Louis, MO): acetaminophen, 1-aminobenzotriazole, dextromethorphan, dextrorphan, diclofenac, glutathione, 4′-hydroxy-diclofenac, ketoconazole, α-naphthoflavone, phenacetin, testosterone, 6β-hydroxy-testosterone, tolbutamide, sulfaphenazole, tranylcypromine, quinidine, trichloroacetic acid, β-nicotinamide adenine dinucleotide phosphate (NADP+), glucose 6-phosphate, and glucose-6-phosphate dehydrogenase. All microsomes, including rat and monkey liver microsomes, Supersomes containing cDNA-expressed P450s, and pooled and individually prepared human liver microsomes, were obtained from BD Gentest (Woburn, MA). (S)-Mephenytoin, 4′-hydroxy-mephenytoin, and 4-hydroxy-tolbutamide were all supplied by Ultrafine Chemicals (Manchester, UK). Stable isotope-labeled glutathione (GSX; γ-glutamyl-cystein-glycin-13C2-15N) was obtained from Cambridge Isotope Laboratories (Andover, MA), and isotopic purity was 94% estimated by the supplier using NMR. Acetonitrile and methanol were obtained from EMD Chemicals (Gibbstown, NJ).
Instruments. MS analyses were performed on a Micromass Quattro Micro triple quadrupole mass spectrometer (Waters, Milford, MA) that was interfaced to an Agilent 1100 HPLC system (Agilent Technologies, Palo Alto, CA). The electrospray ionization ion source was operated in the positive ion mode, and experimental parameters were set as follows: capillary voltage 3.2 kV, source temperature 120°C, desolvation temperature 300°C, sample cone voltage 26 V. Data were processed using Masslynx version 4.0 software (Waters). The NMR spectrum of the diclofenac derivative was obtained on a Bruker 300 NMR spectrometer (Bruker, Newark, DE), and chemical shifts were expressed relative to tetramethylsilane.
Microsomal Incubations. All incubations were performed at 37°C in a water bath. Diclofenac or its derivative was individually mixed with human, monkey, or rat liver microsomal protein in 50 mM potassium phosphate buffer (pH 7.4) supplemented with glutathione. After a 5-min preincubation at 37°C, reactions were initiated by the addition of a NADPH-generating system to give a final volume of 1.0 ml. The final reaction mixture contained 10 μM drug compound, 1 mg/ml microsomal protein, 1 mM GSH or GSX, 1.3 mM NADP+, 3.3 mM glucose 6-phosphate, 0.4 U/ml glucose-6-phosphate dehydrogenase, and 3.3 mM magnesium chloride. After a 60-min incubation, reactions were terminated by the addition of 150 μl of trichloroacetic acid (10%). Incubations with recombinant P450 enzymes were performed similarly, except that liver microsomes were substituted by Supersomes. Samples were centrifuged at 10,000g for 15 min at 4°C to pellet the precipitated protein, and supernatants were subjected to LC-MS/MS for direct analysis of GSH adducts. For the neutral loss MS/MS scanning of GSH adducts, supernatants were concentrated by solid phase extraction as described below, prior to LC-MS/MS analyses.
Inhibition of P450s by Chemical Inhibitors. The effect of specific inhibitors of individual P450 enzymes on the formation of reactive metabolites was investigated using pooled human liver microsomes. Reaction mixtures contained diclofenac, individual P450-specific inhibitors, GSH, and microsomal proteins. Reactions were initiated by the addition of the NADPH-regenerating solution. The final concentration of microsomal protein was 1 mg/ml. Incubations proceeded for 20 min, and reactions were terminated by the addition of trichloroacetic acid. P450-selective inhibitors, α-naphthoflavone (1 μM), sulfaphenazole (5 μM), tranylcypromine (12.5 μM), quinidine (2 μM) and ketoconazole (1 μM), were used to investigate the involvement of CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, respectively. In the control, no chemical inhibitors were included in incubations. Duplicate incubations were performed for each P450 inhibitor.
The effectiveness of individual P450 inhibitors was evaluated using pooled human liver microsomes incubated with P450 marker substrates, as previously described (Yan et al., 2002). Compounds used as P450 marker substrates were 10 μM phenacetin (CYP1A2), 100 μM (S)-mephenytoin (CYP2C19), 500 μM tolbutamide (CYP2C9), 80 μM dextromethorphan (CYP2D6), and 150 μM testosterone (CYP3A4). Individual marker substrates were preincubated with human liver microsomes in the presence and absence of P450-selective inhibitors for 5 min at 37°C prior to the addition of a NADPH-generating solution. After incubation for 20 min, reactions were terminated, and metabolites generated from individual P450 marker substrates were analyzed by the LC-MS/MS method (Yan et al., 2002). A comparison was made relative to the controls (no inhibitors), and activity was expressed as the percentage of control activity remaining.
