


Abstract
The drug-drug interaction (DDI) potential of deleobuvir, an hepatitis C virus (HCV) polymerase inhibitor, and its two major metabolites, CD 6168 (formed via reduction by gut bacteria) and deleobuvir-acyl glucuronide (AG), was assessed in vitro. Area-under-the-curve (AUC) ratios (AUCi/AUC) were predicted using a static model and compared with actual AUC ratios for probe substrates in a P450 cocktail of caffeine (CYP1A2), tolbutamide (CYP2C9), and midazolam (CYP3A4), administered before and after 8 days of deleobuvir administration to HCV-infected patients. In vitro studies assessed inhibition, inactivation and induction of P450s. Induction was assessed in a short-incubation (10 hours) hepatocyte assay, validated using positive controls, to circumvent cytotoxicity seen with deleobuvir and its metabolites. Overall, P450 isoforms were differentially affected by deleobuvir and its two metabolites. Of note was more potent CYP2C8 inactivation by deleobuvir-AG than deleobuvir and P450 induction by CD 6168 but not by deleobuvir. The predicted net AUC ratios for probe substrates were 2.92 (CYP1A2), 0.45 (CYP2C9), and 0.97 (CYP3A4) compared with clinically observed ratios of 1.64 (CYP1A2), 0.86 (CYP2C9), and 1.23 (CYP3A4). Predictions of DDI using deleobuvir alone would have significantly over-predicted the DDI potential for CYP3A4 inhibition (AUC ratio of 6.15). Including metabolite data brought the predicted net effect close to the observed DDI. However, the static model over-predicted the induction of CYP2C9 and inhibition/inactivation of CYP1A2. This multiple-perpetrator DDI scenario highlights the application of the static model for predicting complex DDI for CYP3A4 and exemplifies the importance of including key metabolites in an overall DDI assessment.
Introduction
Deleobuvir (BI 207127) is an inhibitor of hepatitis C virus (HCV) NS5B RNA polymerase and, in combination with faldaprevir and ribavirin, achieved high, sustained viral-load reduction in treatment-naïve and treatment-experienced patients with chronic genotype 1b infection (Zeuzem et al., 2013). In a disposition study in which 14C-deleobuvir was administered to healthy male human volunteers, two major metabolites of deleobuvir were identified, an alkene reduction product (CD 6168) formed by gut bacteria and deleobuvir-acyl glucuronide (deleobuvir-AG) (Fig. 1) (Chen et al., 2015; McCabe et al., 2015). Following a single oral dose of 800 mg deleobuvir to healthy volunteers, CD 6168 and deleobuvir-AG exposure (AUC0–∞) in the systemic circulation was 27 and 43%, respectively, of the parent deleobuvir. Both CD 6168 and deleobuvir-AG are pharmacologically active against HCV replication, albeit 10- and 3-fold less potent, respectively, than deleobuvir (data on file at Boehringer Ingelheim).

Structures of deleobuvir, CD 6168, and deleobuvir-AG.
According to the drug-drug interaction guidance documents from the US Food and Drug Administration (FDA, 2012) and the European Medicines Agency (EMA, 2012), contribution of major metabolites should be considered in assessing overall drug-drug interaction (DDI) liability of the parent compound if a metabolite concentration is >25% of the parent and/or if a metabolite concentration is >10% of total drug-related material in circulation. Higher cut-off values have been proposed in the literature using the logic that polar metabolites lacking structural alerts are typically less potent P450 inhibitors and inactivators than the parent drug (Callegari et al., 2013; Yu and Tweedie, 2013; Yu et al., 2015). These approaches have largely focused on DDI as a result of inhibition and inactivation.
Recent evaluations to estimate clinical drug-drug interaction risk involving a combination of inhibition, inactivation, and induction, using dynamic physiologically based pharmacokinetic models, as well as simpler static models, have shown promising results for CYP3A4 (Fahmi et al., 2008a,b; Zhao et al., 2011; Einolf et al., 2014). In this study, in vitro DDI assessment was performed for parent drug deleobuvir and both of its major metabolites. In vitro, deleobuvir, CD 6168, and/or deleobuvir-AG affected activities of several P450 isoforms by competitive inhibition, inactivation, and/or induction. Dynamic models incorporate temporal changes in drug and enzyme levels in a sophisticated manner, thereby minimizing overprediction of interaction. Static models usually incorporate only the maximal plasma concentrations or the portal vein inlet concentrations, which is not a realistic assumption throughout the time course of co-administration of interacting drugs. In the case of deleobuvir, the complexity of incorporating three perpetrator compounds, with mixed effects, made attempts at dynamic modeling very challenging, and consequently a simpler static model, modified to incorporate multiple perpetrators, was used to evaluate the accuracy of prediction of the effect of deleobuvir on CYP1A2, CYP2C9, and CYP3A4 activity in vivo, which then made feasible a prediction of the effects on other P450 isoforms.
An in vivo study was conducted in HCV-infected patients to assess the net effect of deleobuvir (and generated metabolites) on the pharmacokinetics of caffeine, tolbutamide, and midazolam, used in combination as an in vivo probe substrate cocktail for CYP1A2, CYP2C9, and CYP3A4 activities, respectively (Fuhr et al., 2007; Sabo et al., 2015). The patients in this study also received polyethylene glycol (PEG)–interferon and ribavirin as part of the standard of care. On the basis of information in the Copegus drug label, ribavirin would not have been expected to affect P450 enzymes. However, there had been reports of PEG-interferon α-2b inhibiting CYP1A2, albeit very mildly. These studies had assessed the effect of PEG-interferon α-2b after approximately 1 month of treatment (Gupta et al., 2011; Brennan et al., 2013), which is much longer than the duration reported for our study. Further, in a parallel arm to our study, PEG-interferon α-2b and ribavirin were administered in an identical regimen in combination with faldaprevir and had no effect on CYP1A2 and CYP2C9 activities (Sabo et al., 2015). As such, PEG-interferon α-2b was not expected to have an impact on CYP1A2, and the effects seen on the probe substrates are considered attributable to treatment with deleobuvir and its metabolites. The in vitro DDI predictions for CYP1A2, CYP2C9, and CYP3A4 for deleobuvir and its generated metabolites were compared with the clinical DDI results for the probe substrates.
