


Abstract
Repaglinide is presently recommended by the U.S. Food and Drug Administration as a clinical CYP2C8 probe, yet current in vitro and clinical data are inconsistent concerning the role of this enzyme in repaglinide elimination. The aim of the current study was to perform a comprehensive investigation of repaglinide metabolic pathways and assess their contribution to the overall clearance. Formation of four repaglinide metabolites was characterized using in vitro systems with differential complexity. Full kinetic profiles for the formation of M1, M2, M4, and repaglinide glucuronide were obtained in pooled cryopreserved human hepatocytes, human liver microsomes, human S9 fractions, and recombinant cytochrome P450 enzymes. Distinct differences in clearance ratios were observed between CYP3A4 and CYP2C8 for M1 and M4 formation, resulting in a 60-fold M1/M4 ratio in recombinant (r) CYP3A4, in contrast to 0.05 in rCYP2C8. Unbound Km values were within 2-fold for each metabolite across all in vitro systems investigated. A major system difference was seen in clearances for the formation of M2, which is suggested to be a main metabolite of repaglinide in vivo. An approximately 7-fold higher unbound intrinsic clearance was observed in hepatocytes and S9 fractions in comparison to microsomes; the involvement of aldehyde dehydrogenase in M2 formation was shown for the first time. This systematic analysis revealed a comparable in vitro contribution from CYP2C8 and CYP3A4 to the metabolism of repaglinide (<50%), whereas the contribution of glucuronidation ranged from 2 to 20%, depending on the in vitro system used. The repaglinide M4 metabolic pathway is proposed as a specific CYP2C8 probe for the assessment of drug-drug interactions.
Introduction
Repaglinide is an orally administered insulin secretagog commonly prescribed to patients with type 2 diabetes. The drug is rapidly absorbed and has a half-life of 1 to 2 h, making it a suitable short-acting antidiabetic drug (Bidstrup et al., 2003). Repaglinide metabolism is complex, and six different metabolites have been reported (Fig. 1) (van Heiningen et al., 1999; Bidstrup et al., 2003). The metabolites have no hypoglycemic effect and are mainly excreted via bile into feces (van Heiningen et al., 1999). Involvement of CYP2C8 and CYP3A4 has been reported in vitro, as well as in vivo (Bidstrup et al., 2003; Niemi et al., 2003, 2004; Kajosaari et al., 2005), but the relative contribution of the two enzymes remains unclear.

Structure of repaglinide and its metabolic pathways showing enzymes responsible for the conversion and proposed mechanism for the formation of M2.
Inconsistencies were evident for in vitro data and clinical studies in terms of the importance of the repaglinide metabolic pathways. M1 and M4 are commonly quoted as the main in vitro metabolites (Bidstrup et al., 2003; Kajosaari et al., 2005), whereas a human radiolabel study reported 66% of the administered dose excreted as M2 (van Heiningen et al., 1999). The formation of M2 has been proposed to be a multistep reaction involving initially cytochrome P450 (P450) enzymes, followed by unidentified cytosolic enzymes (Gan et al., 2010). Cytosolic enzymes have recently received increased interest for their role in drug metabolism, and potential misinterpretation of in vitro data as a consequence of ignoring these enzymes has been highlighted (Pryde et al., 2010; Cubitt et al., 2011). Aldehyde oxidase is one of the cytosolic enzymes recognized to be of increasing relevance with newly developed drugs (Zientek et al., 2010; Hutzler et al., 2012), in particular for heterocycle-containing compounds (Garattini et al., 2008; Pryde et al., 2010). In contrast, aldehyde dehydrogenase, another cytosolic enzyme, has shown high affinity toward aliphatic aldehydes (Panoutsopoulos et al., 2004) and has been often involved in tandem with initial P450 oxidation (Salva et al., 2003; Ekhart et al., 2008). Aldehyde oxidase and aldehyde dehydrogenase were both investigated in the current study as potential enzymes involved in the formation of repaglinide M2 metabolite.
Furthermore, acyl glucuronidation has been proposed as an important metabolic pathway of repaglinide with UGT1A1 reported as the major contributing enzyme (Gan et al., 2010). The pharmacokinetics of repaglinide is further complicated by the active hepatic uptake of this drug (Ménochet et al., 2012), mediated by organic anion-transporting polypeptide (OATP) 1B1 uptake transporter (Niemi et al., 2005; Kalliokoski et al., 2008b).
CYP2C8 metabolizes a wide range of drugs, showing overlapping substrate specificity with CYP3A4 (Hinton et al., 2008; Lai et al., 2009; Naraharisetti et al., 2010). Paclitaxel, pioglitazone, verapamil, amiodarone, and cerivastatin represent examples of drugs with a dual contribution of CYP2C8 and CYP3A4 (Lai et al., 2009). Assessing a potential of a new chemical entity to inhibit CYP2C8 during drug development is challenging (Walsky et al., 2005), in particular considering the overlap in specificity of the probe substrates and difficulties in determining an accurate fraction metabolized via P450 metabolism (fmCYP) for these drugs. A number of studies have emphasized the importance of fmCYP estimates for accurate prediction of drug-drug interactions (DDIs) and reduction in the number of false-positive results (Galetin et al., 2006; Hinton et al., 2008). The latest FDA guidance document for DDI studies recommends repaglinide and paclitaxel as in vivo probes for investigating CYP2C8 inhibition (FDA Guidance for Industry 2012: Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf). Because of increased risk of cardiovascular events, the use of rosiglitazone, as was used previously, is now restricted (http://www.fda.gov/Drugs/DrugSafety/ucm255005.htm), highlighting the need for a reliable CYP2C8 probe.
The overall aim of the current study was to perform a systematic analysis of repaglinide metabolic pathways and thereby assess the contribution of specific enzymes to its clearance. The formation of repaglinide metabolites was characterized using a range of in vitro systems, namely pooled cryopreserved human hepatocytes, human liver microsomes (HLM), human S9 fractions and recombinant (r) P450 enzymes. The impact of in vitro systems on the assessment of repaglinide metabolic pathways was studied and the importance of individual metabolic pathways was evaluated. In addition, inhibition experiments in the presence of selective inhibitors (gemfibrozil glucuronide, ketoconazole, disulfiram, and raloxifene) were performed to delineate the contribution of particular metabolic pathways, in particular M2, for which current in vitro data do not support its relevance reported in vivo. The implications of these findings on the appropriateness of repaglinide as a probe substrate for CYP2C8 and interpretation of associated DDIs are discussed.
Materials and Methods
Chemicals.
