


Abstract
Irreversible CYP3A inhibition by drugs constitutes one of the major causes of inhibition-based drug interactions. We evaluated time-dependent inactivation of CYP3A in cryopreserved human hepatocytes for six structurally diverse compounds known to exhibit this property. Inactivation kinetic parameters were also determined using human liver microsomes. Except for diclofenac, which did not cause CYP3A inactivation either in microsomes or in hepatocytes at concentrations up to 100 μM, time-dependent inactivation was observed in hepatocytes for amprenavir, diltiazem, erythromycin, raloxifene, and troleandomycin. The observed inactivation potency in hepatocytes (observed IC50) was compared with the potency predicted using microsomal parameters (predicted IC50). Despite satisfactory prediction for troleandomycin (1.35 and 2.14 μM for the predicted and observed IC50, respectively), over-prediction of inactivation was observed for raloxifene, amprenavir, and erythromycin (observed IC50 values 6.2-, 55-, and 7.8-fold higher, respectively, than the predicted IC50). By contrast, the observed IC50 for diltiazem in hepatocytes was approximately 4-fold lower than the IC50 predicted from microsomal data (under-prediction). After correcting for factors including nonspecific binding and inactivator consumption, prediction was significantly improved for raloxifene (the observed IC50 then became 2-fold higher than the predicted IC50) and for amprenavir to a lesser extent. A specific P-glycoprotein inhibitor, 4-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)-N-[2-(3.4-dimethoxyphenyl)ethyl]-6,7-dimethoxyquinazolin-2-amine (CP-100356), modulated the observed CYP3A inactivation potency by erythromycin and troleandomycin. In summary, these studies reveal three important factors that must be considered when microsomal inactivation parameters are used to predict inhibition-based drug interactions in intact cell systems.
The cytochrome P450 3A (CYP3A) family of enzymes is responsible for the metabolism of more than 50% of marketed drugs and some important endogenous substances. Consequently, drug interactions arising through the inhibition of CYP3A are of significant clinical importance (Lin and Lu, 1998). Mechanisms of CYP3A inhibition include reversible inhibition and time-dependent inactivation (TDI; also known as mechanism-based inhibition), and the latter has been recognized for many drugs and new chemical entities (Ito et al., 1998; Lin and Lu, 1998; Mayhew et al., 2000). Mayhew et al. (2000) showed that the inhibition of CYP3A by N-desmethyl-diltiazem, clarithromycin, and fluoxetine in humans was due to TDI instead of reversible inhibition. Traditionally, TDI is studied in human liver microsomes (HLMs) or cDNA-expressed enzyme systems to obtain inactivation kinetic parameters including the maximum inactivation rate constant (kinact) and the apparent inactivation constant (KI). These parameters, along with the expected inactivator exposure ([I]) and the degradation rate constant of the enzyme, which is usually obtained from animal data (Kanamitsu et al., 2000; Mayhew et al., 2000), are used to predict the extent of enzyme inactivation in vivo.
Cryopreserved hepatocytes are widely used in drug discovery and development in pharmaceutical industries to predict drug interaction and metabolic clearance for new chemical entities. Hepatocytes have the advantage of maintaining a complete set of drug transporters and phase I/II metabolizing enzymes, not fully represented by HLMs. It is anticipated that factors present in hepatocytes that are absent in microsomal systems may influence the prediction of inhibition-based drug interactions in vivo by directly using microsomal kinetic parameters (kinact and KI).
In the present study, we have developed an approach to evaluate the TDI of CYP3A in cryopreserved human hepatocytes. Prediction of the extent of inactivation in hepatocytes was made using kinetic parameters obtained from HLM experiments. The predicted inactivation potency was compared with the values observed in hepatocytes. The reliability of utilizing HLMs to predict TDI in vivo and the feasibility of using hepatocytes to assess TDI will be discussed.

Chemical structures of CYP3A inactivators and CP-100356.
Materials and Methods
Materials. Cryopreserved human hepatocytes and hepatocyte thawing medium were purchased from InVitro Technologies (Baltimore, MD). Pooled human liver microsomes from 60 individuals were obtained from a Pfizer Global R&D (PGRD; a division of Pfizer Inc.) in-house supply. CP-100356 was synthesized at PGRD. Amprenavir was repurified from the commercially available formulations at PGRD. HEPES was purchased from Invitrogen (Carlsbad, CA). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. The chemical structures of various compounds examined are shown in Fig. 1.
