


Abstract
Isolated human hepatocytes are commonly used to predict transporter-mediated clearance in vivo. Such predictions, however, do not provide estimations of transporter contributions and consequently do not allow predictions of the outcome resulting from a change in the activity of a certain transporter, for example, by inhibition or a genetic variant with reduced function. Pitavastatin is a drug that is heavily dependent on hepatic transporters for its elimination, and it is excreted mainly as unchanged drug in the bile. For this reason, pitavastatin was used as a model drug to demonstrate the applicability of a bottom-up approach to predict transporter-mediated disposition in sandwich-cultured human hepatocytes (SCHHs), allowing for the estimation of transporter contributions. Transport experiments in transfected human embryonic kidney cells (HEK293 cell lines) and inverted membrane vesicles overexpressing each of the relevant transport proteins were used to generate parameter estimates for the mechanistic model. By adjusting for differences in transporter abundance between the in vitro systems and individual SCHH batches, the model successfully predicted time profiles of medium and intracellular accumulation. Our predictions of pitavastatin bile accumulation could not be confirmed, however, because of a very low biliary excretion of pitavastatin in relation to the hepatic uptake in our SCHHs. This study is, to our knowledge, the first to successfully simulate transporter-mediated processes in a complex system such as SCHHs at the level of individual transport proteins using a bottom-up approach.
Introduction
Statins (or 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) are dependent on active transporter-mediated uptake and efflux for their distribution and elimination (Catapano, 2010; Maeda et al., 2011). Since the substrate specificity of many hepatic transporters is broad and overlapping, several transport proteins are typically involved in the disposition. The contribution of the involved transporters, however, varies depending on the statin studied (Elsby et al., 2012).
To predict transporter-mediated clearance (CL) in vivo, isolated human hepatocytes are commonly used. A drawback with this experimental system is the difficulty of deconvoluting the role of the various transport proteins in the disposition. This difficulty presents a challenge for assessing the outcome of a change in the activity of a certain transporter (e.g., by inhibition or a reduced function genetic variant). The lack of activity of one transporter could potentially cause a clinical drug-drug interaction (DDI) if the transporter is a major contributor to the disposition. On the other hand, no significant DDI will occur if other transporters with overlapping substrate profiles are able to compensate for the reduced transport. Hence, quantitative assessment of transporter contributions to the overall disposition is essential for accurate predictions of pharmacokinetic changes associated with transporter inhibition or inherited dysfunction.
In previous studies, we introduced a method to assess the contribution of a certain transporter in the disposition of a drug (Karlgren et al., 2012b; Vildhede et al., 2014). This method builds on in vitro experiments in overexpressing systems, allowing the kinetics of a single transporter to be studied in isolation. Using a measure of the difference in transport protein abundance between the in vitro system and the more complex system we aim to model (e.g., human hepatocytes or liver tissue), the kinetics in the complex system can be predicted.
In the present study, experiments in stably transfected human embryonic kidney (HEK) 293 cells and inverted membrane vesicles were integrated using a mechanistic model to predict the time-course of pitavastatin disposition in sandwich-cultured human hepatocytes (SCHHs). Pitavastatin was chosen as a model drug due to its limited metabolism and known transporter-dependent hepatic elimination (Elsby et al., 2012). By studying each transporter individually, we were able to differentiate the role of each transporter in the overall disposition. The model was subsequently used to investigate the influence of decreased or increased transport activity on pitavastatin disposition.
Materials and Methods
Materials
Pitavastatin calcium salt was obtained from AK Scientific (Union City, CA). All other chemicals were of analytical grade and purchased from commercial sources. Inverted Sf9 membrane vesicles overexpressing multidrug resistance-associated protein 3 (MRP3; ABCC3) and HEK293 membrane vesicles overexpressing MRP4 (ABCC4) were purchased from SOLVO Biotechnology (Budapest, Hungary) together with their respective control vesicles. AstraZeneca (Mölndal, Sweden) kindly provided us with inverted membrane vesicles from HEK293 cells overexpressing breast cancer resistance protein (BCRP; ABCG2), MRP2 (ABCC2), or multidrug resistance protein 1 (MDR 1; ABCB1)2, or multidrug resistance protein 1 (MDR1; ABCB1) and their respective control vesicles.