Some of the complexities of evaluating multiple perpetrator compounds, together with differential effects of parent drug versus metabolites, are highlighted. Additionally, an abbreviated incubation of 10 hours was validated in the hepatocyte induction assay to circumvent toxicity observed with deleobuvir and its metabolites; this abbreviated incubation can be applied to other compounds. Data-driven strategies for managing comedication use in larger scale clinical studies with deleobuvir are also discussed.
Materials and Methods
Materials.
Deleobuvir, CD 6168, deleobuvir-AG, 13C6-deleobuvir, 13C6-CD 6168 were synthesized at Boehringer Ingelheim Pharmaceuticals Inc. (Ridgefield, CT). Cryoplateable human hepatocytes were purchased from Invitrogen/ThermoFisher Scientific (Grand Island, NY); donor information can be found in (Supplemental Table 1). Pooled human liver microsomes (HLM; (Supplemental Table 2) and recombinant human cytochromes P450 (rP450; produced in baculovirus-infected insect cells) and control insect cell microsomes, rat type 1 collagen, l-glutamine, penicillin/streptomycin, ITS+ premix, and fetal bovine serum were purchased from Corning (Woburn, MA). NADPH, omeprazole (OME), phenobarbital (PB), rifampicin (RIF), trypan blue, ECM, William’s E media, LDH, and MTT cytotoxicity assay kits and dexamethasone were purchased from Sigma-Aldrich (St. Louis, MO). The mRNA catcher kits, reverse transcription polymerase chain reaction (RT-PCR), and TaqMan primer probes were purchased from Invitrogen/ThermoFisher Scientific. All other reagents and solvents were of analytical grade or higher purity and were obtained from commercial suppliers. Human liver microsomes and rP450s were stored at –80°C until used.
Cytochrome P450 Inhibition.
Each isoform-specific cytochrome P450 (P450) probe substrate (CYP1A2, phenacetin; CYP2B6, bupropion; CYP2C8, amodiaquine; CYP2C9, diclofenac; CYP2C19, (S)-mephenytoin; CYP2D6, dextromethorphan; CYP3A4, midazolam) was incubated in the presence of various concentrations of deleobuvir, CD 6168, or deleobuvir-AG with pooled HLM in potassium phosphate buffer (50 mM, pH 7.4) and respective isoform-specific metabolites (acetaminophen, hydroxybupropion, N-desmethylamodiaquine, 4′-hydroxydiclofenac, 4′ -hydroxymephenytoin, dextrorphan, 1′-hydroxymidazolam) were monitored. HLM, inhibitor, and substrate were preincubated at 37°C for 5 minutes. Incubations with isoform-selective P450 inhibitors (CYP1A2, α-naphthoflavone; CYP2B6, ticlopidine; CYP2C8, montelukast; CYP2C9, sulfaphenazole; CYP2C19, S-(+)-N-3-benzylnirvanol; CYP2D6, quinidine; CYP3A4, ketoconazole) were included as positive controls and showed expected results (data not shown). All experiments were performed under linear conditions with respect to time and protein concentration and were conducted at substrate concentrations equivalent to their Km values determined in HLM [phenacetin (40 μM), bupropion (90 μM), amodiaquine (1.5 μM), diclofenac (4 μM), (S)-mephenytoin (30 μM), dextromethorphan (7 μM), midazolam (3 μM), and testosterone (60 μM)]. Reactions were initiated by the addition of NADPH (2 mM). A 50-μl aliquot of the incubation mixture was removed at desired time points, and reactions were quenched with 40% acetonitrile containing internal standard (nevirapine or stable labeled probe metabolites) and 0.1% acetic acid. For CYP2C8, the reactions were quenched with 40% methanol with 0.3% formic acid. All quenched samples were filtered and analyzed for appropriate P450 reaction-specific probe metabolites by high-performance liquid chromatography–tandem mass spectrometry (LC-MS/MS). Concentrations that achieve a 50% inhibitory effect (IC50) were calculated using Graph Pad Prism and Ki values were calculated as IC50 × 0.5, assuming competitive inhibition.
P450 Inactivation.
Pooled HLM (0.4 mg/ml) and recombinant CYP3A4 (50 pmol/ml, ∼0.6 mg/ml) were incubated with various concentrations of deleobuvir, CD 6168, or deleobuvir-AG in potassium phosphate buffer (50 mM, pH 7.4) at 37°C for 5 minutes. Reactions were initiated by the addition of cofactor NADPH (2 mM). Control samples were included in which deleobuvir, CD 6168, or deleobuvir-AG were incubated at the highest test concentration employed in the respective assay, in the absence of NADPH. At specific time points up to 35 minutes, an aliquot of this primary incubation was removed and diluted 20-fold by adding to a secondary incubation in which the enzyme activity remaining was measured using isoform-specific probe substrates outlined above. Concentration of the probe substrates used were approximately 5- to10-fold their respective Km values. After incubation with NADPH (2 mM) at 37°C for 6 minutes, the reactions were quenched with 40% acetonitrile containing internal standard and 0.1% acetic acid. Samples were filtered and analyzed by LC-MS/MS to monitor the appropriate P450 reaction–specific probe metabolites described above.
Rate constants for loss of P450 activity, normalized to the corresponding controls without the test compound, were used in eq. (1) to obtain inactivation parameters:
 where:
where:
kobs is the observed rate constant for inactivation,
kinact is the maximal inactivation rate constant,
[I] is the concentration of inactivator in the primary incubation, and
KI is the concentration of inactivator at which the rate of inactivation is half maximal.
Samples were analyzed for metabolite production on a 4000 QTRAP (AB Sciex, Thornhill, Ontario, Canada) attached to either a CTC PAL autosampler (Leap Technologies, Carrboro, NC) or a Waters Acquity UPLC system (Milford, MA). The aqueous mobile phase (A) and organic mobile phase (B) consisted of 95:5 (v/v) water/acetonitrile and 95:5 (v/v) acetonitrile/water, respectively. Both mobile phases contained 0.1% acetic acid. For the CYP2C8 assays, the mobile phase (A) consisted of 5 mM ammonium formate, 0.3% (v/v) formic acid in water, and mobile phase (B) consisted of 0.3% (v/v) formic acid in methanol. Samples were eluted through Waters YMC-Pack C4 (50 × 3 mm, 5-μm, CYP2C8) or a Phenomenex Synergi Max RP (150 × 2mm, 4-μm) column, using validated probe substrate analysis methods. The multiple reaction monitoring transitions used were 152.0→ 110.0 (acetaminophen); 155.0→ 110.0 ([13C2, 15N]-acetaminophen); 256.1→ 139.1 (hydroxybupropion); 262.1→ 139.1 (hydroxybupropion-d6); 328.0→ 283.0 (N-desethylamodiaquine); 333.0→ 283.0 (N-desethylamodiaquine-d5); 312.0→230.0 (4′-hydroxydiclofenac); 318.0→236.0 (4′ hydroxydiclofenac-[13C6]); 258.0→157.0 (dextrorphan); 261.0→157.0 (dextrorphan-d3); 342.0→324.0 (1′-hydroxymidazolam); 347.0→327.0 (1′-hydroxymidazolam-[13C3]); 305.0→269.0 (6β-hydroxytestosterone); 312.0→276.0 (6β-hydroxytestosterone-d7); 267.2→226.1 (neviripine); in positive ion mode.