Correlation with Marker Substrate Activities. A panel of 12 human livers was used to assess the formation of reactive metabolites of diclofenac. Microsomes prepared from these livers had been previously characterized for individual P450 marker activities by the supplier (BD Gentest). Marker substrates used to determine specific P450 activities were phenacetin (CYP1A2), coumarin (CYP2A6), (S)-mephenytoin (CYP2C19 and CYP2B6), paclitaxel (CYP2C8), diclofenac (CYP2C9), bufuralol (CYP2D6), chlorzoxazone (CYP2E1), testosterone (CYP3A4), and lauric acid (CYP4A). Because diclofenac is the subject of this study, CYP2C9 activity was internally reevaluated using tolbutamide as a marker substrate, and a good correlation (r = 0.92) was found between 4-hydroxy-tolbutamide activity and 4′-hydroxy-diclofenac activity among the panel of 12 liver microsomal preparations. A correlation study was conducted between the formation of reactive metabolites and enzymatic activities of specific P450 isozymes across the entire human liver microsome panel, under the same incubation conditions.
Synthesis of 2′-Hydroxymonoclofenac. A total of 200 mg of diclofenac was dissolved in 4 ml of NaOH (3 M), and supplemented with 2 ml of dimethylformamide. The diclofenac solution was transferred to a 10-ml CEM reaction vial (CEM Corp., Matthews, NC) containing a magnetic stirring bar. The reaction vial was properly sealed and degassed under vacuum, and then subjected to microwave irradiation for 60 min on a CEM Discovery Microwave Apparatus operated at 200°C and a power level of 150 W. The crude reaction mixture was concentrated by lyophilizing, and the residue was separated by flash chromatography on reverse phase silica gel (gradient elution, H2O/CH3CN from 1:0 to 1:1) to provide 2-hydroxymonclofenac. After lyophilizing, 50 mg of the product was obtained as a white solid. High resolution MS (MH+): m/z 278.0571 (calculated: 278.0584), measured by a Micromass LCT spectrometer. 1H NMR (300 MHz, CDCl3): δ 7.56–7.50 (m, 2H), 7.44–7.34 (m, 2H), 7.23(t, J = 7.8 Hz, 1H), 7.12 (t, J = 7.5 Hz, 1H), 6.42 (d, J = 7.7 Hz, 1H), 3.80 (s, 2H).
Solid-Phase Extractions. Samples resulting from incubations were desalted and concentrated by solid-phase extraction, prior to neutral loss scan MS/MS analyses. Solid-phase extraction was performed using SEP-PAK cartridges packed with 100 mg of sorbent C18 (Waters). Cartridges were first conditioned with 5 ml of methanol and then flushed with 5 ml of water. Samples resulting from centrifugation were loaded into cartridges, and cartridges were washed with 5 ml of water to remove salts and proteins. Components of interest were eluted from cartridges with 1 ml of methanol. Eluted components were dried on a SpeedVac dryer (Thermo Electron, Waltham, MA). The dried samples were resuspended in 150 μl of water/acetonitrile (95:5) and were then subjected to neutral loss scan MS/MS analyses.
LC-MS/MS Analyses. For complete profiling of reactive metabolites, samples were first subjected to chromatographic separations with an Agilent 1100 HPLC system integrated with an autosampler, and eluents were introduced to the Quattro Micro triple quadrupole mass spectrometer that was operated in the neutral loss scanning mode as described above. An Agilent Zorbax SB C18 column (2.1 × 150 mm) was used for chromatographic separations. The starting mobile phase consisted of 95% water (0.5% acetic acid), and the metabolites were eluted using a single gradient of 95% water to 85% acetonitrile over 25 min at a flow rate of 0.3 ml/min. At 25 min, the column was flushed with 85% acetonitrile for 2 min before re-equilibration at initial conditions. LC-MS/MS analyses were carried out on 10-μl aliquots of cleaned samples. Mass spectra collected in the neutral loss scanning mode were obtained by scanning over the range m/z 400 to 700 in 2 s. After a positive peak was detected, a collision-induced dissociation (CID) MS/MS spectrum was subsequently obtained to further elucidate the structure of the glutathione conjugate.
For relative comparison of GSH adduct levels, the mass spectrometer was operated in the multiple reaction monitoring mode. Three transitions were simultaneously monitored in detecting M1 to M3: m/z 617→542, 617→488, and 617→342, and transitions used for detecting M4 were m/z 583→508, 583→454, and 583→308.