TDI of CYP3A in HLMs. For the main body of this study, 1′-hydroxylation of midazolam was used as the probe reaction for CYP3A in both HLMs and cryopreserved human hepatocytes. TDI in HLMs was performed according to methods described elsewhere (Ito et al., 1998). Briefly, inactivators of varying concentrations were preincubated with HLMs in potassium phosphate buffer (0.1 M, pH 7.4) at 37°C for 5 min before the addition of NADPH. The concentrations of microsomal protein and NADPH were 0.2 mg/ml and 1 mM, respectively. The concentrations for the inactivators were 5 to 80 μM erythromycin, 0.25 to 4 μM diltiazem, 0.75 to 12 μM troleandomycin, 100 μM diclofenac, 0.2 to 10 μM amprenavir, and 1.25 to 10 μM raloxifene. The experiments were performed in triplicate. At different times after the addition of NADPH, 10 μl of the inactivator/HLM mixture (first incubation) were transferred to 90 μl of potassium phosphate buffer containing 100 μM midazolam and 1 mM NADPH (10-fold dilution of inactivator in the incubation with midazolam), which had been preincubated at 37°C for 5 min. The reaction lasted for 1 min and was terminated by the addition of 100 μl of a mixture of acetonitrile/methanol (3:1 v/v) containing alprazolam as internal standard (600 nM). To assess the inactivator consumption during first incubation, samples at time zero and at the last time point after the addition of NADPH were quenched with acetonitrile/methanol (3:1, v/v) containing 600 nM alprazolam.
Microsomal protein binding of the inactivators was determined using equilibrium dialysis as described previously (Obach, 1997; Banker et al., 2003). A multiwell Teflon dialysis apparatus was manufactured at PGRD, and the dialysis membrane (molecular weight cutoff 12,000–14,000) was purchased from Spectrum Medical Industries (Los Angeles, CA). An aliquot (150 μl, n = 3) of a mixture containing HLMs (0.2 mg/ml in 0.1 M potassium phosphate buffer, initial incubation condition for TDI studies) and inactivators (nominal concentrations of 0.5 and 5 μM) was placed on one side of the membrane and blank phosphate buffer was added to the other side. The dialysis chamber was maintained at 37°C for 6 h. Samples were quenched with an equal volume of a mixture of acetonitrile/methanol (3:1) containing alprazolam as internal standard (20 nM). In general, equilibrium was reached between 4 and 6 h for all inactivators (data not shown). Sample extraction and analytical methods for the measurement of inactivators and 1-hydroxymidazolam are described below.
TDI of CYP3A in Cryopreserved Human Hepatocytes. Hepatocyte suspensions were prepared according to the procedures provided by the manufacturer. A pool of five individuals (three male and two female) was used. Briefly, hepatocytes were thawed in thawing medium (25 ml per 5 million hepatocytes) and centrifuged at 50g at room temperature for 5 min. The cell pellet was reconstituted in 30 ml of incubation medium (William's Medium E containing 10 mM HEPES) and centrifuged at 50g at room temperature for 5 min. The resulting cell pellet was reconstituted in fresh incubation medium, and cell viability was determined by the trypan blue method (generally between 78 and 82% viability). Cell suspension was placed in a 37°C incubator supplemented with 5% CO2 before use.
TDI in Hepatocytes. Hepatocytes were preincubated with and without time-dependent inactivators in a 48-well plate for 1 h in a 37°C incubator under 5% CO2. The final volume was 250 μl and the final cell concentration was 0.5 × 106 viable cells/ml. The inactivator concentrations were 0.1 to 5 μM amprenavir, 2.5 to 100 μM diclofenac, 0.5 to 4 μM diltiazem, 10 to 80 μM erythromycin, 2.5 to 20 μM raloxifene, and 0.75 to 6 μM troleandomycin. The experiments were performed in triplicate. At the end of preincubation, 200-μl aliquots of the suspension were transferred to 96-well plate microtubes (1.2 ml) and centrifuged at 50g for 5 min at room temperature. One hundred fifty microliters of each supernatant were discarded, and the pellets were resuspended after the addition of 150 μl of fresh medium for washing. The suspension was further centrifuged at 50g for 5 min at room temperature, and 180 μl of each supernatant were discarded. The postwash pellet was resuspended in 100 μl of fresh medium containing midazolam. At this point, the hepatocyte concentration was approximately 1 × 106 cells/ml and midazolam concentration was 100 μM. The suspensions were immediately placed in the incubator (37°C, 5% CO2) and were incubated for 9 min before the addition of a 2× volume of a mixture of acetonitrile/methanol (3:1 v/v) containing 300 nM alprazolam as internal standard. The use of 100 μM midazolam (to ensure saturation condition) and the choice of 9-min incubation time were confirmed by preliminary kinetic studies.