P450 Induction and Cytotoxicity.
To assess induction of CYP1A2, CYP2B6, and CYP3A4 mRNA, three separate lots of cryoplateable human hepatocytes, precharacterized for prototypical induction response (Hu1419, Hu1424, and Hu8123), were used. Cytotoxicity was measured by lactate dehydrogenase (LDH) leakage and the MTT cell-proliferation assay (measuring the reduction of a tetrazolium component to an insoluble formazan product by mitochondria of viable cells), performed in the same donors at 8, 10, and 12 hours. Cytotoxicity for deleobuvir and CD 6168 was assessed at 24 and 48 hours during a separate study. The cells were plated per vendor recommendations and incubated at 37°C with 5% CO2 for 4–6 hours to enable attachment of the cells. After adequate attachment, cells were overlaid with extracellular matrix (ECM). Twenty four hours after preparation of the sandwich cultures, the medium was aspirated and replenished. Between 46 and 48 hours after recovery and plating, the cells were treated with either solvent [0.5% (v/v) equivalent parts acetonitrile and methanol], deleobuvir, CD 6168, deleobuvir-AG (0.1, 0.3, 1, 3, and 10 μM for all three), or a prototypical P450 inducer [omeprazole (0, 0.3, 1, 3, 10, 30, 100, and 300 μM), phenobarbital (0, 3, 10, 30, 100, 300, 1000, and 3000 μM), or rifampicin (0, 0.3, 1, 3, 10, 30, 100, and 300 μM)] for 10 hours. A shorter incubation time (10 hours) was used to avoid cytotoxicity observed upon incubation of hepatocytes with deleobuvir and its metabolites (at >1 μM) for the typical 48–72 hours employed for induction studies. For the precharacterization of the 10-hour time point and donor selection, cells from four donors were treated with 25 μM rifampicin or solvent for 0, 4, 6, 8, 12, 24, and 48 hours. Cells were replenished with fresh media containing rifampicin again at 24 hours. After the incubation, cell lysates were treated with RNA-later and stored at –20°C until isolation of RNA. RNA samples were isolated from these frozen hepatocytes using an mRNA Catcher PLUS kit (cat. no. K1570-03; ThermoFisher Scientific, Grand Island, NY) and stored at –80°C. The mRNA expression for specific gene targets was determined by TaqMan Real-Time RT-PCR using a 7900 Real Time PCR System (Applied Biosystems/ThermoFisher Scientific) and available primer probe sets (Supplemental Table 3). For each study, cells treated from three separate wells were analyzed in duplicate and data for each gene of interest was normalized using housekeeping genes, GAPDH, or β-actin. Statistical analysis was performed to determine any significant difference between drug- and solvent-treated wells using a two-tailed unpaired student t test in Microsoft Excel. Where concentration-dependent induction was observed, the data were fit to determine EC50 values using eq. (2):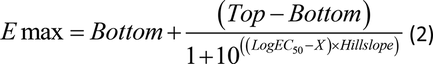 where X is the logarithm of concentration.
where X is the logarithm of concentration.
Protein Binding Estimates in Plasma, HLM, and Induction Culture Media.
Teflon dialysis cells (Spectrum, Rancho Dominguez, CA) and dialysis membranes (Spectra/Por, Spectrum) with 12,000–14,000 molecular weight cut-off were used for equilibrium dialysis to determine binding of [14C]deleobuvir, [14C]CD 6168, and [14C]deleobuvir-AG in pooled human plasma, HLM (0.05–0.5 mg/ml), and induction culture medium (bovine serum albumin, 1.25 mg/ml). Concentration ranges of [14C]deleobuvir, [14C]CD 6168, and [14C]deleobuvir-AG tested were 0.05–100 μM for microsomal binding. For pooled human plasma binding [14C]deleobuvir was tested at 0.15–59 μM, [14C]CD 6168 was tested at 0.52–44 μM, and [14C]deleobuvir-AG was tested at 0.5–70 μM. Binding to induction medium was evaluated for [14C]CD 6168 at concentrations between 0.1 and 30 μM. For the protein binding assessment, five individual dialysis cells were prepared and rotated at 20 rpm using a Spectrum dialysis cell rotator in a water bath maintained at 37°C for 4 hours. At the end of the incubation period, the contents of each side were transferred to scintillation vials and processed for liquid scintillation counting.
DDI Prediction.
Prediction of drug interactions for all enzymes was conducted as described in the regulatory guidances by EMA (2012) and FDA (2012). For determination of DDI potential for CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A4, the static model [eqs. (3) and (4)] was used for predicting area-under-the-curve (AUC) fold change of a sensitive substrate since deleobuvir and/or its metabolites were inhibitors, inactivators, and/or inducers of these P450 isoforms. Substrate and physiologic parameters and explanation of the terms used in the calculations are displayed in (Supplemental Tables 4 and (5).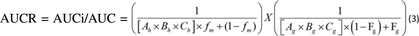 Here, Ah, Bh, Ch refer to inhibition, inactivation and induction effects in the liver, respectively, and the Ag, Bg, and Cg refer to the inhibition, inactivation, and induction effects in the gastrointestinal (GI) track, respectively. The terms fm and Fg refer to fraction metabolized by the pathway under consideration and fraction escaping gut metabolism, respectively. Details regarding each parameter in the equation can be found in the EMA and FDA (draft) guidances and relevant input parameters for deleobuvir DDI predictions are provided in (Supplemental Table 5). This equation was modified to incorporate multiple perpetrators in an additive manner as depicted in eq. (4) (Lutz et al., 2013; Rowland and Yeo et al., 2010).