Molecular Modeling. Superimposition of the structures of CYP2C9-warfarin complex (Protein Data Bank code 1OG5) (Williams et al., 2003) and CYP2C5-diclofenac complex (Protein Data Bank code 1NR6) (Wester et al., 2003) revealed that the heme active sites for CYP2C5 and CYP2C9 are nearly identical, except for a few residues in the CYP2C9-warfarin binding site, which are specific for CYP2C9. We assumed that diclofenac binds to CYP2C9 in a conformation similar to that of CYP2C5. Accordingly, the CYP2C9-diclofenac complex structure was constructed by a molecular docking procedure using CYP2C5-diclofenac complex crystal structure as a reference: 1OG5 and 1NR6 structures were first overlaid; the bound warfarin was removed from 1NR6 structure to obtain the apo-1NR6 protein structure, diclofenac conformation in 1OG5 was subsequently copied to apo-1NR6 structure as a reference molecule. Then, Schrodinger Glide software (Maestro 6.0; Schrödinger, Portland, OR) was used to dock a free diclofenac to the apo-1NR6 structure guided by the reference 1OG5 diclofenac conformation. The energy of the best-scoring pose was then fully minimized using Schrodinger Maestro with OPLS-AA force field (Kaminski et al., 2001).
Results
Characterization of the GSH Adducts Formed in Microsomal Incubations. Previous clinical studies (Degen et al., 1988; Davies and Anderson, 1997) have indicated that peak concentrations (Cmax) of diclofenac are in the range of 1 to 17 μM in human plasma after a single dose of 100 mg given orally. To be clinically relevant, 10 μM diclofenac was used in all incubations performed in this study.
For the initial screening of GSH adducts, samples generated from incubations with human liver microsomes were desalted and concentrated by solid-phase extractions, and resulting samples were subjected to LC-MS/MS neutral loss scan detecting ions that lost both 129 and 75 Da upon CID (Baillie and Davis, 1993). As shown in Fig. 1, two major components that exhibited response to both neutral losses (129 and 75 Da) were detected at retention times of 13.2 (component I) and 13.6 min (component II), respectively, in incubations of diclofenac with human liver microsomes. The peak area ratio of I to II was approximately 1.0 for the neutral loss scan of 129 Da and 0.7 for the neutral loss scan of 75 Da. Neither I nor II was detected when either diclofenac or the NADPH-regenerating system was omitted from incubations. Additionally, formation of both I and II was abated by preincubation of HLMs with 15 μM 1-aminobenzotriazole for 15 min in the presence of the NADPH-generating system (data not shown). These results suggested that GSH adducts were formed from a reactive metabolite of diclofenac via P450-mediated oxidation.