Human Liver Tissue
Normal excess human liver tissue was obtained from liver resections carried out at the Department of Surgery, Uppsala University Hospital (Uppsala, Sweden). The study was approved by the Uppsala Regional Ethical Review Board (ethical approval no. 2009/028). Donors gave their informed consent. The donor demographics are summarized in Table 1.
Demographic data for human liver tissue donors
Cell Culture and Plating
HEK293 Cells.
Mock-transfected HEK293 Flp-In cells and cells stably expressing either sodium taurocholate cotransporting polypeptide (NTCP; SLC10A1), organic anion transporting polypeptide 1B1 (OATP1B1; SLCO1B1), OATP1B3 (SLCO1B3), or OATP2B1 (SLCO2B1) were grown and maintained as described elsewhere (Karlgren et al., 2012a). Cells were seeded in 24-well plates at a density of 600,000 cells per well 2 days (NTCP, OATP1B1, and OATP2B1) or 3 days (OATP1B3) before the experiments. For all cultures in 24-well plates, growth medium without phenol red and hygromycin B was used (Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 2 mM l-glutamate).
Human Hepatocytes.
Primary hepatocytes were isolated using a two-step collagenase perfusion technique described in more detail by LeCluyse and Alexandre (2010). The cells were suspended in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum, penicillin-streptomycin (100 U ml−1, and 100 µg ml−1, respectively), 4 µg ml−1 insulin, and 1 µM dexamethasone. Suspended hepatocytes with a viability >85% were plated on collagen I-coated 24-well plates (BD Biosciences, Franklin Lakes, NJ) at a density of 375,000 cells per well. The cells were allowed to attach for 2–3 hours at 37°C and 5% CO2 atmosphere. Attachment medium was replaced with hepatocyte maintenance medium (Lonza, Basel, Switzerland) supplemented with penicillin-steptomycin, insulin-transferrin-selenium (10 µg ml−1, 5.5 µg ml−1, and 5 ng ml−1, respectively) and 0.1 µM dexamethasone. After overnight incubation, the hepatocytes were overlaid with 0.25 mg/ml Matrigel (BD Biosciences). Cells were maintained in culture for an additional 5 days to allow for bile canaliculi formation. Culture medium was replaced daily.
Kinetic Characterization of Uptake Transporters Using Stably Transfected HEK293 Cells
The transport experiments in HEK293 cells were performed as described previously (Karlgren et al., 2012b). Briefly, cells were washed twice with prewarmed Hanks’ balanced salt solution (HBSS), pH 7.4, followed by incubation with prewarmed substrate solution of increasing concentration for 2 minutes at 37°C. The incubation was terminated by adding ice-cold Dulbecco’s phosphate buffered saline (DPBS), followed by three washes with DPBS. The cells were subsequently dried, and the intracellular drug accumulation was quantified by ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) as described later in this paper.
Active transporter-mediated uptake was calculated by subtracting the accumulation in mock-transfected cells (passive diffusion) from the uptake in the transfected cells. Initial uptake rate was related to substrate concentration by fitting the resulting curve to the Michaelis-Menten equation (eq. 1) using GraphPad Prism v.5.04 (GraphPad Software, La Jolla, CA):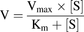 (1)where V is the uptake rate, Vmax is the maximal uptake rate (at saturating substrate concentration), [S] is the substrate concentration, and Km is the substrate concentration at which the uptake rate is half of Vmax.
(1)where V is the uptake rate, Vmax is the maximal uptake rate (at saturating substrate concentration), [S] is the substrate concentration, and Km is the substrate concentration at which the uptake rate is half of Vmax.