2-Despiperidyl-2-amino repaglinide (M1), 2-despiperidyl-2-(5-carboxypentylamine) repaglinide (M2), 3′-hydroxy repaglinide (M4) (mixtures of diastereomers), repaglinide acyl-β-d-glucuronide, and gemfibrozil 1-O-β-glucuronide were purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). All solvents were obtained from BDH Laboratory Supplies (VWR International, Lutterworth, Leicestershire, UK). All other compounds and reagents were purchased from Sigma-Aldrich Company Ltd. (Poole, Dorset, UK).
Source of In Vitro Systems.
Recombinant human CYP3A4, CYP3A5, and CYP2C8 expressed with cytochrome P450 oxidoreductase in baculovirus-infected insect cells (Supersomes) were purchased from BD Gentest (Woburn, MA). Pooled HLM equal gender mix from 150 donors was obtained from BD Gentest. CYP3A4-catalyzed testosterone 6β-hydroxylation, CYP2C8-catalyzed paclitaxel 6α-hydroxylation, and UGT1A1-catalyzed estradiol 3-glucuronidation activities reported in this HLM pool were 5300, 260, and 930 pmol · min−1 · mg protein−1, respectively. Pooled human S9 fractions were purchased from BD Gentest and obtained from the same donor pool as HLM. CYP3A4-catalyzed testosterone 6β-hydroxylation and CYP2C8-catalyzed paclitaxel 6α-hydroxylation activity in S9 fractions were 1200 and 70 pmol · min−1 · mg protein−1, respectively. No information on UGT activity was provided in S9 fractions. Single-freeze pooled cryopreserved human hepatocytes from 20 donors were obtained from XenoTech, LLC (Kansas City, KS). Reported enzyme activities in hepatocytes were as follows: CYP3A4 testosterone 6β-hydroxylation, CYP2C8 amodiaquine N-dealkylation, and UGT 7-hydroxycoumarin glucuronidation activities were 367, 295 and 733 pmol · min−1 · 106 cells−1, respectively.
General Incubation Conditions.
All experiments were performed in duplicate on three separate occasions unless stated otherwise. The final concentration of the organic solvent (methanol) in the incubation mixes did not exceed 1% v/v. All reactions were terminated by addition of ice-cold methanol containing internal standard (indomethacin), and control incubations without enzyme or without cofactors were performed in parallel. Formation of repaglinide metabolites was measured by liquid chromatography (LC)-tandem mass spectrometry (MS/MS) as described below. For formation studies, time and protein linearity studies were performed before kinetic studies to optimize the final conditions used. Details on protein concentrations, estimated unbound fraction and incubation time used in the current study are listed in Table 1.
Incubation conditions for experiments performed in pooled human hepatocytes, human liver microsomes, human S9 fractions, and recombinant P450 enzymes
Repaglinide Metabolite Formation in Recombinant Enzymes, HLM, and S9.
Individual incubation mixtures consisting of repaglinide at various concentrations (eight concentrations ranging from 0.5 to 150 μM) were prepared in a 96-deep-well microtiter plate together with either (A) 0.1 M phosphate buffer, pH 7.4, 10 mM MgCl2, and an NADPH-regenerating system (1 mM NADP+, 7.5 mM isocitric acid, and isocitric dehydrogenase at 1 unit/ml) for P450 metabolite formation or (B) 0.1 M phosphate buffer, pH 7.1 (containing 3.45 mM MgCl2, 1.15 mM EDTA, and 115 μM saccharic acid lactone), UDP-glucuronic acid (final concentration of 5 mM) for UGT metabolite formation. The plate was placed on a shaker at 37°C, and the appropriate enzyme source was thawed and put on ice. Thawed HLM and S9 were activated by incubation on ice with alamethicin (50 μg/mg protein) for 15 min before the incubation for the UGT formation experiment. Reactions were initiated by the addition of the enzyme. Conditions for S9 were adapted from Dalvie et al. (2009) and UGT formation from Kilford et al. (2009).
Repaglinide Metabolite Formation in Human Hepatocytes.
Pooled cryopreserved human hepatocytes (lot 1010270) were stored in liquid nitrogen until required. Before experiments, cells were thawed rapidly in a water bath at 37°C. Thawed cells were carefully poured and resuspended into prewarmed Williams' Medium E at pH 7.4 and 37°C. The cell suspension was centrifuged at 50g for 5 min at room temperature. The formed supernatant was gently removed, leaving only the cell-containing pellet. The cells were resuspended in an appropriate volume of Krebs-Henseleit buffer at pH 7.4 and 37°C. Total number of cells, concentration of the suspension, and cell viability were determined using a hemocytometer after staining with trypan blue; minimum viability for all experiments was 65%. The cell suspension was subsequently diluted with Krebs-Henseleit buffer to the required concentration for the experiment (giving a final incubation concentration of 0.4 × 106 cells/ml) and thereafter preincubated at 37°C for 5 min. Repaglinide solutions (in methanol) were diluted in Krebs-Henseleit buffer and preincubated at 37°C for 5 min. Incubations were initiated by the addition of hepatocyte suspension to repaglinide solutions in a 96-well flat-bottom polystyrene microplate, followed by gentle swirling in an atmosphere of 5% CO2. All incubations were performed in triplicate on three separate occasions.
Effect of Inhibitors on Metabolite Formation in Pooled S9 Fractions.
Individual incubation mixtures were prepared with S9 fractions in a manner similar to that described in Repaglinide Metabolite Formation in Recombinant Enzymes, HLM, and S9, with the addition of inhibitors. The rationale for inhibitor selection and corresponding concentrations was to ensure inhibition of one metabolic pathway of repaglinide at the time. The selected inhibitors were gemfibrozil glucuronide (75 μM) (Ogilvie et al., 2006), ketoconazole (0.2 μM, selected concentration is expected to have minimal inhibitory effect on CYP2C8) (Ong et al., 2000; Shitara et al., 2003), disulfiram (10 μM) (Hu et al., 1997; Salva et al., 2003), and raloxifene (0.05 μM) (Obach et al., 2004). Raloxifene has also been shown to be a time-dependent inhibitor of CYP3A4, resulting in KI estimates approximating 10 μM (Chen et al., 2002). A raloxifene concentration of 0.05 μM was therefore chosen to avoid any confounding effects because of CYP3A4 inhibition. For gemfibrozil glucuronide, preincubation with NADPH was performed for 30 min before the addition of repaglinide to allow formation of the hydroxylated glucuronide metabolite and irreversible inhibition of CYP2C8 as reported previously (Ogilvie et al., 2006).
Disulfiram IC50 Determination.