LC-MS/MS detection of GSH adducts derived from diclofenac by constant scanning for neutral losses of 129 Da (a) and 75 Da (b).
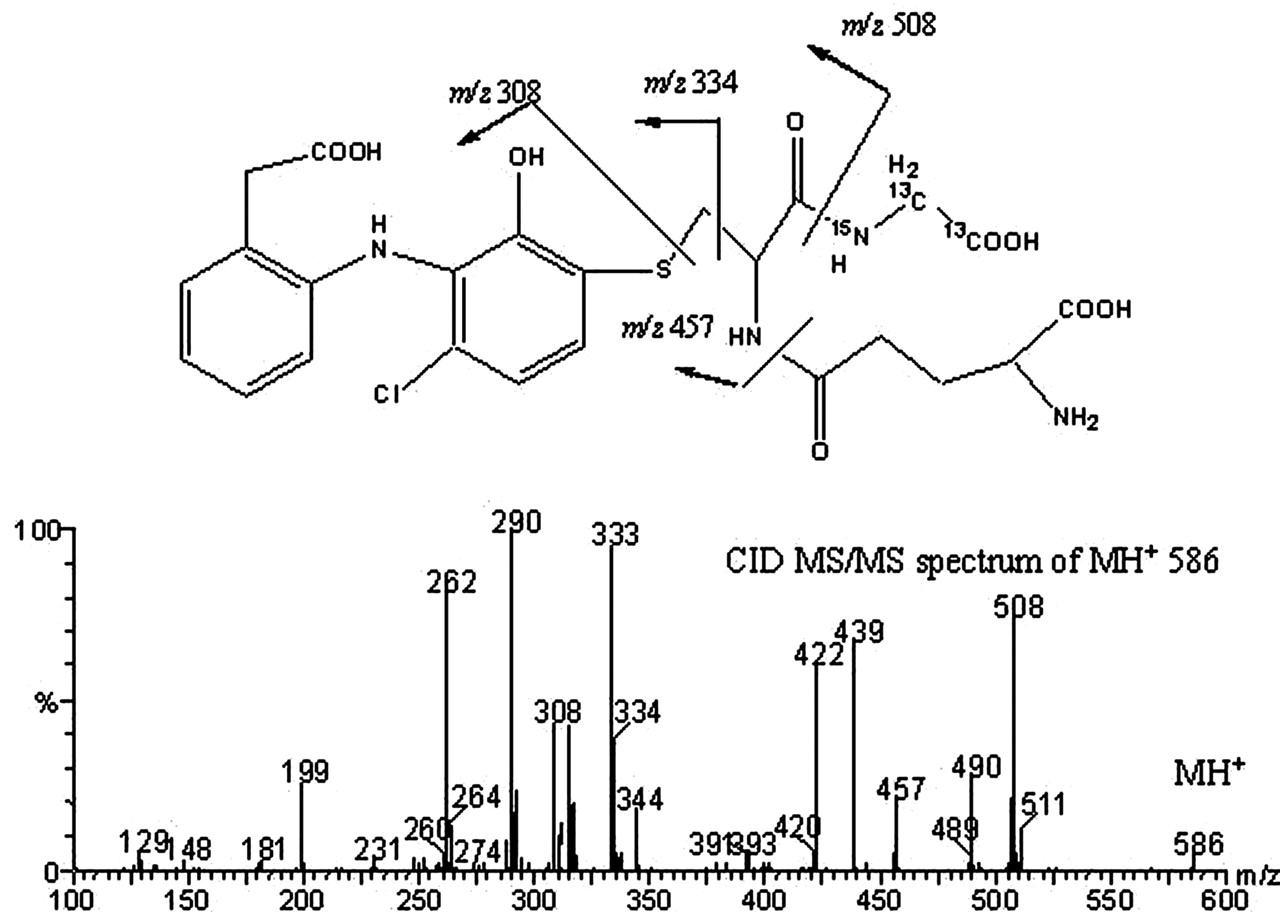
The parent ion of component I at retention time 13.3 min was at m/z 583 (Fig. 2, a and b). A chlorine isotope peak was observed at m/z 585 (∼37% of the M + 1 ion), which clearly indicated the formation of a new GSH adduct that contained only one chlorine atom. Subsequent CID MS/MS spectrum of the MH+ at m/z 583 gave product ions at m/z 508 and 454, resulting from neutral losses of glycine (75 Da) and γ-glutamate (129 Da), respectively, which confirmed the presence of a GSH moiety in the metabolite (Fig. 3). A GSH adduct, either 2′-hydroxy-3′-(glutathione-S-yl)-monoclofenac or 3′-hydroxy-2′-(glutathione-S-yl)-monoclofenac, was tentatively proposed, which is consistent with the chlorine isotope cluster and cleavage of CID product ions. The plausible fragmentation pathways are shown in Scheme 2. The molecular ions of the GSH adduct underwent a loss of water to give rise to ions at m/z 565; double cleavages at the α-carbon of glycine formed product ions at m/z 334, and cleavage adjacent to the thioether moiety led to the product ion at m/z 308 that subsequently lost a water to form ions at m/z 290; a further loss of CO from ions at m/z 290 gave rise to m/z 262. Product ions at m/z 508 and at m/z 454 resulting from the neutral losses underwent a further loss of water to form ions at m/z 490 and m/z 436, respectively; a loss of NH3 from ions at m/z 436 resulted in product ions at m/z 419. The newly identified GSH adduct was assigned as M4 in this study, following M1 to M3 previously identified by others (Tang et al., 1999a).

CID-MS/MS spectra of component I (M4). a, MS/MS spectrum of neutral loss of 129 Da; b, MS/MS spectrum of neutral loss of 75 Da.

CID-MS/MS spectra of component I (M4) at m/z 583.

CID fragmentation pathways of the GSH adduct M4.
The parent ion associated with component II at retention time 13.6 min was at m/z 617 with a chlorine isotope peak at m/z 619 (∼75% of the M + 1 ion) (S1), suggesting that this component was one of the three GSH adducts previously identified (Tang et al., 1999a). This was confirmed by the CID spectrum of the MH+ ions at m/z 617 that showed product ions at m/z 599, 542, 488, 470, 342, and 324. Component II was subsequently identified as M2 by comparing the retention time of the GSH adduct formed by 4′-hydroxydiclofenac incubated with CYP2C9 (Tang et al., 1999b).