Passive clearance in the mock-transfected HEK293 cells was determined by linear regression analysis. All transport experiments in HEK293 cells were run in duplicate on at least two separate occasions.
Kinetic Characterization of Efflux Transporters Using Inverted Membrane Vesicles
Concentration-dependent transport by MDR1, MRP2, MRP3, MRP4, and BCRP was assessed using inverted membrane vesicles. Transport experiments were performed using the rapid filtration technique modified from Pedersen et al. (2008). Briefly, vesicles were quickly thawed at 37°C and diluted in prewarmed transport buffer (10 mM Tris-HCl, pH 7.4, 250 mM sucrose, 10 mM MgCl2, and 10 mM phosphocreatine). A mixture of substrate, ATP, and creatine kinase (final concentrations of 4 mM, and 90 U/ml, respectively) was added to initiate transport. After incubation for 5 minutes at 37°C, transport was stopped by addition of ice-cold stop solution (10 mM Tris-HCl, pH 7.4, 250 mM sucrose, and 0.1 M NaCl). Incubation solutions were immediately transferred to and filtered through a 96-well glass fiber filter plate with a pore size of 0.65 µm (Millipore, Bedford, MA). Vesicles on the filter were rinsed four times with ice-cold stop solution and allowed to dry. Drug accumulation in the vesicles was quantified by UPLC-MS/MS as follows.
Active transport was calculated as the difference in uptake in transporter-expressing vesicles and control vesicles. Initial transport rate was plotted against substrate concentration. The Michaelis-Menten kinetic parameters (Km and Vmax) were determined by nonlinear regression to eq. 1 in GraphPad Prism v.5.04 (GraphPad Software, La Jolla, CA). Transport experiments in inverted membrane vesicles were run in triplicate on at least two separate occasions.
Time-Dependent Disposition in Sandwich-Cultured Human Hepatocytes
Time-dependent disposition of pitavastatin in SCHHs was assessed using a method adapted from the originators to the B-CLEAR technology (Qualyst Transporter Solutions, Research Triangle Park, NC) (Liu et al., 1999). At start of the experiment, cells were washed twice with either standard HBSS, pH 7.4, or Ca2+-and Mg2+-free HBSS (disrupting tight junctions, thereby opening up bile canaliculi), followed by preincubation in the same buffer for 10 minutes at 37°C. Transport was initiated by adding standard HBSS containing 0.1, 0.6, 1.0, or 2.5 µM of pitavastatin. Preliminary experiments showed that bile canaliculi did not reseal during incubations in standard HBSS of up to 45 minutes (data not shown). After incubation, substrate solution was removed and saved for analysis. Any further uptake/efflux was stopped by addition of ice-cold DPBS, followed by three rinses with the same. The amount of drug accumulated in the SCHHs and the remaining amount in the medium samples were quantified using UPLC-MS/MS as follows.
Intracellular drug accumulation was determined from the experiments with preincubation in Ca2+-and Mg2+-free HBSS (disrupted bile canaliculi); combined accumulation in cells and bile canaliculi was measured in the experiments using standard HBSS. Transport experiments in SCHHs were run in triplicate. The mass balance was greater than 90% in all experiments.
Determination of Protein Concentration
In all cell experiments, total protein content was measured in representative wells using the BCA Protein Assay according to manufacturer’s instructions (Pierce Biotechnology, Rockford, IL).
UPLC-MS/MS Analysis
Substrate accumulated in cells and membrane vesicles was extracted with 200 µl acetonitrile/water (60/40) spiked with 50 nM warfarin as internal standard. The samples were centrifuged at 2465g and 4°C for 20 minutes. Substrate concentration was determined using a Waters Xevo triple quadrupole mass spectrometer with electrospray ionization coupled to an Acquity UPLC system (Waters, Milford, MA). UPLC separation was performed on a reversed-phase BEH C18 column (2.1 × 50 mm, particle size 1.7 µm) at a flow rate of 0.5 ml/min. The linear mobile gradient consisted of acetonitrile, formic acid and water.