To assess the effect of competitive inhibition of the inactivators, hepatocytes were preincubated in the absence of inactivator for 1 h and then coincubated with midazolam (100 μM) and each inactivator. The final inactivator concentrations were 2.5 μM amprenavir, 50 μM diclofenac, 1 μM diltiazem, 10 μM erythromycin, 5 μM raloxifene, 2.5 μM troleandomycin, and 2 μM ketoconazole (positive control). The experiments were performed in triplicate. The effect of inactivators on cell viability was evaluated before and after 1-h preincubation by determining glutathione S-transferase activities in the medium of the cell suspension (Habig et al., 1974).
Inactivator Levels during 1-h Preincubation with Hepatocytes. Free inactivator concentration (Iu) was equivalent to total inactivator concentration in hepatocyte medium since no protein was added in this study. Hepatocyte suspensions were centrifuged at 50g for 5 min at 0, 15, 40, and 60 min. The supernatant was quenched with an equal volume of a mixture of acetonitrile/methanol (3:1 v/v) containing alprazolam as internal standard (20 nM final concentration) for the analysis of Iu. In the cases where metabolic consumption and nonspecific binding were significant, a mean time-averaged inhibitor concentration ([Ī) was obtained to represent average Iu during the 1 h of preincubation. Total inactivator levels in the cell suspension before and after the 1-h preincubation were also analyzed.
Effect of Coincubation with Efflux Transport Inhibitor in Hepatocytes. CP-100356, a P-glycoprotein inhibitor, was used to assess the possible involvement of this transporter in the modulation of CYP3A inactivation by the compounds tested in hepatocytes. CP-100356 (0.5 μM) was coincubated with the time-dependent inactivator during the 1-h preincubation of hepatocytes. The cells were washed and the activity of CYP3A was measured as described above.
Liquid Chromatography-Tandem Mass Spectrometry Methods. Samples quenched with organic solvent containing internal standard were vortexed and centrifuged at 1900g for 20 min. The supernatant was analyzed using a Micromass Quattro LC mass spectrometer (Quattro II) equipped with a Waters 2790 high-performance liquid chromatography system (Waters, Milford, MA). Chromatographic separation was performed by a Zorbax C18 column (2 × 50 mm, 5 μm; Agilent Technologies, Palo Alto, CA) coupled with a Keystone Javelin C18 guard column (2 × 20 mm; Western Analytical Products, Inc. Lake Elsinore, CA), at a flow rate of 0.2 to 0.3 ml/min using a linear gradient elution consisting of A, 50 mM ammonium acetate in water; and B, acetonitrile/methanol (89:11, v/v). Table 1 shows the analytical parameters for each analyte described in this study. Alprazolam is the internal standard for all the analytes. The m/z transition for alprazolam is 309→205. The retention time of alprazolam varied from 4.9 to 6.4 min due to different high-performance liquid chromatography gradient condition of each analyte (Table 1).
LC-MS-MS analytical parameters of 1-hydroxymidazolam and time-dependent inactivators
Data Analysis. The percentage of inactivator unbound (% Free) in HLMs was calculated according to eq. 1: 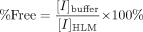 where [I]buffer and [I]HLM were the inactivator concentrations in the buffer side and the microsomal side at equilibrium, respectively.
where [I]buffer and [I]HLM were the inactivator concentrations in the buffer side and the microsomal side at equilibrium, respectively.
Kinetic parameters (kinact and KI) of CYP3A inactivation in HLMs were obtained by plotting the logarithm of the ratio of the remaining enzyme activity in the presence of inactivator to the activity in the absence of inactivator against the preincubation time. The apparent inactivation rate constant (λ) was determined from the slope of the initial linear phase on the semi logarithmic plots at each inactivator concentration. kinact and KI were obtained from the double-reciprocal plots of inactivator concentration [I] versus λ according to eq. 2 (Kitz and Wilson, 1962). 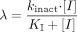
TDI of CYP3A in hepatocytes was predicted using kinetic parameters obtained from microsomal incubations according to eq. 3, in which Ei and E0 were CYP3A activities after 1-h preincubation (t = 60 min) with and without inactivator, respectively.
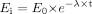

TDI of CYP3A in human liver microsomes. Legend shows the inhibitor concentrations in μM.