Stable Isotope Labeling of GSH Adducts in Microsomal Incubations. To further verify the identity of GSH adducts, GSX, a triply labeled glutathione (two 13C and one 15N at glycine), was used to trap reactive metabolites generated in human liver microsomal incubations (Yan and Caldwell, 2004). Neutral loss scanning MS analyses of samples generated from HLM incubations revealed a mass shift of 3 Da for molecular ions of both M4 (m/z 586) and M2 (m/z 620). Additionally, stable isotope-labeled M4 and M2 displayed chlorine isotope clusters identical to their natural counterparts. The tandem MS spectrum of M4 at m/z 586 is shown in Fig. 4. As expected, product ions at m/z 508, 490, 334, 308, 290, and 262 were detected; new product ions at m/z 457, 439, and 422 appeared, which apparently correspond to product ions at m/z 454, 436, and 419 of unlabeled M4 (Fig. 3).
GSH Conjugate Formation in Human, Monkey, and Rat Liver Microsomes. LC-MS/MS was used to detect GSH adducts M1 to M4 formed in incubations of diclofenac with human, monkey, and rat liver microsomes. Interestingly, M4 was detected only in the incubation with human liver microsomes (Fig. 5). Species difference was also found for M1 to M3. M2 was the most abundant GSH conjugate formed in human liver microsomes; small amounts of M1 were detected in the incubation with monkey liver microsomes; both M1 and M3 were detected in incubations with rat liver microsomes, and the former conjugate is more predominant (S2).
GSH Adduct Formation in Incubations with Recombinant P450s. The formation of the GSH adducts was investigated in microsomes derived from insect cell-expressed recombinant human CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4. As seen in Fig. 6, at the same enzyme concentration (100 pmol/ml), CYP2C9 generated the greatest amount of M4; although CYP2C19 also catalyzed M4 formation, the level of M4 was less than 2% of that formed by CYP2C9. No M4 was detected in incubations with other P450 enzymes including CYP1A2, CYP2C8, CYP2D6, CYP2E1, and CYP3A4.
GSH adduct M2 was also detected in the same incubations. Consistent with observations by others (Tang et al., 1999b), it was found that formation of M2 was specifically mediated by CYP2C9, and no M2 was detected in incubations with other P450s including CYP2C19 (S3). M1 and M3 were primarily derived from CYP3A4-mediated oxidation of the phenyl acetic acid ring and, thus, were not the focus of this study.
Inhibition of P450-Mediated Diclofenac Bioactivation. The inhibitory effect of specific inhibitors of individual P450 enzymes on the formation of M2 and M4 was examined using pooled human liver microsomes. Since the concentration of inhibitors used in the inhibition study varied widely among different laboratories, the effective concentrations were determined in house using pooled human liver microsomes and P450 marker substrates (Yan et al., 2002). The formation of both M2 and M4 was strongly inhibited (>70%) by the CYP2C9-selective inhibitor sulfaphenazole (5 μM) in human liver microsomal incubations (Table 1). However, the inhibitory effect on M2 and M4 formation was minimal (<10%) for other P450-selective inhibitors including α-naphthoflavone (CYP1A2), tranylcypromine (CYP2C19), quinidine (CYP2D6), and ketoconazole (CYP3A4), although significant inhibition (>60%) by these selective inhibitors was observed in the metabolism of corresponding P450 marker substrates (Table 1).
Effect of P450 isoform-selective inhibitors on the formation of M2 and M4 in HLM incubations
Formation of M2 and M4 in a Panel of Human Liver Microsomes. Studies were carried out to investigate the correlation between formation of M2 and M4 and enzymatic activities of individual P450 enzymes (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4). As shown in Table 2, a strong correlation was observed between 4-hydroxy-tolbutamide activity, a CYP2C9 marker activity, and the extent of formation of M2 (r = 0.89, p < 0.005) and M4 (r = 0.84, p < 0.005). Correlations were insignificant between the extent of formation of M2 and M4 and the marker activity of other P450s in the entire panel of human liver microsomes (Table 2).
Correlation between M2 and M4 formation and P450 marker activity in HLMs
Structural Modeling of CYP2C9-Diclofenac Complex. Although a crystal structure of CYP2C9-diclofenac complex has not been reported yet, the recently published structures for CYP2C5-diclofenac complex (Williams et al., 2003) and CYP2C9-warfarin complex (Wester et al., 2003) provide excellent templates for modeling CYP2C9-diclofenac complex structure. CYP2C5 and 2C9 share high sequence identity (77%) and similarity (83%), and also exhibit the same regioselectivity and similar catalytic efficiency for oxidation of diclofenac (Williams et al., 2003). Therefore, it is reasonable to assume that diclofenac binds to CYP2C9 and CYP2C5 in a very similar bonding mode.