IC50 values were determined in S9 fractions for disulfiram to further confirm the involvement of aldehyde dehydrogenase in the formation of M2. An NADPH-regenerating system (as described above), 0.1 M phosphate buffer, pH 7.4, and repaglinide at concentrations of 10, 25, and 50 μM were prepared in a 96-deep-well microtiter plate. Repaglinide concentrations were selected to include a range of concentrations (1/2 Km − 2 × Km) encompassing the Km for the formation of M2 metabolite in S9 (see Results). Seven disulfiram concentrations evenly spread on a log scale (ranging from 0.1 to 100 μM) were added to the incubation. The plate was placed on a shaker at 37°C, and the reaction was initiated by the addition of S9.
LC-MS/MS Method.
Incubation samples (methanol quenched) were centrifuged for 10 min at 2500 rpm, the supernatants transferred to LC autosampler vials, and aliquots (10 μl) were injected into an LC-MS/MS system. Metabolites were separated on a Luna C18 column ((3 μm, 4.6 × 50 mm; Phenomenex, Macclesfield, UK) by gradient elution at 1 ml/min using an Alliance 2795 liquid chromatograph (Waters, Watford, UK). Run conditions were as follows: 100% solvent A (90% water and 1 mM ammonium acetate with 10% methanol) for the first 2 min, 90% solvent B (10% water and 1 mM ammonium acetate with 90% methanol) and 10% solvent C (10% water and 0.05% formic acid with 90% methanol) between minute 2 and 3, followed by 100% solvent B for another minute. The system was allowed to re-equilibrate back to initial run conditions for 1 min. Split eluent (0.25 ml/min) was analyzed by electrospray atmospheric pressure ionization combined with multiple reaction monitoring of manually optimized product ions using a Micromass Quattro Ultima tandem mass spectrometer (Waters). Probe voltage was 3.25 kV (bases/neutrals) or 3.0 kV (acids); desolvation and source temperatures were 350 and 125°C, respectively; desolvation gas (nitrogen) and cone gas (nitrogen) flow rates were 600 and 150 l/h, respectively; collision gas (argon) pressure was 1.5 × 10−3 mbar. Multiple reaction monitoring conditions for individual analytes are given in Table 2. For each assay, two calibration standards, including a blank, were prepared in a matrix identical to the incubation extracts (to compensate for matrix interference), which included analyte at above and below the experimental concentrations (calibration range, 0.02–5 μM). Calibration standards were analyzed at the start and end of each analysis (to verify satisfactory stability and lack of any potential carryover). The ion chromatograms were integrated and quantified by quadratic regression of standard curves using MassLynx 4.1 (Waters). Concentration values were accepted if the internal standard ratio was greater than a value equal to the calibration regression intercept plus approximately 10 times the estimated between-run S.D. of the intercept (lower limit of quantification). Repeatability precision was considered adequate if duplicate sample values were within 10% of each other.
Summary of LC-MS/MS conditions for repaglinide metabolites investigated and internal standard with details on mass transitions and retention times
The LC-MS/MS method monitored M1, M2, M4, and repaglinide glucuronide simultaneously; quantification of actual concentrations of formed metabolites was possible because of the recent availability of metabolic standards for major repaglinide metabolites in contrast to earlier studies that relied on quantification of metabolites using arbitrary units (Kalliokoski et al., 2008a; Tornio et al., 2008). However, certain analytical challenges were evident. M0-OH is a minor metabolite formed via hydroxylation on the isopropyl moiety of repaglinide and had mass transition identical to that of M4 (formed by β-hydroxylation in the piperidine ring system of repaglinide). No reference compound was available for M0-OH at the time of the experiment, and there is a possibility of contamination of the estimation of the parameters for M4, because the separation of two metabolites was not possible. However, this was considered unlikely, because M0-OH has been reported to be a repaglinide metabolite of minor importance in vitro as well as in vivo (van Heiningen et al., 1999; Bidstrup et al., 2003).
Analysis of Repaglinide Metabolite Formation Data.
Obtained in vitro data for all four systems investigated were analyzed by nonlinear regression analysis using GraFit 5.0.10 (Erithacus Software Ltd., Horley, UK). Vmax and Km were estimated from metabolite formation experiments by fitting a standard Michaelis-Menten equation to the data obtained; the Vmax/Km ratio was used to calculate intrinsic clearance (CLint) for each metabolic pathway. To allow a direct comparison between in vitro systems, CLint values obtained were expressed per gram of liver using the following scaling factors; 120 × 106 cells/g liver for hepatocytes, 40 mg protein/g liver for HLM (Brown et al., 2007), and 96.1 mg protein/g liver for S9 (Watanabe et al., 2009).
Estimation of the Contributions of CYP2C8, CYP3A4, and Glucuronidation to the Metabolism of Repaglinide.
Clearance estimates for each metabolite in HLM, S9, and hepatocytes were incorporated in eqs. 1 to 3 to calculate fraction metabolized via CYP3A4 (fmCYP3A4), CYP2C8 (fmCYP2C8), and UGT enzymes (fmUGT) in vitro. The relative contribution of CYP3A4 (0.74) and CYP2C8 (0.26) to the formation of M2 was estimated from inhibition data obtained with ketoconazole and gemfibrozil glucuronide, assuming complete inhibition of CYP3A4 and CYP2C8, respectively, by these inhibitors:

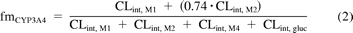
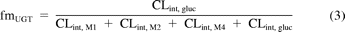 where CLint, gluc is the intrinsic clearance by glucuronidation, CLint, M1 is the intrinsic clearance via the repaglinide M1 pathway, CLint, M2 is the intrinsic clearance via the repaglinide M2 pathway, and CLint, M4 is the intrinsic clearance via the repaglinide M4 pathway.
where CLint, gluc is the intrinsic clearance by glucuronidation, CLint, M1 is the intrinsic clearance via the repaglinide M1 pathway, CLint, M2 is the intrinsic clearance via the repaglinide M2 pathway, and CLint, M4 is the intrinsic clearance via the repaglinide M4 pathway.
Analysis of Effect of Inhibitors on Repaglinide Metabolite Formation.
In vitro data obtained in the presence of four inhibitors were analyzed by nonlinear regression as described under Analysis of Repaglinide Metabolite Formation Data. The effects of ketoconazole, gemfibrozil glucuronide, disulfiram, and raloxifene were subsequently assessed by comparing CLint values obtained for individual metabolic pathways in control condition with estimates obtained in the presence of inhibitors investigated. In addition, estimated velocities for specific pathways were analyzed with and without selected inhibitors in relation to repaglinide concentration.
Analysis of Disulfiram Inhibition Data.
Equation 4 was fitted to the inhibition data obtained in S9 fractions to determine IC50 using GraFit 5.0.10. The formation of the metabolites at different disulfiram concentrations was expressed as a percentage of the control activity (incubations in the absence of inhibitor).