The IC50, Predicted was defined as the inactivator concentration that is required to cause Ei = 0.5E0 in hepatocytes after 1 h preincubation according to eq. 3. Theoretically, the value of λ that results in this effect is the same for all inhibitors (0.0115 min-1, at t = 60 min) according to eq. 3. Assumptions were made to aid the prediction of hepatocyte inactivation potency using kinetic parameters generated from microsomal experiments. These assumptions are as follows: a) Iu is represented by inactivator concentration in the hepatocyte medium; b) equilibrium was reached instantaneously for the inactivators throughout the preincubation period in hepatocytes; and c) the entry of the inactivator into the cell is mediated mainly by diffusion.
For inactivators that showed incomplete recovery or significant depletion in hepatocyte incubation, a time-averaged inactivator concentration in the medium ([Ī]) was used to represent the Iu. [Ī] was calculated by trapezoidal integration using monoexponential eq. 4 by WinNonlin software (Version 3.01; Pharsight, Mountain View, CA). 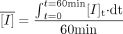
The observed CYP3A inactivation in hepatocytes was fitted to an inhibitory effect sigmoid Emax model (eq. 5;1 using WinNonlin software, in which Emax, IC50, and γ are the maximum enzyme activity, inactivator concentration at which 50% of activity was inhibited, and Hill's coefficient, respectively. 
Using eq. 5, the remaining CYP3A activity was fitted against both nominal inactivator concentrations and the concentrations corrected for nonspecific binding and metabolic consumption (Iu or [Ī]) to obtain IC50, Nominal and IC50, Corrected.
Simulations of the inactivation of CYP3A were performed using Microsoft Excel (Microsoft, Redmond, WA). HLM-predicted inactivation was simulated according to eq. 3, and the observed inactivation was simulated according to eq. 5 with either IC50, Nominal or IC50, Corrected.
Results
TDI in HLMs.Figure 2 shows the time-dependent loss of CYP3A activity in HLMs by six compounds reported to exhibit this property. The inactivation rate constants were obtained from the linear portion of the logarithmic ratio Ei/E0 versus time at each inactivator concentration, as described under Materials and Methods. No apparent CYP3A inactivation was observed for diclofenac at concentrations up to 100 μM. The parameters from this study and literature reports are summarized in Table 2. In general, our findings are in agreement with those reported previously. Amprenavir and raloxifene rapidly inactivated CYP3A under the experimental conditions of this study, exhibiting kinact values approximately 1 order of magnitude higher than those of diltiazem, erythromycin, and troleandomycin (Table 2).
Kinetic parameters of TDI of CYP3A in HLM
Inactivator levels during the inactivation incubation (first incubation) were analyzed at time 0 and at the last time point after the addition of NADPH. Table 3 shows that inactivator consumption was not significant for diltiazem, erythromycin, and troleandomycin. In contrast, loss of inactivator was observed for amprenavir and diclofenac at lower concentrations, and for raloxifene at all concentrations. Table 4 shows the inactivator microsomal free fraction at nominal 0.5 and 5 μM concentrations. The unbound fraction of raloxifene was 15 and 23% at 0.5 and 5 μM, respectively. Diclofenac also showed 47% microsomal binding at 0.5 μM. No significant microsomal protein binding was observed for the other compounds. Therefore, the apparent KI for raloxifene and amprenavir (Table 2) are overestimates due to potential microsomal binding and/or inactivator consumption (Tables 3 and 4).
Inactivator depletion in HLMs during first incubation Values are mean (S.D.), n = 3 incubations.
Microsomal-free fractions (% free) of inactivators at 0.2 mg/ml microsomal protein at 37°C Values are mean (S.D.), n = 3 incubations.
TDI of CYP3A in Cryopreserved Human Hepatocytes. The use of cryopreserved human hepatocytes for the study of TDI was validated as described under Materials and Methods. Cell viabilities were not different before and after 1-h preincubation in the absence or presence of the highest concentrations of inactivators (data not shown). CYP3A activities before and after 1-h preincubation were 56.3 ± 3.4 and 56.6 ± 3.0 pmol of 1-hydroxymidazolam per min per 106 cells, respectively (n = 3 separate incubations). As shown in Fig. 3 (open diamonds), amprenavir, diltiazem, erythromycin, raloxifene, and troleandomycin showed concentration-dependent CYP3A inactivation in hepatocytes after 1-h preincubation. Diclofenac had minimal effect on CYP3A activity at concentrations up to 100 μM (data not shown).
To discern the effect of reversible inhibition, inactivators and probe substrate midazolam were coincubated with hepatocytes, and the effect on CYP3A inhibition was compared with TDI experiments (Table 5). For amprenavir, diltiazem, erythromycin, raloxifene, and troleandomycin, coincubation had minimal effect on CYP3A activity, as opposed to the inactivation observed in the TDI experiments in which equal or lower inactivator concentrations were used in the preincubation. Coincubation with diclofenac (50 μM) did not inhibit CYP3A activity. Ketoconazole is a potent noncompetitive inhibitor of CYP3A (Gibbs et al., 1999b) that served as a positive control in this study. CYP3A activity was inhibited by 81% when hepatocytes were coincubated with 2 μM ketoconazole.