Figure 7 depicts the binding mode of diclofenac in the CYP2C9 heme active site. Similar to what is seen in CYP2C5-diclofenac complex, the phenyl acetic acid ring distal from the heme group can form π-π interaction with Phe114 and Phe476. Together with the hydrogen bond network formed by the carboxylate group to Asp293 and Asn107, this π-π interaction is the key interaction to anchor the diclofenac in the active site; in agreement with the mutagenesis experiments (Melet et al., 2003), the structure shows that Phe114 and Phe476 of CYP2C9 are key residues in diclofenac recognition. The dichlorophenyl ring is proximate to the heme group, consistent with the fact that the oxidation of diclofenac by CYP2C9 occurs in this part of the molecule. Most noticeably, the orientation of the dichlorophenyl ring places the 3′-carbon atom closest to the heme iron atom; and the distance between Fe and C-3′ is 4.4 Å, and C-4′ is located 4.7 Å from the heme iron atom. This structure clearly suggests that the active high-valent iron-oxo likely attacks both the C-3′ and C-4′ during the oxidation reaction, since two carbons are close to the oxo atom. Based on the homology modeling, Melet et al. (2003) published a CYP2C9-diclofenac complex structure also revealing that C-3′ in the dichlorophenyl ring is in the closest proximity to the heme iron atom. It is our current belief that the CYP2C9-diclofenac binding mode in Fig. 6 provides a structural rationale for the formation of both M2 and M4.

CID-MS/MS spectra of stable isotope labeled component I (M4) at m/z 586.

M4 formation in liver microsomes derived from human (top), monkey (middle), and rat tissues (bottom). Signal responses are normalized to compare the level of GSH adduct.

Formation of GSH adduct M4 in incubations with recombinant CYP2C9 (a) and CYP2C19 (b), respectively. Signal responses are not normalized to show M4 generated by CYP2C19.
Discussion
Chemically reactive metabolites have been implicated in the biochemical mechanisms of idiosyncratic toxicity of diclofenac, and great efforts have been made to characterize reactive intermediates formed in rats and in vitro incubations (Tang et al., 1999a,b; Poon et al., 2001). However, the proof of reactive metabolites as a causative agent for drug-related hepatotoxicity still remains elusive (Tang, 2003). It is essential to completely understand bioactivation pathways of diclofenac to address its mechanism of toxicity.

The modeling structure of CYP2C9-diclofenac complex. Two aromatic rings of diclofenac are in orange, and the heme iron is in purple. Substitutes of diclofenac are shown in green for Cl and blue for the NH group.
In this study, a new GSH adduct (M4) was detected using LC-MS/MS in incubations of diclofenac with human liver microsomes. Glutathione moiety of M4 was further confirmed by tandem MS spectrum of the stable isotope-labeled conjugate that exhibited a mass shift of 3 Da in neutral loss MS scanning. It was also found that M4 formation is specifically mediated by P450s. Additionally, it is interesting to observe that M4 was formed only in incubations with human liver microsomes. This observation may provide a likely explanation for the fact that M4 was not previously detected in rat bile (Tang et al., 1999a) after dosing with diclofenac.
Similar to that of M2, formation of M4 in human liver microsomes is mediated specifically by CYP2C9. This conclusion is supported by the following observations: 1) recombinant CYP2C9 catalyzed formation of M2 and M4 in incubations; 2) formation of both M2 and M4 was strongly inhibited by sulfaphenazole, a CYP2C9-selective inhibitor; and 3) a strong correlation was observed between M2 and M4 formation and CYP2C9 marker activity in a panel of human liver microsome preparations. CYP2C19, an isoform sharing 88% homology with CYP2C9, also catalyzed the formation of M4, but the conversion rate was significantly lower than that of CYP2C9. Therefore, CYP2C19 is unlikely to play a major role in the bioactivation of diclofenac in human liver microsomes. This argument is also supported by the finding that tranylcypromine, a CYP2C19-selective inhibitor, did not inhibit M4 formation in human liver microsomes but effectively suppressed (S)-mephenytoin 4′-hydroxylation, a CYP2C19 marker activity (Table 1); in addition, M4 formation was not correlated with CYP2C19 marker activity as measured by (S)-mephenytoin 4′-hydroxylation in a panel of human liver microsomes (Table 2).

Proposed bioactivation pathways leading to the formation of M4.