Here, Ah, Bh, Ch refer to inhibition, inactivation and induction effects in the liver, respectively, and the Ag, Bg, and Cg refer to the inhibition, inactivation, and induction effects in the gastrointestinal (GI) track, respectively. The terms fm and Fg refer to fraction metabolized by the pathway under consideration and fraction escaping gut metabolism, respectively. Details regarding each parameter in the equation can be found in the EMA and FDA (draft) guidances and relevant input parameters for deleobuvir DDI predictions are provided in (Supplemental Table 5). This equation was modified to incorporate multiple perpetrators in an additive manner as depicted in eq. (4) (Lutz et al., 2013; Rowland and Yeo et al., 2010).

Clinical Drug-Drug Interaction Study.
The effect of deleobuvir on CYP1A2, CYP2C9, and CYP3A4 was evaluated in the clinic in the initial phase of a larger open label Phase II study. Caffeine, tolbutamide, and midazolam were used as probe substrates for CYP1A2, CYP2C9, and CYP3A4, respectively. This multicenter study (seven sites in Canada) was conducted in HCV-infected patients. The study was conducted in accordance with the International Conference on Harmonization guideline for Good Clinical Practice and the principles of the Declaration of Helsinki, and was approved by the local ethics committee and is registered at ClinicalTrials.gov under registration number NCT01525628.
Patients (11 males and 8 females) were administered single oral doses of caffeine (200 mg), tolbutamide (500 mg), and midazolam (2 mg) prior to initiation of HCV therapy. Blood samples for pharmacokinetic analysis of drug concentrations were collected over 24 hours. Therapy was then initiated with deleobuvir (600 mg TID; 6-hour/6-hour/12-hour dosing schedule) in combination with weight-based QD ribavirin (Copegus) and weekly pegylated-interferon α-2a (Pegasys). The probe substrates were administered after 8 days of dosing with deleobuvir, ribavirin, and pegylated-interferon α-2a, and blood samples were again collected over 24 hours. Patients that required any medications that are moderate or potent inhibitors or inducers of CYP1A2, CYP2C9, or CYP3A were excluded from the study from up to 5 days prior to day 1 through to day 19 of the treatment phase. Plasma concentrations of caffeine, tolbutamide, and midazolam and metabolites of interest were measured by LC-MS/MS (Tandem Laboratories, Salt Lake City, UT). Samples containing analyte and dueterated internal standard were extracted and analyzed in an API 4000 or API 5000 mass spectrometer (AB Sciex, Framingham, MA). Quantification was performed using linear (midazolam, 1-OH midazolam) or quadratic (caffeine, tolbutamide, 4-OH tolbutamide) weighted regression analysis (1/×2) of peak area ratios of analytes and internal standards. Quantification ranges (ng/ml) were: caffeine, 10.0–10,000; midazolam and 1-OH midazolam, 0.2–10.0; tolbutamide and 4-OH tolbutamide, 5.0–5,000. Pharmacokinetic parameters [maximum plasma concentration (Cmax), AUC0–tz, or AUC0–∞] were calculated for the probe substrates using WinNonLin version 5.2.
Plasma concentrations and pharmacokinetics of deleobuvir, deleobuvir-AG, and CD 6168 were monitored to ensure exposure to the study medication compounds. Ex vivo, 40% loss of deleobuvir-AG was observed from spiked plasma; however, deleobuvir-AG was highly stable under acidified conditions. Hence, plasma samples were mixed with citric acid at collection to stabilize the acyl glucuronide metabolites from spontaneous chemical degradation. Deleobuvir and its metabolites were measured using validated bioanalytical assays and authentic standards as described previously (Chen et al., 2015). After day 9, the dosing continued with deleobuvir and faldaprevir in combination with pegylated-interferon and ribavirin until the end of treatment (24 weeks) (Sabo et al., 2015). The pharmacokinetic and pharmacodynamics endpoints of the complete study are beyond the scope of this discussion. The statistical model used for the analysis of the in vivo data was an analysis of variance (ANOVA) model on the logarithmic scale. This model included effects accounting for the following sources of variation: “subject” and “treatment.” The effect “subject” was considered as random, whereas the other effect was considered as fixed.
Results
P450 Inhibition and Inactivation.
Of the seven isoforms evaluated, deleobuvir, CD 6168, and/or deleobuvir-AG inhibited CYP2B6, CYP2C8, CYP2C9, and CYP3A4 and the respective Ki values for inhibition are presented in Table 1. CYP2C8 was inhibited most potently by deleobuvir-AG with a Ki value of 0.022 μM. Deleobuvir and CD 6168 also inhibited CYP2C8 with Ki values of 0.13 and 0.27 μM, respectively. Deleobuvir-AG also inhibited CYP1A2, CYP2C19, and CYP2D6 (Table 1), whereas no inhibition of CYP1A2, CYP2C19, and CYP2D6 was observed for deleobuvir and CD 6168 up to 50 μM.
Ki (μM)a values for competitive inhibition of P450 isoforms by deleobuvir, CD 6168, and deleobuvir-AG
Out of the seven isoforms evaluated, deleobuvir mildly inactivated CYP1A2 and CYP3A4 and potently inactivated CYP2C8 (Table 2). CD 6168 also inactivated CYP2C8 more potently than CYP3A4. Deleobuvir-AG inactivated CYP2C8 more potently than both deleobuvir and CD 6168. Figure 2, A–C shows relative loss of CYP2C8 activity by deleobuvir, CD 6168, and deleobuvir-AG at roughly equimolar concentrations, and Fig. 2D shows the fitting of kobs and kinact for CYP2C8 inactivation by these three perpetrators. Deleobuvir and deleobuvir-AG were also mild inactivators of CYP1A2 (Table 2).
Inactivation parameters KI (μM) and kinact (min−1) for time-dependent P450 inactivation by deleobuvir, CD 6168, and deleobuvir-AG

Time- and concentration-dependent inhibition of CYP2C8 by deleobuvir (A), CD 6168 (B), and deleobuvir-AG (C); kinetics of CYP2C8 inactivation by deleobuvir, CD 6168, and deleobuvir-AG at various concentrations (D) (one representative experiment from n = 2).
Time Course of mRNA Induction by Rifampicin.