The effect of coincubation with inhibitor and 1-h preincubation with inactivator on CYP3A activities in cryopreserved human hepatocytes Values are mean (S.D.), n = 3 incubations.
Calculation of Iu in Hepatocyte Experiments.Table 6 summarizes the inactivator levels measured in the hepatocyte incubations. Inactivator levels in the medium are equal to Iu since no protein was added in the medium. Active uptake and/or nonspecific binding can be evaluated by assessing the recovery of the inactivators (Iu as a percentage of total concentration) at time 0. The recovery of the inactivator in the medium at time 0 was complete (near unity) for erythromycin and troleandomycin. However, the recovery ratios for raloxifene, amprenavir, diltiazem, and diclofenac were approximately 0.1, 0.4, 0.75, and 0.8, respectively. Inactivator consumption can be evaluated by measuring the percentage remaining of both Iu and total inactivator concentration after 1-h preincubation. Table 6 shows significant inactivator consumption for amprenavir (at lower concentrations), raloxifene, and diclofenac. The time courses of the Iu change for amprenavir and raloxifene are shown in Fig. 4. Taken together, the data demonstrated that it was not appropriate to use Iu at any single time point to represent the effective inactivator concentration for amprenavir and raloxifene. Instead, a time-averaged free inactivator concentration ([Ī]) (eq. 4) was used. Therefore, for inactivators displaying low recovery in hepatocyte incubation medium, the nominal concentrations were replaced by Iu (for diltiazem) and [Ī] for amprenavir and raloxifene (Fig. 4) for further evaluation (Table 7; Fig. 3).
Inactivator depletion and recovery from hepatocyte incubation media Values are mean (S.D.), n = 3 incubations.
Comparison of CYP3A inactivation between HLM-predicted potency and the potency observed in cryopreserved human hepatocytes
Prediction of TDI of CYP3A in Cryopreserved Hepatocytes using Microsomal Parameters. Comparison between the observed inactivation and HLM-predicted inactivation in hepatocytes was conducted. The practice was only to provide a descriptive relationship for the limited number of inactivators used in this study and was not meant to generate a quantitative correlation between the two systems. Diclofenac was excluded because of the absence of TDI under the conditions used in this study. Table 7 summarizes the observed inactivation (IC50, Nominal and IC50, Corrected) and HLM-predicted inactivation (IC50, Predicted). Under-prediction was defined when IC50, Predicted was higher than IC50, Nominal in hepatocytes, whereas over-prediction was defined when IC50, Predicted was lower than IC50, Nominal. Microsomal parameters of diltiazem apparently under-predicted the inactivation by approximately 4-fold (IC50, Nominal and IC50, Predicted of 3.22 and 12.32 μM, respectively). Correction of the observed inactivation using Iu in hepatocytes tends to magnify the under-prediction (IC50, Corrected became 2.41 μM). On the other hand, over-prediction was observed for the remaining four inactivators. The IC50, Nominal was 55-, 6.2-, and 7.8-fold higher than IC50, Predicted for amprenavir, erythromycin, and raloxifene, respectively. Predicted inactivation by troleandomycin appears to be the most accurate (IC50, Nominal and IC50, Predicted of 2.14 and 1.35 μM, respectively). Correction of the observed inactivation using [Ī] markedly narrowed the difference between the observed and predicted IC50 values for raloxifene and amprenavir. The observed IC50, Corrected for raloxifene was 1.01 μM as compared with the IC50, Predicted of 0.62 μM. The IC50, Corrected for amprenavir became 0.36 μM (versus IC50, Nominal = 1.09 μM) as compared with the IC50, Predicted of 0.02 μM. Iu or [Ī] correction was not made for troleandomycin and erythromycin due to their relative complete recoveries and the absence of metabolic consumption (Table 7). The differences between the observed inactivation and the predicted inactivation can also be visualized by the simulated inactivation curves in Fig. 3. Observed inactivation was simulated using parameters generated according to eq. 5 using both IC50, Nominal and IC50, Corrected, whereas the predicted inactivation was simulated using HLM parameters (eq. 3, t = 60 min) for each compound tested.