Quantification of Transporter Expression in Inverted Membrane Vesicles and Sandwich-Cultured Human Hepatocytes
Day 5 sandwich-cultured human hepatocytes in a 24-well plate were washed twice with ice-cold DPBS. DPBS was added to the wells, and the cells were scraped off and combined. The cells were centrifuged at 300g and 4°C for 5 minutes. The supernatant was aspirated, and the cell pellet was frozen down and stored at −80°C until further analysis.
Membrane fractions from the SCHH pellets were extracted with the native membrane protein extraction kit as described elsewhere (Li et al., 2008b). Total protein content of the membrane fractions was determined using the BCA Protein Assay Reagent Kit according to the manufacturer’s instructions.
Proteins in the SCHH membrane fractions and inverted membrane vesicles were denatured and digested with trypsin as described (Li et al., 2008a; Balogh et al., 2012; Qiu et al., 2013). Abundances of surrogate peptides of the transporters were determined by peptide-based LC-MS/MS measurements with corresponding isotope-labeled peptides as internal standards (Li et al., 2008a; Balogh et al., 2012; Kimoto et al., 2012; Qiu et al., 2013).
Mechanistic Mathematical Model for the Prediction of Time-Dependent Disposition in SCHHs
A mechanistic pharmacokinetic model was developed using the free software R version 3.0.0 (R Core Team, 2013, Vienna, Austria; (http://www.R-project.org) with the package deSolve (Soetaert et al., 2010). The model included compartments describing medium, intracellular and bile content as depicted in Fig. 1. In the model, distribution of drug between the compartments was described by saturable active uptake/efflux and unsaturable passive diffusion. The change of drug amount in the various compartments was described by differential equations (eq. 2–4). Mixing in the three compartments was assumed to be instantaneous.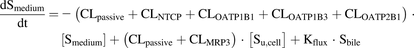 (2)
(2) (3)
(3)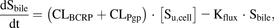 (4)where Sx is amount of drug in compartment X (medium, cell, or bile), [Smedium] is substrate concentration in the incubation solution (Vmedium=200 µl), CLpassive is the passive clearance, CLtransporter is the active uptake or efflux clearance of each transporter, Kflux describes the flux from bile networks into the medium due to periodical contractions of the bile canaliculi, and [Su,cell] is the intracellular unbound concentration.
(4)where Sx is amount of drug in compartment X (medium, cell, or bile), [Smedium] is substrate concentration in the incubation solution (Vmedium=200 µl), CLpassive is the passive clearance, CLtransporter is the active uptake or efflux clearance of each transporter, Kflux describes the flux from bile networks into the medium due to periodical contractions of the bile canaliculi, and [Su,cell] is the intracellular unbound concentration.

Schematic representation of the three-compartment mechanistic model describing pitavastatin disposition in SCHHs. The model included a medium, a cellular, and a bile canalicular compartment. The distribution of drug between the compartments was described by nonsaturable passive diffusion (CLpassive) and saturable active transport (CLtransporter). The dynamic pulsing of the bile canaliculi, causing release of accumulated drug in the bile compartment to the medium, was described by the Kflux parameter.
The passive clearance of pitavastatin was determined to be 15.8 ± 3.00 µl/min from experiments in mock-transfected HEK293 cells. The periodical contractions of the bile canaliculi were considered an inherent property of the SCHHs, and the experimentally determined Kflux-value of 0.044 for rosuvastatin (Pfeifer et al., 2013) was used as input in our simulations. [Su,cell] was defined according to eq. 5:
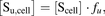 (5)
(5)where fu is the intracellular fraction unbound of pitavastatin. The fu-value of 0.053 reported by Menochet and colleagues was used as input (Menochet et al., 2012). The cellular volume was estimated from protein content using the reported value of 7.4 µl/mg protein (Lee, Brouwer, 2010a).