The time course for maximal CYP3A4 mRNA induction was determined to validate the approach of using a shortened time course for generating induction parameters (EC50 and Emax) from human hepatocytes. At the prototypical incubation times of 48 or 72 hours, the Emax determination was limited by cytotoxicity of deleobuvir and CD 6168 (data not shown). The magnitude of mRNA induction for three lots of human hepatocytes treated with rifampicin (25 μM), a prototypical CYP3A4 inducer, after 2-, 4-, 8-, 12-, 24-, and 48-hour incubation is displayed in Fig. 3. In general, the donors reached maximal level of induction between 8 and 12 hours. The magnitude of CYP3A4 mRNA increases at these time points was consistent with the magnitude observed at 48 hours. To further validate this approach, induction parameters and EC50 and Emax values were determined for prototypical inducers [omeprazole (0, 0.3, 1, 3, 10, 30, 100, and 300 μM), phenobarbital (0, 3, 10, 30, 100, 300, 1000, and 3000 μM), and rifampicin (0, 0.3, 1, 3, 10, 30, 100, and 300 μM)] after 10-hour incubation (Fig. 4), and DDI for a CYP3A4 substrate was predicted by the net effect (static) model. The predicted AUCi/AUC ratio was similar to that reported using induction parameters from 48- to 72-hour incubations (calculations not shown) (Fahmi et al., 2008a,b; Shou et al., 2008).

Time course of mRNA induction after rifampicin (25 μM) treatment.

Concentration-dependent increase in mRNA by prototypical P450 inducers after 10-hour incubation (squares, donor Hu8123; diamonds, donor Hu1424; circles, donor 1419).
Cytotoxicity Results.
No notable changes in LDH or MTT assay (<15% of control values) were seen at 8, 10, and 12 hours at concentrations up to 100 μM of deleobuvir, CD 6168, or deleobuvir-AG. At later time points (24 and 48 hours) morphologic changes, increased LDH leakage (>50% of control values), and decreased expression of housekeeping gene β-actin were noted for deleobuvir and its metabolites at concentrations above 3 μM. Specifically, at 24 hours, 100 μM CD 6168 caused an 80% reduction in mitochondrial activity and at 48 hours a >30% reduction was observed at concentrations ≥3 μM.
mRNA Induction by Deleobuvir, CD 6168, and Deleobuvir-AG.
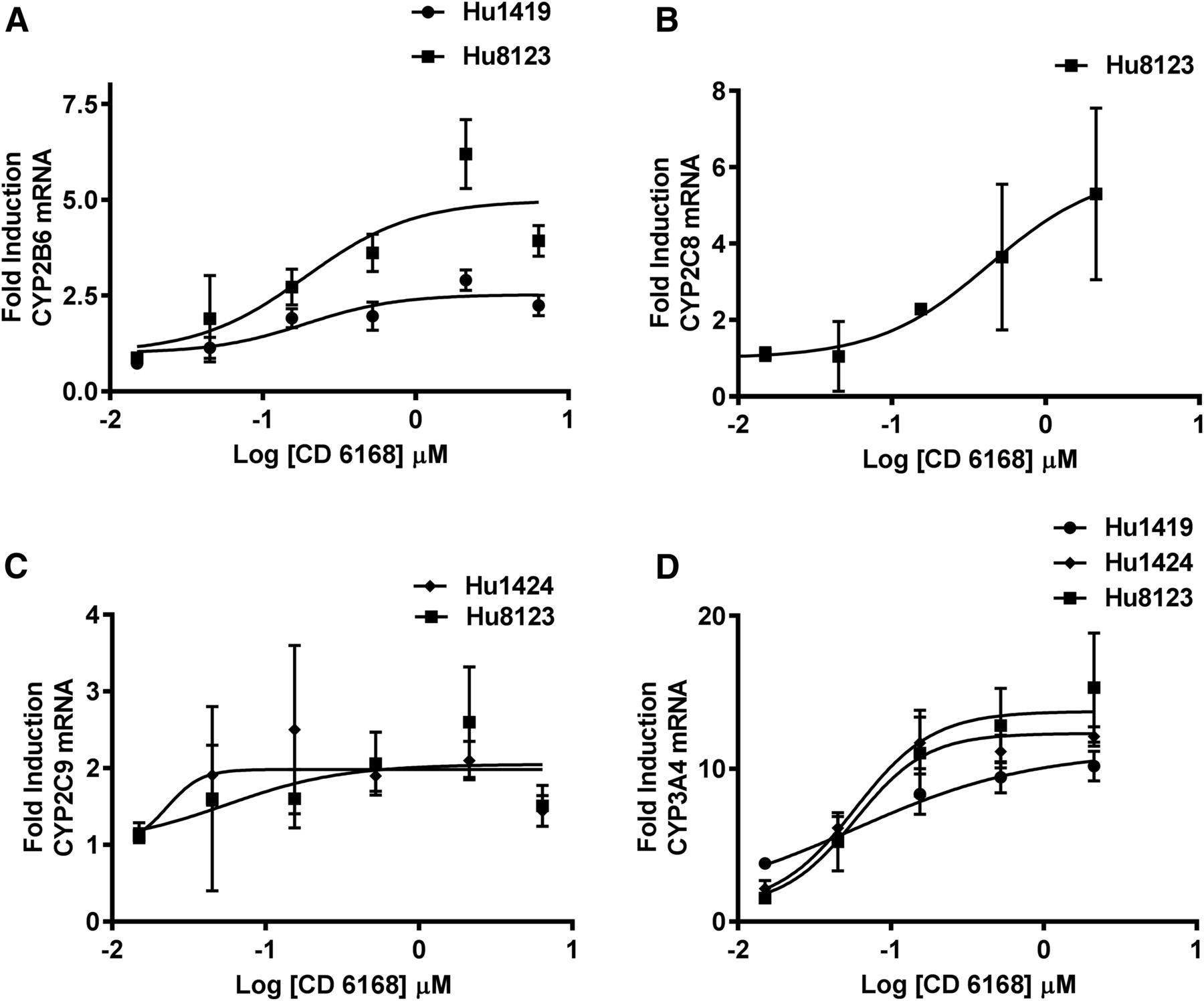
The induction potential of various concentrations of deleobuvir, CD 6168, and deleobuvir-AG toward CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A4 mRNA was investigated after incubation for 10 hours. Deleobuvir did not cause an increase in mRNA for any of the P450 isoforms tested, with the possible exception of CYP3A4, although increases were mild, inconsistent, not concentration-dependent, and not statistically significant and so are not considered clinically relevant. CD 6168 treatment resulted in concentration-dependent increases in CYP2B6, CYP2C8, CYP2C9, and CYP3A4 mRNA in one, two, or all three of the hepatocyte donors evaluated, as shown in Fig. 5, A–D. The increase was statistically significant (α = 0.05) at concentrations ≥1 μM for CYP2B6 and CYP2C8 in hepatocyte donors who showed induction. For CYP2C9, statistical significance was achieved at concentrations ≥0.3 μM for donor 1424 and at ≥ 3μM for donor 8123. For CYP3A4, all donors exhibited statistically significant induction at concentrations ≥0.3 μM. There was no induction observed for CYP1A2 by CD 6168. Deleobuvir-AG resulted in a statistically significant but mild increase in CYP1A2 mRNA in hepatocytes from one out of three donors. Maximal induction response in this donor compared with the prototypical inducer (omeprazole, 50 μM) was 30%. Table 3 summarizes Emax and EC50 parameters for deleobuvir metabolites as well as the prototypical inducers in hepatocyte donors where data fitting was possible. Induction parameters were derived using the unbound concentrations of the compounds in induction culture media.