Effect of 1-h preincubation of inactivators on CYP3A activities in cryopreserved human hepatocytes. Open square, HLM-predicted inactivator concentration that caused the same inactivation observed in hepatocytes. Open diamond, observed CYP3A activity at nominal concentration of the inactivator (n = 3, CV = 2–18%). Closed diamond, observed CYP3A activity at corrected concentration using Iu (diltiazem) or [Ī] (amprenavir and raloxifene). Solid lines, simulated inactivation using eq. 4, Emax, IC50, and γ. Broken lines, predicted inactivation using HLM parameters according to eq. 2 (Table 2).
Effect of Efflux Transporter on TDI of CYP3A in Hepatocytes. To assess the effect of P-glycoprotein on the inactivation potency of the compounds tested, hepatocytes were coincubated with 0.5 μM CP-100356 and each inactivator during the 1-h preincubation. The concentrations chosen for each inactivator (0.5, 2, 20, 5, and 1.5 μM for amprenavir, diltiazem, erythromycin, raloxifene, and troleandomycin, respectively) are close to the observed respective IC50, Nominal values in this study (Fig. 3; Table 7). The concentration of CP-100356 (0.5 μM) was approximately 20-fold lower than its IC50 for CYP3A and about 5-fold higher than the IC50 for P-glycoprotein as reported by Wandel et al. (1999). CYP3A activity was not affected when 0.5 μM CP-100356 was present in the 1-h preincubation (data not shown). Figure 5 shows the effect of coincubation of CP-100356 on the inactivation potency for each compound tested. CP-100356 significantly enhanced CYP3A inactivation potency of the macrolides, troleandomycin and erythromycin (p < 0.01 and p < 0.05, respectively). CP-100356 did not significantly alter the inactivation potencies of raloxifene, diltiazem, and amprenavir.
Discussion
The major findings of this study include: 1) observation of TDI of CYP3A in cryopreserved hepatocytes by inhibitors known to exhibit this property in HLMs; 2) discovery that discrepancies between the predicted inactivation using microsomal kinetic parameters and those observed in hepatocytes can be partially explained by factors governing the accessibility of inactivators to CYP3A in hepatocytes. These factors include, but are not limited to, nonspecific binding, metabolic consumption, and active transport. To date, studies directed toward the prediction of TDI of CYP3A in hepatocytes using microsomal parameters have not been performed. The information from this work should therefore improve our understanding of the discrepancies associated with the prediction of drug interaction in vivo using HLMs.

Inactivator concentrations of raloxifene (left panel) and amprenavir (right panel) in hepatocyte incubation medium during 1-h preincubation. Legends show the nominal concentrations of each inactivator.
Time-dependent cytochrome P450 inactivation results from the formation of reactive metabolites or intermediates, which bind tightly or irreversibly to the heme or apoprotein, leading to a catalytically inactive protein. Consequently, the concentration of active enzyme is controlled by the rate of enzyme inactivation as well as endogenous rates of de novo synthesis and degradation (Lin and Lu, 1998). The generally accepted approach for the prediction of the magnitude of TDI in vivo has been to apply microsomal kinetic parameters and the projected [I] (eq. 2), together with estimates of the endogenous degradation rate constant of the “victim” enzyme (Mayhew et al., 2000). Although successful for some compounds (Kanamitsu et al., 2000; Mayhew et al., 2000; Ito et al., 2003), direct use of microsomal parameters may fail to predict in vivo drug-interactions for marketed drugs known to have TDI properties (Liras, 2003).
In this report, we evaluated the TDI of CYP3A in two widely used systems, HLMs and cryopreserved hepatocytes, for six compounds from different therapeutic classes. Pooled liver preparations were used to take into account the interindividual variability associated with the CYP3A family. Raloxifene, a selective estrogen receptor modulator, was shown to inactivate CYP3A4 via a reactive intermediate from the active site, which can be attenuated by glutathione (Chen et al., 2002). Macrolide antibiotics, erythromycin and troleandomycin, are believed to form complexes with heme through a nitroso intermediate initiated by CYP3A through N-dealkylation (Periti et al., 1992). Similarly, TDI caused by the vasodilator diltiazem appears to be due to further metabolism of its N-desmethyl-diltiazem metabolite (Jones et al., 1999; Mayhew et al., 2000). The HIV protease inhibitor amprenavir was recently shown to inhibit CYP3A by binding to the heme (Ernest et al., 2003). The nonsteroidal anti-inflammatory agent diclofenac was reported to be a time-dependent inactivator of CYP3A with a rather high KI of 1.6 mM (Masubuchi et al., 2002).