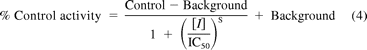 where “Control” is the fitted uninhibited value in the absence of inhibitor, “Background” is the activity observed with maximum inhibition, [I] is the inhibitor concentration, and S is the slope factor.
where “Control” is the fitted uninhibited value in the absence of inhibitor, “Background” is the activity observed with maximum inhibition, [I] is the inhibitor concentration, and S is the slope factor.
Correction for Nonspecific Protein Binding.
Relevant kinetic parameters were corrected for nonspecific protein binding. The binding in HLM, S9, and rP450 was estimated by eq. 5 using a value of 0.58 as the binding constant (Ka) (Gertz et al., 2008), assuming that binding in S9 and rP450 was comparable to that in HLM at the same protein concentrations. Fraction unbound in hepatocytes (fuhep) was estimated by eq. 6 (Kilford et al., 2008) where volume ratio (VR) accounted for binding to both viable and dead cells. Estimated fractions unbound for each in vitro system are shown in Table 1.
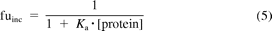 where fuinc is the fraction unbound from protein in the incubation and Ka is the microsomal binding constant.
where fuinc is the fraction unbound from protein in the incubation and Ka is the microsomal binding constant.
 where VR represents the volume ratio of hepatocytes to medium (0.005 for 1 × 106 cells/ml) and repaglinide logD7.4 is 2.6 (Mandic and Gabelica, 2006).
where VR represents the volume ratio of hepatocytes to medium (0.005 for 1 × 106 cells/ml) and repaglinide logD7.4 is 2.6 (Mandic and Gabelica, 2006).
Database of Repaglinide in In Vivo Inhibition Drug-Drug Interaction Studies.
Reported clinical DDI studies were collated, considering only studies after multiple oral administration of an inhibitor or inhibitor combination (assuming steady state was reached) before a single oral dose of repaglinide. The change in the area under the curve (AUC) in the presence and absence of the inhibitor was used as a metric to evaluate the magnitude of DDI. The dosing interval between the last dose of inhibitor and administration of repaglinide was noted. When reported, information on the changes in the metabolite AUC ratios as result of concomitant administration of an inhibitor was collated, as shown in Table 3. Herbal interaction studies were excluded from the database.
Repaglinide drug-drug interactions in vivo with details on main P450 enzymes involved, administered repaglinide dose, number of subjects, change in repaglinide AUC, and change in metabolite AUC ratios
Results
Formation of Repaglinide P450 Metabolites in a Range of In Vitro Systems.
Formation of repaglinide metabolites was characterized over a range of substrate concentrations in recombinant CYP2C8, CYP3A4, and CYP3A5 (Fig. 2). Kinetic profiles in rP450s followed standard Michaelis-Menten kinetics. Distinct differences in pathway ratios between CYP3A4 and CYP2C8 were observed for M1 and M4. Formation of the M4 hydroxy metabolite was 35-fold more pronounced in CYP2C8 than in CYP3A4. The opposite trend was seen for M1, resulting in 32-fold greater formation of this metabolite in CYP3A4 relative to that in CYP2C8. The results of this study confirm that CYP3A4 is the major enzyme contributing to the formation of M1, whereas M4 is predominantly metabolized by CYP2C8. Formation of M2 was below the limit of quantification (approximately 1 nM) in the recombinant P450 enzymes investigated. The contribution of CYP3A5 was negligible in comparison with that of the other enzymes (CYP2C8/CYP3A5 and CYP3A4/CYP3A5 CLint ratios were 360- and 85-fold for M4 and M1, respectively).

Representative formation plots for repaglinide metabolites in rCYP2C8 (A), rCYP3A4 (B), and rCYP3A5 (C) over a range of repaglinide concentrations. □ represents repaglinide M1; ▴ represents M4 metabolite. Results are mean ± S.D. from three separate experiments.
Kinetic profiles for the formation of repaglinide metabolites were also obtained in pooled HLM, S9, and human hepatocytes (Fig. 3). All formation profiles followed standard Michaelis-Menten kinetics. Repaglinide was not metabolized to any appreciable extent in the absence of cofactors. Estimated kinetic parameters for the formation of repaglinide metabolites in the in vitro systems investigated are summarized in Table 4. Unbound Km for M4 metabolite (predominantly CYP2C8) was the lowest in all the systems investigated (6.9–12 μM). In contrast, Km, u estimates for M1 were the highest, ranging from 31 to 51 μM in S9 and hepatocytes, respectively. Km, u values were comparable for each metabolite across the different systems (all within 2-fold).

Representative formation plots for repaglinide metabolites in human liver microsomes (A), human S9 fractions (B), and human hepatocytes (C) over a range of repaglinide concentrations. □ represents M1; ▾, M2; ▴, M4 and ○, repaglinide glucuronide. Results are mean ± S.D. from three separate experiments.
In vitro kinetic characteristics of repaglinide metabolites in pooled human hepatocytes, human liver microsomes, human S9 fractions, and recombinant P450 enzymes
Clearance estimates obtained are corrected for nonspecific binding using the estimated unbound fractions shown in Table 1. Results are expressed as mean ± S.D. (n = 3).
Clearance estimates were converted to per gram of liver using scaling factors (see Materials and Methods) to allow direct comparison of different in vitro systems. The formation of M2 resulted in major differences between systems and a 7.2-fold higher unbound intrinsic clearance (CLuint) in hepatocytes in comparison with HLM. A similar trend was seen between S9 and HLM. The rank order for formation of repaglinide P450 metabolites was M2 > M4 > M1 in both S9 and hepatocytes. In contrast, M4 was the major metabolite in HLM and the formation of M2 was minor. The formation of M1 in human hepatocytes was found to be very variable across experiments (coefficient of variation >100%). Formation of M2, M4, and repaglinide glucuronide was monitored simultaneously, and variations in parameter estimates for these pathways were less pronounced (<25%).
Involvement of Non-P450 Mediated Metabolism.
Contribution of repaglinide glucuronidation.
The contribution of glucuronidation was assessed by monitoring formation of repaglinide acyl glucuronide in HLM, S9, and hepatocytes (Fig. 3; Table 4). Km, u values were within 2-fold for all three in vitro systems. Scaled CLuint was estimated to be 15.6, 173, and 224 μl · min−1 · g liver−1 based on the data from hepatocytes, S9, and HLM, respectively. Limited information is available on the effect of cryopreservation on the activity of UGT enzymes, which may contribute to some extent to differences in activity between hepatocytes and other systems, in addition to donor differences. Stability of repaglinide acyl glucuronide was not considered to be an issue, considering the short incubation time (15 min) used in hepatocyte experiments.