Transporter-mediated clearance of each hepatic transporter was scaled from in vitro experiments in HEK293 cells/membrane vesicles to SCHHs using eq. 6: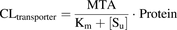 (6)where [Su] is the unbound substrate concentration that each transporter experiences, that is, [Smedium] for the uptake transporters and [Su,cell] for the efflux transporters, and MTA is the maximal transport activity, calculated according to eq. 7 for the uptake transporters and eq. 8 for the efflux transporters:
(6)where [Su] is the unbound substrate concentration that each transporter experiences, that is, [Smedium] for the uptake transporters and [Su,cell] for the efflux transporters, and MTA is the maximal transport activity, calculated according to eq. 7 for the uptake transporters and eq. 8 for the efflux transporters: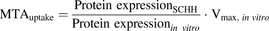 (7)
(7) (8)where finverted is the fraction of membrane vesicles that are inverted, and fmembrane is the fraction of membrane protein in relation to total protein in hepatocytes. The fraction of inverted membrane vesicles from a successful preparation is typically around one-third (Keppler et al., 1998), but it varies from batch to batch, depending on preparation techniques. The fraction of membrane protein in hepatocytes is also approximately one-third (Peng et al., 2015), although this value may vary for different hepatocyte batches. For our simulations, finverted and fmembrane values of one-third were used.
(8)where finverted is the fraction of membrane vesicles that are inverted, and fmembrane is the fraction of membrane protein in relation to total protein in hepatocytes. The fraction of inverted membrane vesicles from a successful preparation is typically around one-third (Keppler et al., 1998), but it varies from batch to batch, depending on preparation techniques. The fraction of membrane protein in hepatocytes is also approximately one-third (Peng et al., 2015), although this value may vary for different hepatocyte batches. For our simulations, finverted and fmembrane values of one-third were used.
The initial condition was stated and the model was subsequently used to simulate the time course in SCHHs. The differential equations were solved using the deSolve package.
Statistical Analysis
Data are presented as means with standard deviation. Differences in drug accumulation in overexpressing HEK293 cells/membrane vesicles compared with mock-transfected cells/control vesicles were assessed by unpaired t test. Probability values (P values) are symbolized by *P < 0.05, **P < 0.01, and ***P < 0.001. P values below 0.05 were considered statistically significant.
The predictive capability of the model was assessed by determining the average fold error (AFE) and the absolute average fold error (AAFE). The fold errors were calculated as the ratio of the simulated to the observed amount of pitavastatin (S) in each compartment for the time points when samples were taken as shown in eq. 9: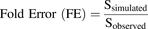 (9)The AFE, which is a measure of the bias of the prediction, was calculated according to eq. 10, where n is the number of time points for which samples were taken:
(9)The AFE, which is a measure of the bias of the prediction, was calculated according to eq. 10, where n is the number of time points for which samples were taken: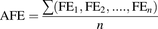 (10)The AAFE, which is a measure of the accuracy of the prediction, was calculated in eq. 11 as follows:
(10)The AAFE, which is a measure of the accuracy of the prediction, was calculated in eq. 11 as follows: (11)Our simulations rely on parameter values determined experimentally; each parameter determined with uncertainty. This uncertainty in the experimental data was not included in our simulations. Instead, we investigated the impact of the parameter uncertainty on the outcome through sensitivity analysis by varying the transporter Vmax, Kflux, and fu parameters 10 times above and below their experimental values.
(11)Our simulations rely on parameter values determined experimentally; each parameter determined with uncertainty. This uncertainty in the experimental data was not included in our simulations. Instead, we investigated the impact of the parameter uncertainty on the outcome through sensitivity analysis by varying the transporter Vmax, Kflux, and fu parameters 10 times above and below their experimental values.
Results
In Vitro Characterization of Pitavastatin Transport in HEK293 Cells and Inverted Membrane Vesicles.