Effect of P-glycoprotein inhibitor CP-100356 (0.5 μM) on the CYP3A inactivation potency by time-dependent inactivators in cryopreserved hepatocytes (mean ± S.D., n = 4 incubations). Inactivator concentrations: amprenavir, 0.5 μM; diltiazem, 2 μM; erythromycin, 20 μM; raloxifene, 5 μM; troleandomycin, 1.5 μM. *, p < 0.05; **, p < 0.01.
We did not observe TDI by diclofenac in either HLMs or hepatocytes at concentrations up to 100 μM. Further investigation at higher concentrations was not conducted because 100 μM is already much higher than free diclofenac exposure observed in humans. Indeed, diclofenac was not likely to cause clinically relevant CYP3A inhibition, given its high microsomal KI and high plasma protein binding (Masubuchi et al., 2002). Thus, diclofenac was excluded from the HLM-hepatocyte prediction exercise. The remaining five inactivators demonstrated a concentration-dependent and time-dependent CYP3A inactivation in hepatocytes after 1-h preincubation (Table 5).
In general, HLM-based prediction overestimates the inactivation potency observed in hepatocytes for troleandomycin, erythromycin, raloxifene, and amprenavir by approximately 2- to 60-fold (IC50, Predicted lower than IC50, Nominal, Table 7). The discrepancy may be due to metabolic stability, nonspecific binding, active transport, or the combination of these factors. The depletion of the unbound inhibitor concentration available to the enzyme may introduce serious error into a quantitative prediction of an in vivo drug-drug interaction (Gibbs et al., 1999a; Xu et al., 2003). Among the inactivators tested, amprenavir and raloxifene had significantly low recoveries in the hepatocyte medium after the compounds were spiked into the cell suspension (approximately 10–40% recoveries, Table 6), suggesting concentrative uptake of these two compounds into the hepatocytes, mediated either by uptake transport or high lipophilicity. Indeed, the calculated logD7.4 values for amprenavir and raloxifene were 4.2 and 5.5, respectively (ACD Labs, Toronto, ON, Canada), which are the highest among the compounds tested in this study. The significant microsomal binding for raloxifene may imply strong nonspecific binding of this compound to cellular constituents in hepatocyte incubations (Tables 4 and 6). Metabolic consumption during the 1-h preincubation in hepatocytes further decreased the availability of inactivator to the enzyme. The major biotransformation pathway for raloxifene is glucuronidation (Kemp et al., 2002). Effects of phase II enzyme are often not represented in HLM experiments. The discrepancy between Iu and nominal concentration for amprenavir may be partly attributed to the metabolic consumption by CYP3A (Treluyer et al., 2003). Correction using [Ī] improved the prediction markedly for raloxifene and to a lesser extent for amprenavir. One should also note that the apparent KI values are likely overestimates for raloxifene and amprenavir, given that microsomal binding and/or HLM metabolic consumption (Tables 3 and 4) would theoretically widen the gap between the predicted IC50 and the observed IC50.
The major assumption of these predictions is that entry of inactivator into the cell is not rate-limiting and is mediated by diffusion only (free drug hypothesis). However, if the inactivator is a substrate of active transporter(s), Iu in the medium will no longer represent the intracellular inactivator concentration. Lam and Benet (2004) have demonstrated that the metabolism of digoxin (a substrate of both uptake and efflux transporters) in freshly isolated rat hepatocyte suspension can be modulated by inhibitors of both uptake and efflux transporters.
In this study, we chose CP-100356, a P-glycoprotein inhibitor (Wandel et al., 1999), to address the potential influencing factors associated with active transporters. If P-glycoprotein were functionally active in cryopreserved hepatocytes by reducing intracellular inactivator levels, or by efficiently removing obligatory metabolites of the pathway, one would observe decreased inactivation. To date, little is known about the expression, function, and orientation of P-glycoprotein after cryopreservation. In intact liver, hepatocytes are polarized, with transporters preferentially expressed on either the basolateral membrane or the bile canalicular membrane. The disruption of polarity upon collagenase isolation of hepatocytes may lead to the loss of P-glycoprotein. Roelofsen et al. (1995) have demonstrated the endocytosis of canalicular multispecific organic anion transporter after isolation of rat hepatocytes, which was confirmed by the disappearance of transport activity from the cell surface and the appearance of intracellular vesicles that accumulate the substrate. Assuming P-glycoprotein undergoes similar changes, one would expect the decreased availability of a P-glycoprotein substrate to cytoplasmic components (such as CYP3A) due to efflux by the remaining cell membrane P-glycoprotein activity and/or accumulation into the intracellular vesicles by P-glycoprotein. We demonstrated that coincubation of 0.5 μM CP-100356 (a concentration that