Concentration-dependent increase in mRNA by CD 6168 after 10-hour incubation.
Median induction parameters EC50 (μM) and Emax from mRNA levels after treatment with CD 6168 in three human hepatocyte donors
Protein Binding Determinations in HLM, Human Intestinal Microsomes, Hepatocyte Medium and Human Plasma.
Deleobuvir and its major metabolites demonstrated significant plasma protein binding that was not saturable up to the concentrations evaluated. Average human plasma protein binding was 99.3% for deleobuvir and CD 6168 and 99.6% for deleobuvir-AG. Deleobuvir and its metabolites exhibited similar binding in HLM, which ranged from 25 to 36% at 0.05 mg/ml to 67 to 78% at 0.5 mg/ml of microsomal protein. At the concentrations tested, CD 6168 was ∼85% bound in the induction assay medium. Bound concentration of deleobuvir-AG in the induction assay medium was calculated to be ∼89% on the basis of the plasma protein binding data and interpolation to albumin content in the induction assay medium. This approach was only used for deleobuvir-AG after evaluation with CD 6168 and confirmation that such extrapolated protein binding values were close to the experimentally determined values. Owing to the low magnitude of induction observed with deleobuvir-AG, this was considered to be an acceptable approximation of protein binding for in vivo DDI prediction. Inhibition, inactivation, and induction parameters were corrected for protein binding using the values for the appropriate matrix.
Safety Profile of the Clinical Study.
Deleobuvir plus ribavirin and two doses of pegylated-interferon α-2a within the first 8 days of dosing were relatively well tolerated with no deaths or serious adverse events. Following the 8-day DDI assessment, faldaprevir was added to the treatment, with treatment continuing over 24 weeks. Two patients interrupted dosing prematurely owing to adverse events (febrile neutropenic dermatosis at dosing day 9; grade 2 hyperbilirubinemia and abdominal discomfort at dosing day 24). Both participants fully recovered. Nausea (36.8–37.5%), headache (15.8–18.8%), fatigue (12.5–15.8%), and photosensitivity reaction (10.5–31.1%) were the most commonly observed adverse events over the 24-week duration of dosing. As recognized in previous studies, there is considerable overlap in side effect profile between faldaprevir and deleobuvir (SOUND C-2; Zeuzem et al., 2013). Furthermore, it is challenging to disentangle the contribution of ribavirin and early pegylated interferon exposure to the observed side effect profile.
In Vivo DDI with Caffeine, Tolbutamide, and Midazolam and Plasma Exposure of Deleobuvir, CD 6168, and Deleobuvir-AG.
When caffeine was coadministered with deleobuvir on day 9, caffeine Cmax increased approximately 39% and AUC0–∞ increased approximately 64%, compared with day 1 (P450 probes alone) (Table 4). When tolbutamide was coadministered with deleobuvir, tolbutamide Cmax and AUC0–∞ decreased approximately 8 and 14%, respectively, compared with day 1. There was a small increase in 4-OH-tolbutamide/tolbutamide AUC ratio compared with day 1 (Table 4). When midazolam was coadministered with deleobuvir, midazolam Cmax and AUC0–∞ increased approximately 24 and 23%, respectively, compared with day 1. There was an ∼10% decrease in 1-OH-midazolam/midazolam AUC ratio compared with day 1 (Table 4).
Adjusted geometric means (%gCV) for pharmacokinetic parameters and relative bioavailability of caffeine, tolbutamide, 4-OH tolbutamide, midazolam, and 4-OH midazolam in HCV-infected patients before (day 1) and after (day 9) treatment of 8 days with 600 mg TID deleobuvir
Net Effect Modeling and Comparison with Clinical Outcome.
The static model described in the EMA (2012) and FDA (2012) guidances on DDI was used to predict the clinical outcome of deleobuvir treatment on sensitive substrates of CYP1A2, CYP2C9, and CYP3A4 and the predictions were retrospectively compared with the in vivo results from the clinical DDI study described above. The maximal plasma concentrations obtained in this study (Table 5) were used to calculate the inlet portal concentrations for deleobuvir and CD 6168, which were then used in the net effect prediction. Maximal plasma concentrations for deleobuvir-AG were directly used in the predictions. This distinction was made because the glucuronide metabolite is formed in the hepatocytes and thus portal vein concentrations were not expected to be higher than maximal plasma concentrations. Since CD 6168 is formed presystemically in the gastrointestinal tract by gut bacteria, it was regarded as administration of a second parent drug (Chen et al., 2015; McCabe et al., 2015). The fraction absorbed for deleobuvir was considered to be 0.5, as an absorption, distribution, metabolism, and excretion (ADME) study showed ∼50% of the radioactivity in feces was accounted for by CD 6168 and its metabolites (Chen et al., 2015). Since deleobuvir-AG is predominantly formed in the liver and any amount eliminated in the bile is hydrolyzed back to deleobuvir in the GI tract, the contribution of deleobuvir-AG to DDI in the GI tract was deemed to be negligible. The in vitro inhibition, inactivation, and induction parameters used were corrected for protein binding. The resulting projections and corresponding in vivo results are displayed in Table 6. There was an overprediction of AUC changes for both CYP1A2 and CYP2C9 when considering deleobuvir, CD 6168, and deleobuvir-AG together. For CYP3A4, the prediction using parameters for deleobuvir alone indicated a significant increase in AUC (6.15-fold), whereas including CD 6168 and deleobuvir-AG predicted no change in AUC (0.97-fold), which agree well with the actual clinical data (1.23-fold).