Contribution of cytosolic enzymes to repaglinide metabolism.
Experiments were conducted to investigate the effect of disulfiram and raloxifene on the formation of repaglinide metabolites in S9 fractions. Disulfiram (10 μM) resulted in a 89% reduction in CLint, M2, whereas the effect on the formation clearance of repaglinide M1 and M4 was less pronounced (10 and 22%, respectively). Raloxifene (0.05 μM), a potent inhibitor of aldehyde oxidase, reduced the CLint, M2 by 10% despite the concentration of raloxifene exceeding the previously identified IC50 estimate by >10-fold (Obach et al., 2004). A 16 and 5% reduction in CLint for M1 and M4, respectively was observed when repaglinide was incubated with raloxifene.
On the basis of preliminary single concentration inhibition data, the IC50 of disulfiram was obtained to confirm further the contribution of aldehyde dehydrogenase in the formation of M2. Disulfiram inhibited repaglinide M2 formation in a concentration-dependent manner, resulting in complete inhibition (>97%) at the highest nominal disulfiram concentration (Fig. 4). The noncompetitive nature of inhibition was indicated by overlapping IC50 plots obtained at three repaglinide concentrations; estimated IC50 was 1.2 μM, confirming involvement of aldehyde dehydrogenase in the formation of M2. Minor inhibition of repaglinide M1 and M4 by disulfiram was observed, but IC50 curves could not be defined (data not shown). Previous studies have suggested that the formation of M2 is a two-step process. The authors further hypothesized the following mechanism. Repaglinide undergoes α-hydroxylation in the piperidine ring system (P450-mediated pathway), followed by tautomerization of the molecule to a ring-opened aldehyde. The intermediate formed would then be converted to the corresponding carboxylic acid by aldehyde dehydrogenase. The proposed two-step formation of repaglinide M2 metabolite is shown in Fig. 1.
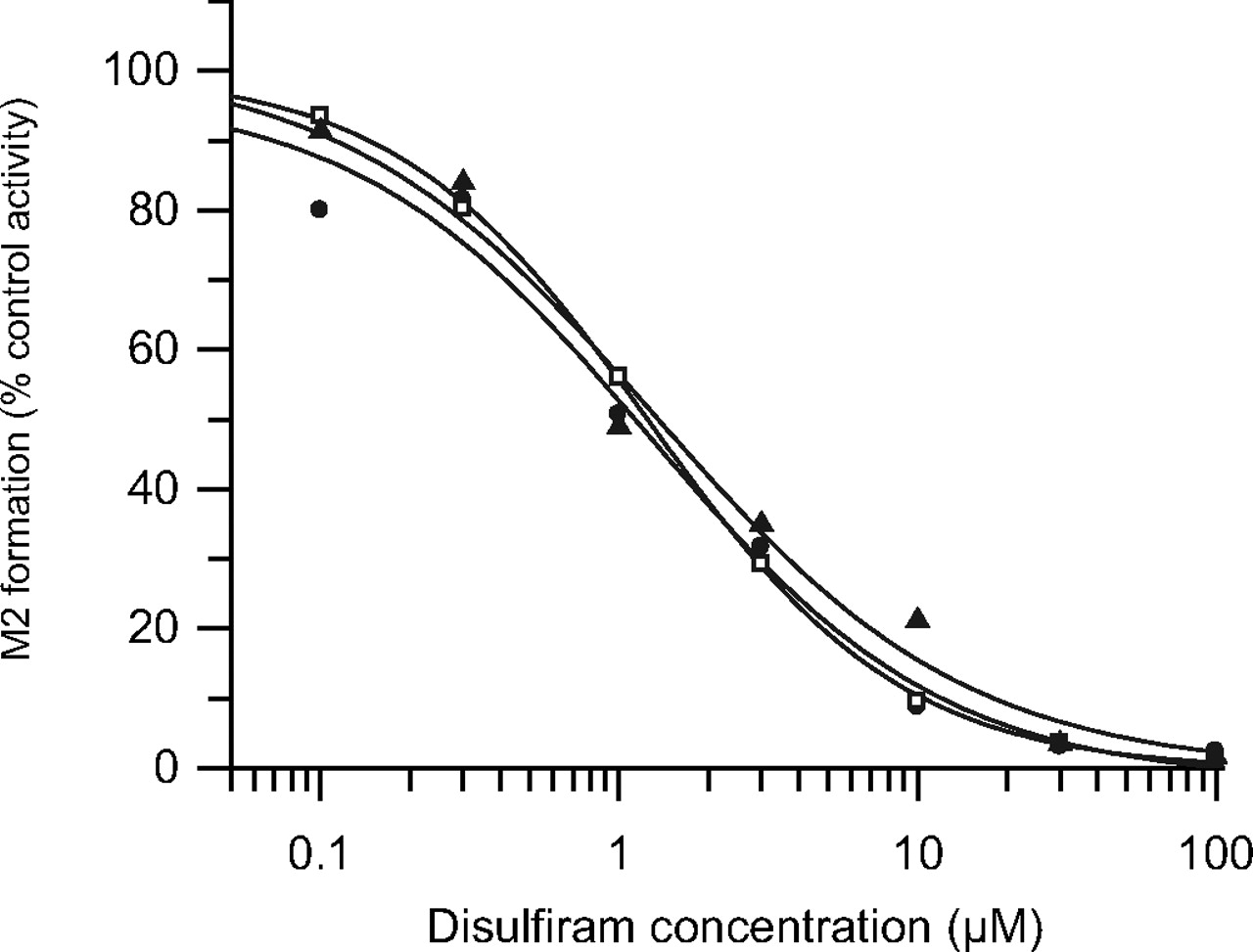
Nominal disulfiram concentration versus percent remaining formation of M2 in pooled S9 fractions. Experiments were performed using three different repaglinide concentrations: ● represents 10 μM; □, 25 μM and ▴, 50 μM. Data points represents the mean of three separate experiments.
Contribution of CYP3A4 and CYP2C8 to the Formation of M2 in S9 Fractions.
The effects of ketoconazole and gemfibrozil glucuronide on CLint, M2 were examined to fully delineate the contribution of CYP3A4 and CYP2C8 in the initial P450-mediated metabolic step. Results obtained in recombinant P450 enzymes showed distinct roles of CYP3A4 and CYP2C8 for M1 and M4 formation, respectively. Therefore, the effect of ketoconazole and gemfibrozil glucuronide on the formation of M1 and M4, respectively, was consequently monitored as a positive control. Complete CYP3A4 inhibition was confirmed because the presence of ketoconazole resulted in a 89% reduction in CLint, M1. Analogous to the effect of ketoconazole, preincubation with gemfibrozil glucuronide resulted in a time-dependent reduction of 92% for the CYP2C8-mediated M4 formation. The inhibitory effect of these inhibitors on the other pathway investigated (ketoconazole on M4 and gemfibrozil glucuronide on the formation of M1) was <30%. Experiments with gemfibrozil glucuronide resulted in a 24% reduction in CLint, M2 in comparison with control incubations. In contrast, the effect of ketoconazole on M2 was more pronounced, resulting in the remaining 27% of the control. Therefore, the estimated contribution of CYP3A4 and CYP2C8 in the initial P450-mediated step of M2 formation was 0.74 and 0.26, respectively, on the basis of averaged values of the two inhibition studies.