A two-step oxidation mechanism has previously been proposed for the bioactivation of the dichlorophenyl ring, direct oxidation at the C-4′ position resulting in 4′-hydroxydiclofenac, followed by further oxidation to form a benzoquinone imine (Tang et al., 1999a). CYP2C9-mediated formation of the new GSH adduct M4 suggests that an additional metabolism pathway may also be responsible for the bioactivation of diclofenac. Unlike M2, M4 contains one chlorine atom, clearly indicating that P450-mediated epoxidation between the C-2′ and C-3′ position of the dichloroaniline ring is the most reasonable mechanism by which a loss of chlorine could result after conjugation with GSH. Two GSH adducts, 2′-hydroxy-3′-(glutathione-S-yl) monoclofenac and 3′-hydroxy-2′-(glutathione-S-yl) monoclofenac, conform the CID MS/MS spectrum of M4 (Fig. 3). Another isomer, 4′-hydroxy-2′-(glutathione-S-yl) monoclofenac, would also give a similar CID MS/MS spectrum, but formation of this isomer requires P450-mediated oxidation at the C-4′ position and substitution of chlorine with glutathione catalyzed by glutathione-S-transferases. A separate incubation of 4′-hydroxydiclofenac with NADPH-fortified HLMs did not produce M4 (data not shown), which ruled out the involvement of glutathione-S-transferases in the formation of M4. Attempts were made to isolate M4 directly from incubations of diclofenac with CYP2C9 using preparative LC, but failed to obtain enough of the conjugate in the desired purity for NMR analyses, due to the low abundance of GSH adducts and coeluting of M2 and M4 on preparative columns. Efforts to synthesize M4 have currently been hampered by the lack of a feasible chemistry strategy.
Regardless, formation of M4 is indicative of a unique reactive metabolite formed by diclofenac. We rationalized a metabolic pathway, in attempt to explain M4 formation in human liver microsomal incubations. As shown in Scheme 3, diclofenac is first oxidized to diclofenac-2′,3′-oxide that is trapped in incubations as 2′-hydroxy-3′-(glutathione-S-yl)-2′,3′-dihydro-diclofenac; the GSH adduct further undergoes a loss of HCl, resulting in a stable conjugate, 2′-hydroxy-3′-(glutathione-S-yl)-monoclofenac. Alternatively, another isomeric adduct could form by conjugating GSH to diclofenac-2′,3′-oxide at the C-2′ position, resulting in 3′-hydroxy-2′-(glutathione-S-yl)-2′,3′-dihydrodiclofenac, which subsequently lost HCl to form 3′-hydroxy-2′-(glutathione-S-yl)-monoclofenac. However, the C-2′ position of the arene oxide would be somewhat sterically hindered by both chlorine and the aniline ring, and thus, 3′-hydroxy-2′-(glutathione-S-yl)-monoclofenac was not formed. It is our current speculation that M4 detected in this study is 2′-hydroxy-3′-(glutathione-S-yl)-monoclofenac. Obviously, it is highly desirable to further confirm the proposed structure using a synthetic compound.

CYP2C9-mediated oxidation of the dichlorophenyl ring of diclofenac via the proposed epoxidation mechanism (solid arrow) and direct hydroxylation pathway (dashed arrow) (Tang et al., 1999a).
Considering the fact that hydroxylation of diclofenac-2′,3′-oxide would result in 3′-hydroxydiclofenac, this mechanism is in agreement with the following observations: 1) 3′-hydroxydiclofenac was detected in humans after dosing with diclofenac (Stierlin and Faigle, 1979; Stierlin et al., 1979; Sawchuk et al., 1995); 2) similar to that of M2, formation of 3′-hydroxydiclofenac is mediated specifically by CYP2C9 in human liver microsomes (Bort et al., 1999).
Another likely pathway is that CYP2C9 mediates ipso substitution of chlorine to form a phenol metabolite that is further oxidized to a benzoquinone imine. This intermediate also conjugates with GSH to produce M4. To investigate this potential bioactivation mechanism, the proposed phenol metabolite, 2′-hydroxymonoclofenac, was chemically synthesized. However, M4 was not detected in incubations of 2′-hydroxymonoclofenac with either human liver microsomes or recombinant CYP2C9 (data not shown).