did not inhibit CYP3A) significantly increased the CYP3A inactivation potency by troleandomycin and erythromycin (Fig. 5). Interestingly, amprenavir has been shown to be a P-glycoprotein substrate (Polli et al., 1999; Choo et al., 2000; Huang et al., 2001); however, CP-100356 did not alter its inactivation potency (Fig. 5). The strong concentrative effect observed for amprenavir may have masked the P-glycoprotein-mediated efflux. Like many P-glycoprotein inhibitors, the selectivity of CP-100356 toward other transporters is not known, and the modulation of the CYP3A inactivation potency for macrolide antibiotics may have arisen from the inhibition of basolateral efflux transporter(s), which does not recognize amprenavir as a substrate. Our findings of effects of CP-100356 on macrolide-induced inactivation suggested that P-glycoprotein (or other efflux proteins that can be inhibited by CP-100356) may have survived and maintained its efflux functionality during the hepatocyte cryopreservation and resuspension. It has to be noted that cryopreserved hepatocytes are widely used to predict drug interaction and in vivo metabolic clearance of a new chemical entity. Thus, for compounds that are substrates for active transporters, care should be taken when interpreting data collected from cryopreserved hepatocytes to predict in vivo outcome. It is also recognized that other transporters (both uptake and efflux) that survived cryopreservation and resuspension can also contribute to the discrepancy between HLMs and hepatocytes observed in this study. Further investigation of the mechanism through which efflux transporters alter the inactivation potency of macrolide antibiotics will be conducted.
The apparent discrepancy of the 4-fold difference between IC50, Nominal and IC50, Predicted for diltiazem (Table 7) is difficult to address. The low microsomal kinact (0.012 min-1) value of diltiazem is very close to the predicted λ value that would cause 50% inhibition (0.0115 min-1 at 60 min; eq. 3). Mathematically, predicting inactivator concentration that causes more than 50% inactivation in hepatocytes results in a negative value (eq. 3). It is also noted that the IC50, Predicted is greater than microsomal KI (Table 7), implying that the prediction of 50% inactivation in hepatocytes is operated at λ = kinact (eq. 2). Together, one would expect a large degree of uncertainty of IC50, Predicted under the conditions set up in this report. For compounds with low kinact such as diltiazem, it appears to be very difficult to detect the existence of TDI from microsomal experiments (Fig. 3), whereas individuals on diltiazem (60 mg, three times a day for 3 days) had a 2-fold increase of triazolam exposure (Kosuge et al., 1997). Our observation of CYP3A inactivation in hepatocytes for compounds with low kinact such as diltiazem shows the potential advantage of using hepatocytes as a means to evaluate the projected in vivo potency of a time-dependent inactivator obtained from microsomal experiments.
In conclusion, when potential factors such as metabolic consumption, nonspecific binding, and active transport are considered, cryopreserved hepatocytes can augment the use of HLMs as a tool to evaluate a compound's potential to cause TDI in vivo.
Acknowledgments
We thank David Neul for hepatocyte preparations. The valuable suggestions and advice from Yun Xu, Dr. Rene H. Levy (University of Washington) regarding inhibitor depletion, and Dr. Alfin Vaz (Pfizer Groton) regarding assessment of TDI in hepatocytes were sincerely appreciated. We also thank Drs. Ellen Y. Wu, James L. Ferrero, William Pool, and Wei-Zhu Zhong for carefully reviewing the manuscript and valuable comments.
Footnotes
-
↵1 We have chosen the inhibitory Emax model to estimate the observed inactivation IC50 based on a limited number of observations (four for each inactivator). However, one should note that this is not an appropriate model to quantitatively assess the time-dependent event in hepatoctye incubations.
-
This work was partially supported by National Institutes of Health Grant GM32165.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.002832.
-
ABBREVIATIONS: TDI, time-dependent inactivation; HLM, human liver microsome; PGRD, Pfizer Global R&D; CP100356, 4-(6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-yl)-N-[2-(3,4-dimethoxyphenyl)ethyl]-6,7-dimethoxyquinazolin-2-amine; IC50, Nominal, the nominal concentration of inactivator to cause 50% inactivation observed in hepatocytes; IC50, Corrected, IC50 corrected for nonspecific binding and time-dependent loss due to metabolism in hepatocytes; IC50, Predicted, predicted inactivator concentration in hepatocytes that produced 50% inactivation of CYP3A activity using microsomal parameters; Iu, free inactivator concentration in hepatocyte incubation medium; [Ī] time-averaged inactivator concentration in the hepatocyte incubation medium; KI, apparent inactivation constant; kinact, maximum inactivation rate constant; λ, apparent inactivation rate constant.
- Received November 2, 2004.
- Accepted February 24, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}