Repaglinide Pathway Ratios in S9 Fractions.
Figure 5 illustrates repaglinide metabolite pathway ratios obtained in the control state and in the presence of inhibitors. The relationship between the repaglinide concentration and the relative importance of metabolic pathways was investigated in both scenarios. Because of complete M1 inhibition, ketoconazole increased the M2 to M1 pathway ratio in comparison to the control; however, the extent of increase was substrate concentration-dependent, ranging from 11 to 5 at low and high repaglinide concentrations, respectively (Fig. 5A). Figure 5B shows M2 to M4 pathway ratios for control incubations and in the presence of gemfibrozil glucuronide. Irreversible inhibition of CYP2C8 by gemfibrozil glucuronide resulted in increased M2/M4 ratio (4- to 9-fold in comparison to control over the range of repaglinide concentrations). Disulfiram inhibited the formation of M2, resulting in an increased M4/M2 ratio in comparison with control incubations (Fig. 5C); the difference was less pronounced at higher repaglinide concentrations for which the M4/M2 ratio was 2-fold. The inhibitor effects of raloxifene on repaglinide pathways were not pronounced, and therefore pathway ratios obtained in the presence of raloxifene (Fig. 5D) showed a minor difference in comparison to the control state.

Pathway ratios for M2/M1 (A), M2/M4 (B), M4/M2 (C), and M2/M1 (D) in human S9 fractions over a range of repaglinide concentrations. ○ represents the control ratio without inhibitor, and ● represents the effect of inhibitor where (A) displays the effect of ketoconazole, (B) gemfibrozil glucuronide, (C) disulfiram, and (D) raloxifene.
Relative Contribution of Metabolic Pathways to Repaglinide Metabolism In Vitro.
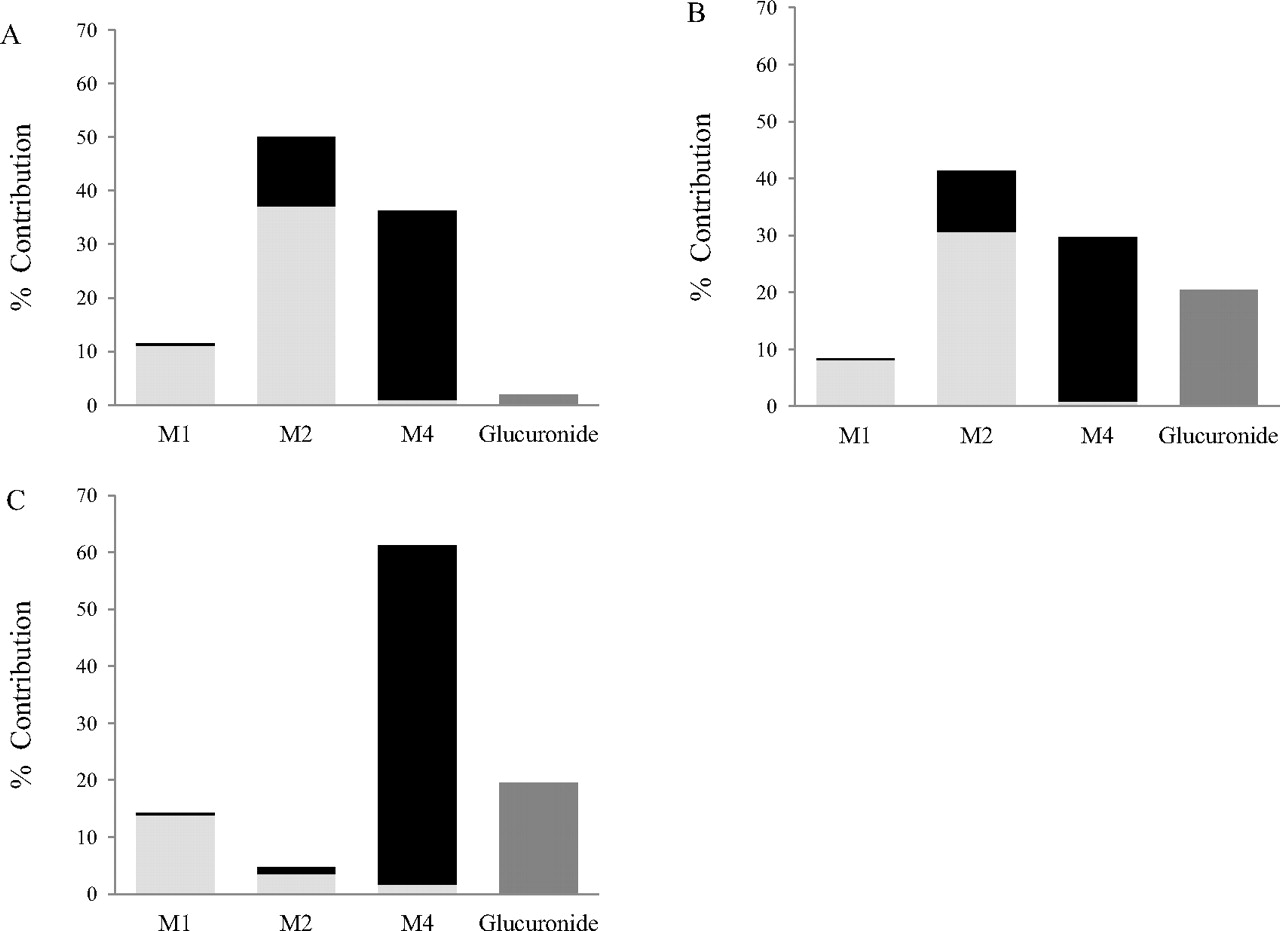
Figure 6 shows the percentage contribution of each pathway based on the data obtained in hepatocytes, S9, and HLMs. In addition, the individual contribution of CYP2C8, CYP3A4, and UGT enzymes to the formation of each metabolite is illustrated. The contribution of M2 to the overall CLint was 41 and 50% in S9 and hepatocytes, respectively. In contrast, the contribution of M2 to the overall clearance in HLM was minor (<5%). An increased importance of M1 and M4 was observed in HLM, leading to differential contribution of CYP2C8 and CYP3A4 in this system in comparison with hepatocytes and S9. The significantly higher glucuronidation activity obtained in S9 and HLM resulted in this pathway contributing up to 20% in both systems in contrast with 2% in hepatocytes.