P450-mediated epoxidation reactions represent a common biotransformation pathway for aromatic hydroxylation (Guengerich, 2002). Several aromatic compounds such as bromobenzene (Jollow et al., 1974) and lamotrigine (Maggs et al., 2000) are metabolized by P450s via formation of reactive arene oxides. Although the exact mechanism leading to formation of arene oxides has not been completely understood yet, many kinetic and theoretical studies of benzene hydroxylation by P450 enzymes support a model in which a cationic σ-complex (or radical σ-complex) is first produced, prior to the formation of arene oxides (Ortiz de Montellano, 1995; de Visser and Shaik, 2003). Because diclofenac-3′,4′-oxide has previously been proposed to be the precursor molecule of 4′-hydroxydiclofenac (Blum et al., 1996), formation of which was mediated specifically by CYP2C9 (Leemann et al., 1993), we hypothesized that both diclofenac-3′,4′-oxide and diclofenac-2′,3′-oxide are formed via a common intermediate. As depicted in Scheme 4, oxidation at the 3′-C position of diclofenac generates a cationic σ-complex (or radical σ-complex) that subsequently forms two isomeric oxides, diclofenac-3′,4′-oxide and diclofenac-2′,3′-oxide. Because the NH is a strong para-directing group, diclofenac-3′,4′-oxide is converted to 4′-OH-diclofenac as the major metabolite. Also, 3′,4′-oxide conjugates with GSH to form M2. It should be noted that formed 4′-OH-diclofenac can be further oxidized to a benzoquinone imine that can be trapped by GSH as M2 (Tang et al., 1999a). Similarly, diclofenac-2′,3′-oxide forms 3′-OH-diclofenac and can be trapped as M4 in the presence of GSH. This epoxidation mechanism is supported by the structure of CYP2C9-diclofenac complex, which was constructed using crystal structures of CYP2C5-diclofenac complex (Williams et al., 2003) and CYP2C9-warfarin complex (Wester et al., 2003) as our modeling templates. As seen in Figure 6, the C-3′ is proximate to the heme iron and in a favorable position for CYP2C9-mediated oxidation. Because of high homology between CYP2C9 and 2C5, the structure of CYP2C9-diclofenac complex is considered to be highly reliable. In addition, this structure is consistent with modeling results reported by others (Melet et al., 2003).
In the two-step bioactivation model (Tang et al., 1999a), a direct hydroxylation occurs at the C-4′ position to give rise to 4′-hydroxydiclofenac as the major metabolite. This mechanism is also supported by the structure of the CYP2C9-diclofenac complex, which reveals that the C-4′ is close to the heme group. However, the two-step mechanism alone could not elaborate the formation of 3′-OH-diclofenac and M4 in incubations of diclofenac with CYP2C9. The two-step model is primarily substantiated by the finding that CYP2C9 catalyzed oxidation of 4′-OH-diclofenac to form M2 in incubations in the presence of GSH (Tang et al., 1999a). However, conversion of 4′-OH-diclofenac to M2 is not conclusive evidence to suggest that 4′-OH-diclofenac is an obligatory intermediate for the formation of M2 from diclofenac.
In this study, we proposed epoxidation as an additional mechanism leading to the bioactivation of diclofenac, in an attempt to provide a likely explanation for the formation of 3′-OH-diclofenac and 4′-OH-diclofenac, M2, and M4 in the presence of GSH. It is likely that both mechanisms, direct hydroxylation at the C′-4 and epoxidation at the C′2-C3′ (Scheme 4), are responsible for the bioactivation of diclofenac, given the fact that both the C-4′ and C′-3 are proximate to the heme, and 4′-OH-diclofenac is a major metabolite that can be further oxidized to diclofenac-1′,4′-quinone imine.
In conclusion, a novel reactive intermediate was detected in incubations of diclofenac with human liver microsomes. Consistent with formation of the glutathione adduct, a bioactivation pathway leading to the formation of diclofenac-2′,3′-arene oxide is rationalized. The precursor of 2′-hydroxy-3′-(glutathione-S-yl)diclofenac was proposed to be diclofenac-2′,3′-arene oxide, formation of which is specifically mediated by CYP2C9. It is our hypothesis that, in addition to the two-step mechanism proposed by others (Tang et al., 1999a), bioactivation of diclofenac occurs at the C-3′ position, resulting in 3′,4-oxide and 2′,3′-oxide, respectively. Given the fact that arene oxides are known causative agents in the toxicity of many toxicants (Guengerich, 2001), identification of diclofenac arene oxides further substantiates the speculation that oxidative bioactivation may play a significant role in the toxicity of diclofenac. Considering the association of arene oxides with hepatotoxicity (Guengerich, 2001), the present finding is of significance in understanding the toxicology mechanism of diclofenac, and more mechanistic studies are warranted in the future. One would expect that a crystal structure of CYP2C9-diclofenac complex would provide more conclusive information on this subject.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.003095.
-
ABBREVIATIONS: P450, cytochrome P450; GSX, isotope-labeled glutathione; GSH, glutathione; LC, liquid chromatography; HLM, human liver microsome; MS, mass spectrometry; MS/MS, tandem MS; CID, collision-induced dissociation.
- Received November 22, 2004.
- Accepted March 3, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}