Pharmacokinetic parameters for deleobuvir, CD 6168, and deleobuvir-AG on day 9
N = 17 patients. AUCτ,ss and Cmax,ss were derived from concentration-time data for the first dosing interval of the day, where τ = 6 hours
Predicted exposure changes using the net effect model and observed exposure changes for CYP1A2, CYP2C9, and CYP3A4 probe substrates
Discussion
Regulatory guidances from the FDA and EMA have proposed that metabolites present at greater than 25% of the parent molecule or greater than 10% of drug-related material (EMA) should be investigated in vitro for DDI potential (EMA, 2012; FDA, 2012). In the current clinical study, in HCV-infected patients, following 8 days of BID dosing at 600 mg of deleobuvir, CD 6168, a reduction product formed by gut bacteria (McCabe et al., 2015), and an acyl glucuronide of deleobuvir (deleobuvir-AG) (Chen et al., 2015) were circulating at peak exposures of 59 and 32% of the parent deleobuvir, respectively. CD 6168 exposure is higher upon multiple dosing despite a short half-life (Chen et al., 2015), probably owing to the unique site of formation in the GI tract. In vitro studies were conducted to determine the inhibition, inactivation, and induction of P450s by deleobuvir and its two major metabolites. A number of contrasting outcomes were found. First, several P450 isoforms were affected in opposing directions, i.e., increases in enzyme activity by induction or decreases in activity by inhibition or inactivation. Second, deleobuvir metabolites were more potent than deleobuvir, especially in the case of CYP2C8 inactivation (deleobuvir-AG) and P450 induction (CD 6168). Deleobuvir and CD 6168 (reduction of an alkene), which are structurally very similar, inhibited and inactivated P450 enzymes with similar potencies (Tables 1 and 2). Deleobuvir-AG inactivated CYP2C8 more potently than parent, providing another example of an acyl glucuronide that is a potent inactivator of CYP2C8, as has been shown with gemfibrozil (Ogilvie et al., 2006) and clopidogrel (Tornio et al., 2014). The exact mechanism of the inactivation with deleobuvir-AG is not known at this time. A comparison of the kinact/KI ratios (shown in parentheses) indicates that deleobuvir-AG (1.00 minute−1 ⋅ μM−1) is a more potent inactivator than gemfibrozil (0.0105 minutes−1 ⋅ μM−1) and clopidogrel (0.0047 minutes−1 ⋅ μM−1) in vitro. However, additional factors have to be considered to translate this to their potential for in vivo inactivation, including relevance of individual parameters of the ratio, fm of the comedication and perpetrator concentration, and fm for the relevant pathway.
Additionally, deleobuvir-AG also inhibited CYP1A2 and CYP2D6 more potently than deleobuvir and inhibited CYP2B6 with a similar potency as parent (Table 1). The competitive inhibition of CYP1A2 by deleobuvir-AG was somewhat surprising since CYP1A2 is considered to have an active site suitable for planar molecules (Zhou et al., 2009). However, on the basis of the static model for DDI prediction, the overall impact on CYP1A2 activity in vivo was driven by inactivation by deleobuvir, rather than inhibition by deleobuvir-AG (Table 6).
Alternative perspectives to regulatory guidances on the potential of a metabolite to inhibit or inactivate P450s have been discussed (Callegari et al., 2013; Yu and Tweedie, 2013; Yu et al., 2015). However, these do not address induction of P450s by metabolites. Structurally similar metabolites have been shown to possess similar or lower potency of induction (Petzer et al., 2003; Medina-Diaz et al., 2009). One recent report highlighted carboxymefloquine as being a pregnane X receptor (PXR) ligand, whereas the parent mefloquine was not (Piedade et al., 2015). With deleobuvir, reduction of an alkene to form CD 6168 resulted in a potent inducer. In an hepatocyte induction assay, under standard experimental conditions, metabolites can be formed from parent drug, and as such the induction liability of the metabolite is also being considered, with the obvious caveat of differential levels in vitro and in vivo. However, CD 6168 is formed presystemically by gut bacteria and is not generated in the liver (McCabe et al., 2015) and would therefore not be represented by adding deleobuvir alone in these induction studies. Deleobuvir-AG would be formed during incubation of hepatocytes with deleobuvir itself, but the extent may be lower than that observed in vivo (Chen et al., 2015). Therefore, it was rational to conduct induction studies in which deleobuvir, deleobuvir-AG, and CD 6168 were individually assessed.
The generally accepted standard methodologies for evaluation of induction in human hepatocytes employ incubation times of 48 or 72 hours (Lin, 2006; Sinz et al., 2008; Chu et al., 2009). Deleobuvir and its metabolites were cytotoxic to sandwich cultured hepatocytes at concentrations above 1 μM upon incubation for 48 hours. Elevation of mRNA through upregulation by nuclear receptors is the initial event leading to increases in protein and can be detected within 4–6 hours (Zhang et al., 2010). Validation of shorter incubation times was done using a two-step approach. First, the optimal time course was established by monitoring CYP3A4 mRNA increases in hepatocytes by rifampicin at various time points after treatment (Fig. 3). Second, at the optimal time point selected (10 hours), induction parameters for prototypical inducers (omeprazole for CYP1A2, phenobarbital for CYP2B6, and rifampicin for CYP3A4) were found to be comparable to those reported in the literature (Fahmi et al., 2008a,b, 2009, 2010; Shou et al., 2008; Zhang et al., 2010). Under these validated experimental conditions, deleobuvir and deleobuvir-AG did not induce P450 enzymes. Strikingly, CD 6168, which is structurally very similar to deleobuvir, was a potent inducer of CYP3A4 (Table 3). CD 6168 also induced other PXR/constitutive androstane receptor (CAR) target genes, namely CYP2B6, CYP2C8, and CYP2C9 (Fig. 5) with a rank order of induction similar to that observed with the PXR agonist rifampicin, suggesting that induction via CD 6168 may also be via PXR (and possibly CAR). Additional studies are required to fully define the differential interactions of deleobuvir and CD 6168 with upstream effectors such as PXR and CAR. The dearth of reports of induction by metabolites, but not the parent drug, such as observed with CD 6168 and carboxymefloquine, suggests the need for additional consideration of metabolite DDI via induction, beyond those proposed by pharmaceutical researchers (Yu et al., 2015) and regulators.