Percentage contribution of each metabolite to total repaglinide metabolism in human hepatocytes (A), human S9 fractions (B), and human liver microsomes (C). Each bar graph shows percentage contribution of individual enzymes: light gray bar, CYP3A4; black bar, CYP2C8; dark gray bar, UGT enzymes.
Formation of M1 and M4 was associated with CYP3A4 and CYP2C8, respectively, based on the distinctive pathway ratio obtained in recombinant enzymes. Inhibition by ketoconazole and gemfibrozil glucuronide was used to calculate the relative contribution of CYP3A4 and CYP2C8 to the metabolism of M2. Estimations of fmCYP values resulted in an overall comparable contribution of CYP2C8 and CYP3A4 in S9 and hepatocytes. The fraction metabolized via CYP2C8 (63%) was larger than the fraction metabolized via CYP3A4 (18%) in HLM. A summary of fractions metabolized via individual enzymes in hepatocytes, S9, and HLM is shown in Table 5.
Repaglinide fraction metabolized via CYP2C8, CYP3A4, and UGT enzymes in pooled human hepatocytes, human liver microsomes, and human S9 fractions
Repaglinide In Vivo Inhibition Drug-Drug Interaction Studies.
Reported in vivo studies on repaglinide DDIs were collated, together with details on the effect of various inhibitors on repaglinide P450 metabolites. In total, 17 repaglinide DDIs were collated on the basis of the inclusion criteria stated previously. The dataset included 12 different perpetrators affecting CYP2C8, CYP3A4, or a combination of the two. The change in repaglinide AUC ranged from no significant effect for a number of inhibitors to a 19-fold increase when gemfibrozil was coadministered with itraconazole (Table 3). Some of the studies in the dataset represented the interactions with the same perpetrator (gemfibrozil) after different dosing intervals between its last dose and administration of repaglinide (Tornio et al., 2008; Backman et al., 2009). No effect on repaglinide AUC and <1.2-fold change in the ratio of M2 to either M1 or M4 was observed when repaglinide was given 96 h after the last dose of gemfibrozil (Backman et al., 2009). In contrast, simultaneous administration resulted in an 8.1-fold increase in repaglinide AUC (range 5.5- to 15-fold) (Niemi et al., 2003). Using the classification system proposed for CYP3A4 inhibitors (Bjornsson et al., 2003) indicated that 14% of the DDIs investigated were strong (AUC increased by 5-fold or more), 7% classified as moderate DDIs (AUC increased between 2- and 5-fold), and 36% were weak (AUC increased between 1.25- and 2-fold). Studies including montelukast, pioglitazone, ketoconazole, bezafibrate, and fenofibrate as perpetrators resulted in no interaction (<1.25-fold change in repaglinide exposure) (Hatorp et al., 2003; Kajosaari et al., 2004, 2006a,b). Seven DDI studies reported the effect on M1 in addition to the effect on repaglinide, whereas the effect on M2 and M4 was stated in five. Analysis of the effects of inhibitors on repaglinide metabolites showed more than 6-fold increase in the M2/M4 AUC ratio in comparison with control conditions when repaglinide was administered within 6 h after gemfibrozil (Tornio et al., 2008; Backman et al., 2009). The same study resulted in no fold change in the M2/M4 AUC ratio when the dosing interval was >24 h, reflecting the CYP2C8 half-life. The CYP3A4 inhibitor telithromycin led to a 4-fold increase in the M2/M1 AUC ratio in comparison with the control (Kajosaari et al., 2006b).
Discussion
Repaglinide is currently recommended as an in vivo CYP2C8 probe by the FDA (FDA Guidance for Industry 2012: Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf), but the kinetic characterization and enzymes involved in the metabolism of this drug have not been fully delineated. The current study characterized the formation of four repaglinide metabolites using recombinant P450 enzymes, HLM, S9, and human hepatocytes, supported by additional experiments with specific inhibitors. The contribution of CYP2C8, CYP3A4, and UGT enzymes to the overall clearance was assessed in all systems, with the exception of recombinant enzymes. In addition, metabolite ratios in relation to repaglinide concentrations were investigated in the presence and absence of selected inhibitors.
The current study demonstrated clear system-dependent differences in the formation of repaglinide M2 metabolite in HLM compared to S9 and human hepatocytes. The majority of earlier in vitro studies investigating repaglinide metabolism have been performed using either HLM or recombinant P450 enzymes (Bidstrup et al., 2003; Kajosaari et al., 2005). Bidstrup et al. (2003) reported M1 and M4 as the major metabolites in vitro, which is in line with the result obtained in HLM in the current study (Figs. 3 and 6). The formation of M2 in microsomes is restricted because of the lack of cytosolic enzymes, and hence this in vitro system was unable to confirm relevance of M2 suggested from in vivo data (66% of administered dose excreted as this metabolite in feces and urine). In contrast, S9 and hepatocytes identified M2 as the major contributing pathway and can therefore be seen as preferable in vitro systems for characterizing compounds such as repaglinide. The formation of the M2 metabolite contributes 41 and 50% to the overall CLint in S9 and hepatocytes, respectively, in contrast to HLM in which the contribution of this pathway was minimal (5%, as expected because of its metabolic origin). Such inadequate characterization of a major pathway in vitro can lead to inaccuracies in estimating the fmCYP. As illustrated in Table 5 and Fig. 6, the fmCYP estimates in HLM were skewed towards a larger CYP2C8 contribution because of the increased importance of the M4 pathway. The discrepancy in conclusions drawn from microsomes compared with those from S9 and hepatocytes emphasizes the importance of an appropriate in vitro system selection. S9 are advantageous compared with hepatocytes, considering their practicality and cost implications. In addition, S9 fractions are currently available as large 150 donor pools, consisting of an equal donor mix on a per milligram of microsomal protein basis, giving a representative “average” individual in terms of drug metabolism activity, whereas cryopreserved hepatocytes were available in pools of 20 donors at the time of the experiments.
Estimated Km, u values for the formation of repaglinide acyl glucuronide obtained in this study were comparable in HLM, S9, and hepatocytes, consistent with the trends seen for Km u for P450 metabolic pathways. However, variations were seen in scaled Vmax, resulting in a disparity in contribution of glucuronidation to the overall clearance. In addition to donor differences, lower formation of the acyl glucuronide observed in pooled cryopreserved hepatocytes in comparison with S9 and HLM could be a result of potential degradation of endogenous UDP-glucuronic acid during the experiment, as suggested previously (Swales and Utesch, 1998; Wang et al., 2005). Further investigation into the impact of supplementing pooled human hepatocytes with exogenously added cofactors is required to assess the potential limitations of this in vitro system in the assessment of glucuronidation clearance.