Thus, the overall DDI prediction of deleobuvir required an evaluation of the DDI potential of the metabolites. On the basis of in vitro data for deleobuvir alone, the inhibition potential would have been markedly higher than the actual DDI observed in the clinic for CYP3A4 (Table 6). Inactivation of CYP3A4 clearly dominated the net effect prediction for deleobuvir since it is not an inducer. Induction of CYP3A4 by CD 6168 counteracted the inactivation effect of deleobuvir. As a result, the net effect at steady state was predicted to be a lack of an overall effect on CYP3A4. The situation was somewhat different for CYP2C9, as deleobuvir and its metabolites do not inactivate CYP2C9. While the tolbutamide exposure tended to be lower after deleobuvir dosing, the effect was not statistically significant. Thus, the model overpredicted the induction potential of CYP2C9. For CYP1A2, there was a mild overprediction of the inhibitory effects, but the DDI prediction was not very different when deleobuvir was considered with or without metabolites. This is not surprising as induction of CYP1A2 was not observed with CD 6168 and only a weak induction was observed with deleobuvir-AG. Thus, the overall DDI prediction for CYP3A4, when all perpetrators were considered, was close to that observed in vivo, whereas the model overpredicted the induction effect for CYP2C9 and the inhibitory effect for CYP1A2. The net effect model, developed primarily using in vitro and in vivo data for CYP3A4 (Fahmi et al., 2008b), may need further refinement for other P450 isoforms. Alternative approaches using systemic concentrations for inhibition and inactivation, or more realistic fm and Fg values, may improve the prediction for CYP2C9 and CYP1A2. While the use of a static model has not been frequently reported for highly complex DDI predictions involving multiple perpetrators, particularly with opposing effects on the enzymes, the model appeared to be highly valuable for deleobuvir. Additional examples will further increase the confidence in this approach. Deleobuvir and its metabolites exhibited a tendency to accumulate in cultured hepatocytes by up to 20-fold (Chen et al., 2015), suggesting a potential role for organic anion-transporting polypeptide (OATP)–mediated hepatic uptake. While FDA DDI guidance only recommends the use of unbound concentrations in the portal vein for DDI predictions, EMA DDI guidance recommends that the liver accumulation be taken into consideration for the predictions. Assuming intracellular protein binding is the same as plasma protein binding when ∼20-fold higher perpetrator concentrations were used in the predictions, inhibition and inactivation of caffeine (CYP1A2) and midazolam (CYP3A) were significantly overpredicted, but tolbutamide (CYP2C9) and CYP2C8 substrates repaglinide and cerivastatin had only minor differences (calculations not shown). This difference may be attributable primarily to somewhat lower fm values of tolbutamide, repaglinide, and cerivastatin compared with caffeine and midazolam (fm values are referenced in (Supplemental Table 5). Owing to the mild nature of the DDI observed in the clinical study, comedications that are CYP1A2, CYP2C9, and CYP3A4 substrates were not restricted from use in the Phase 3 studies. Although the prediction for CYP1A2 and CYP2C9 was less accurate than for CYP3A4, the predictions for the former isoforms were not unacceptable. Also, using this approach, a moderate-to-high increase in AUC of 2.2-fold and 5.5-fold, respectively, was predicted for CYP2C8 substrates repaglinide [fm,CYP2C8 ∼0.51 (Säll et al., 2012)] and cerivastatin [fm CYP2C8 ∼0.82; considered as 1 – fm,CYP3A4 (Ohno et al., 2007)], whereas a net induction was predicted for CYP2B6 substrate efavirenz, a known autoinducer. The effect on these enzymes was not evaluated clinically because relatively few drugs are metabolized by them. Appropriate restrictions were implemented during larger patient studies for comedications that are highly cleared by CYP2B6 and CYP2C8. Further consideration is being given to the possibility that deleobuvir and/or its metabolites could be substrates and inhibitors of OATP1B1 and OATP1B3. If deleobuvir and its metabolites show a potential for an OATP-based interaction, an even higher interaction is possible with dual substrates of CYP2C8 and OATPs, such as repaglinide and simvastatin, than that predicted by the static model, which only considers interaction via CYP2C8.
Owing to ethical considerations, the current study was conducted in HCV-infected patients, which imposed inherent design limitations. As such, the effect of a single dose of deleobuvir on P450 probe substrates was not evaluated. Consequently, the impact of only competitive inhibition was not determined. This limited the validation of the static model for predictability of inhibition, which would have been determined following one or two doses, since deleobuvir has a relatively short half-life of 2.84 hours (Chen et al., 2015). Induction and inactivation require steady state to be reached as well as optimization of biologic processes (de novo generation of protein for induction) for a net steady-state effect, which was evaluated in this study after 8 days of dosing with deleobuvir.
In conclusion, there are two interesting aspects of deleobuvir in which metabolites significantly contribute to the DDI liability of the parent drug. First, deleobuvir-AG provides another example of an acyl glucuronide that inactivates CYP2C8 more potently than the parent. Second, CD 6168 represents a structurally similar presystemic metabolite that induced CYP2C9 and CYP3A4, whereas the parent deleobuvir exhibited no induction. Combining the DDI liabilities of deleobuvir, such as inactivation of CYP3A4 and inhibition of CYP2C9, together with the DDI liabilities of the metabolites was essential for determining the net effect on these P450 isoforms.
Acknowledgments
The authors thank Monica Keith-Luzzi and Kathy Phelan for conducting protein binding studies, Dr. Timothy Tracy for scientific advice, Karen Kimura for operational aspects of the clinical study, investigating physicians and last but not the least, the patients who participated in this study.
Authorship Contributions
Participated in research design: Sane, Ramsden, Sabo.
Conducted experiments: Ramsden, Rowland, Whitcher, Cooper.
Performed data analysis: Ting, Ramsden.
Wrote or contributed to the writing of the manuscript: Sane, Ramsden, Sabo, Tweedie.
Footnotes
- Received September 4, 2015.
- Accepted December 17, 2015.
↵This article has supplemental material available at dmd.aspetjournals.org.
Abbreviations
- AUC
- area under the concentration time curve
- Cmax
- maximum plasma concentration
- CAR
- constitutive androstane receptor
- DDI
- drug-drug interaction
- deleobuvir-AG
- deleobuvir-acyl glucuronide
- GI
- gastrointestinal
- HCV
- hepatitis C virus
- HLM
- human liver microsomes
- LC-MS/MS
- high-performance liquid chromatography–tandem mass spectrometry
- LDH
- lactate dehydrogenase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- OATP
- organic anion-transporting polypeptide
- P450
- cytochromes P450
- PEG
- polyethylene glycol
- PXR
- pregnane X receptor
- QD
- every day (or daily)
- r
- recombinant
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}