Repaglinide is initially metabolized via CYP2C8 and CYP3A4 to form an aliphatic aldehyde intermediate before being converted to the carboxylic acid metabolite (M2), as shown in Fig. 1. Involvement of aldehyde oxidase and aldehyde dehydrogenase in the second formation step of M2 was investigated by assessing the effect of known inhibitors of these enzymes, raloxifene and disulfiram. The almost 90% reduction in the formation of M2 in the presence of disulfiram and the minor effect of raloxifene (CLint, M2 reduced by 10%) strongly indicated involvement of aldehyde dehydrogenase in the formation of M2. In addition, aldehyde oxidase and aldehyde dehydrogenase have different subcellular localization in the liver. Aldehyde dehydrogenase has been identified in the cytosol and mitochondria and also to some extent in microsomes (Yoshida et al., 1998), whereas aldehyde oxidase has been found in the cytosol (Pryde et al., 2010). Repaglinide metabolite formation studies performed in microsomes in the current study resulted in minor formation of M2 (Fig. 3), supporting the presence of some isoforms of aldehyde dehydrogenase. Finally, previously identified substrates for the two enzymes differ markedly in their chemical structure. Aldehyde oxidase more often oxidizes complex or aromatic aldehydes (Panoutsopoulos et al., 2004; Pryde et al., 2010), whereas aldehyde dehydrogenase shows high affinity toward aliphatic aldehydes and has been associated with two-step metabolic processes involving initial P450 metabolic activation, supporting further its involvement in repaglinide M2 formation.
The relative importance of repaglinide metabolic pathways varied with substrate concentration, as seen in Fig. 5; this trend was also apparent for inhibited pathway ratios, in particular, obtained in the presence of ketoconazole and disulfiram (Fig. 5, A and C). The impact of substrate concentration on metabolite ratio is related to the in vitro characteristics for the pathways investigated and illustrates the benefit of obtaining full kinetic characterization of specific pathways. The intracellular concentration of repaglinide available for metabolism will be a result of an interplay of multiple processes, namely hepatic uptake, passive permeability, and intracellular binding (Gertz et al., 2010; Ménochet et al., 2012) and is likely to vary, in particular, in the case of OATP1B1 inhibition/polymorphism or dual transporter/metabolism inhibition, leading to potential differences in the relative importance of individual metabolic pathways of repaglinide. In vitro experiments in the current study showed increased metabolite ratios in the presence of ketoconazole, gemfibrozil glucuronide, and disulfiram in comparison with the control state as a reflection of the inhibition of the respective enzymes, CYP3A4, CYP2C8, and aldehyde dehydrogenase. The increase was particularly evident in the case of gemfibrozil glucuronide (M2/M4 ratio increased up to 9-fold compared with that for the control); this reduced M4 formation was within the fold change in the M2/M4 AUC ratio reported in vivo (6–18 when gemfibrozil was administered either 6 or 1 h before repaglinide, respectively) (Tornio et al., 2008; Backman et al., 2009).
Analysis of various collated repaglinide DDI studies shows that the majority of repaglinide DDIs (≥75%) were classified as weak or nonsignificant, consistent with the existence of multiple metabolic pathways and the fmCYP estimates obtained in this study (fmCYP2C8, fmCYP3A4, and fmUGT estimated in S9 were 0.41, 0.39, and 0.20, respectively). The most pronounced effect was seen in the interaction studies with gemfibrozil (up to 8.1-fold increase in repaglinide AUC) (Niemi et al., 2003; Kalliokoski et al., 2008a; Tornio et al., 2008; Backman et al., 2009). The fraction metabolized via CYP2C8 estimated in this study was <50% and would not account for the observed increase in repaglinide AUC caused by gemfibrozil and its glucuronide, strongly indicating that inhibition of repaglinide uptake into the cell in addition to irreversible inhibition of CYP2C8 needs to be considered. The complexity of this interaction requires a dynamic mechanistic model incorporating in vitro data on repaglinide active uptake kinetics, passive diffusion, intracellular binding, as well as metabolism. The impact of multiple inhibitors acting via different pathways can be of serious consequences, as illustrated also in the case of the repaglinide-gemfibrozil-itraconazole interaction (Niemi et al., 2003).
The ability to assess CYP2C8 inhibition in vivo is currently restricted because of the lack of a substrate predominantly metabolized by this enzyme. Repaglinide metabolite standards are now commercially available, allowing definitive effects on specific metabolic pathways to be investigated. In the current study formation of repaglinide M4 hydroxy metabolite was found to be a specific CYP2C8-mediated pathway. Clinical data on monitoring M4 and changes in this metabolite, either on its own or relative to other repaglinide metabolites, as an indication of CYP2C8 inhibition, are currently limited to gemfibrozil studies (Table 3). Future clinical DDI studies monitoring the effect on repaglinide metabolites are required for further evaluation of the use of M4 as a proposed novel specific CYP2C8 probe.
In summary, the systematic assessment of repaglinide metabolic pathways performed in the current study showed system-dependent differences in the contribution of P450, cytosolic and glucuronidation enzymes. Metabolite kinetic formation profiles showed distinct pathway differences for M1 and M4 in CYP3A4 and CYP2C8. The metabolism of M2 represents a complex multistep formation process involving initially CYP3A4 and CYP2C8, followed by aldehyde dehydrogenase. CYP2C8 appears to play a less prominent role in repaglinide clearance than previously believed. However, monitoring of repaglinide M4 formation and changes in metabolite ratios could aid in interpretation of the effect of potential perpetrators of CYP2C8 in vivo.
Authorship Contributions
Participated in research design: Säll, Houston, and Galetin.
Conducted experiments: Säll.
Performed data analysis: Säll.
Wrote or contributed to the writing of the manuscript: Säll, Houston, and Galetin.
Acknowledgments
We acknowledge Sue Murby (University of Manchester) for valuable assistance with the LC-MS/MS and Dr. Michael Gertz (University of Manchester) for useful discussions.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
ABBREVIATIONS:
- P450
- cytochrome P450
- UGT
- UDP-glucuronosyltransferase
- OATP
- organic anion-transporting polypeptide
- DDI
- drug-drug interaction
- FDA
- U.S. Food and Drug Administration
- HLM
- human liver microsomes
- S9
- supernatant fraction from liver homogenate
- r
- recombinant
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- AUC
- area under the curve.
- Received February 27, 2012.
- Accepted March 26, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}