Prior to pitavastatin kinetic studies, transport activity in the transporter-transfected HEK293 cells was confirmed by uptake studies using atorvastatin as probe substrate (Fig. 2, A–D). The uptake of pitavastatin in HEK293 cells was linear for at least 3 minutes up to a concentration of 800 µM (data not shown). The NTCP-, OATP1B1-, OATP1B3-, and OATP2B1-mediated uptake followed Michaelis-Menten kinetics as shown in Fig. 2, E–H. OATP2B1 is reported to have two different binding sites, but pitavastatin interacted with only one of the binding sites (Supplemental Fig. S1). Kinetic parameters of the transporter-mediated uptake are summarized in Table 2.
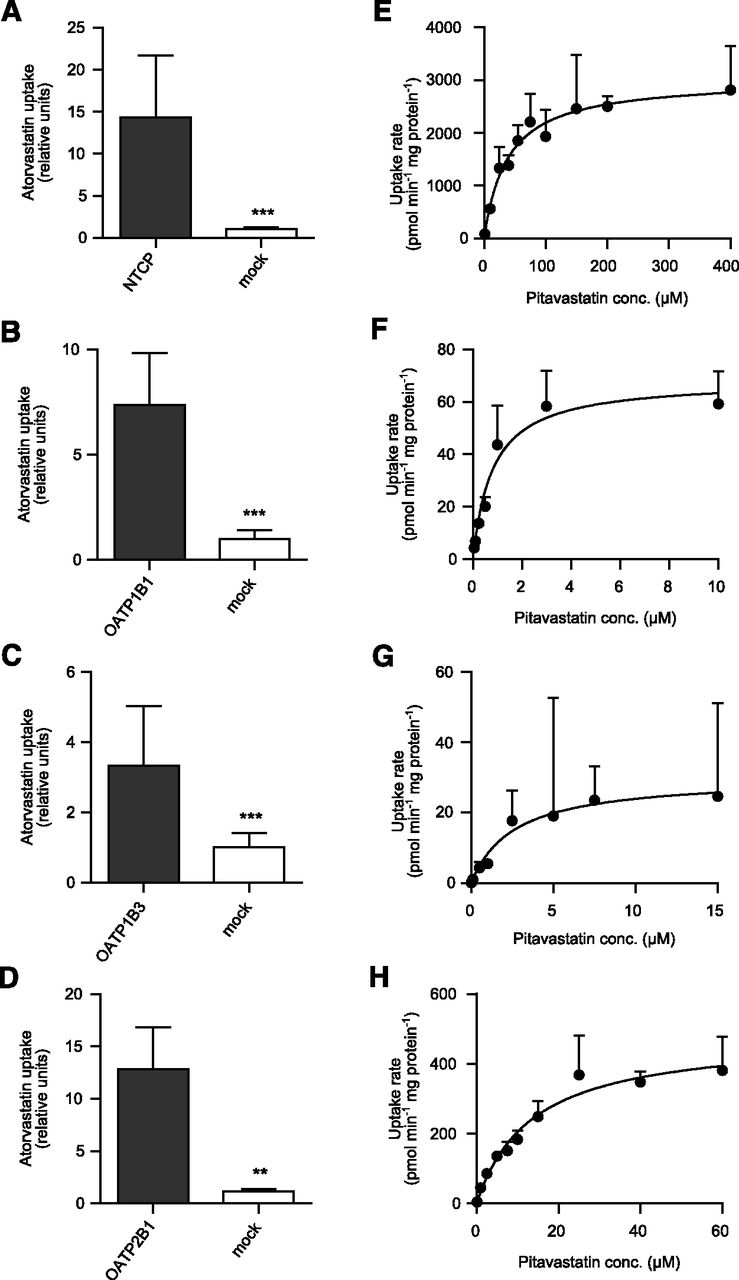
Uptake of 1 µM atorvastatin acid (probe substrate) in HEK293 cells stably expressing NTCP (A), OATP1B1 (B), OATP1B3 (C), or OATP2B1 (D) compared with the passive uptake in mock-transfected cells. ***P < 0.001; **P < 0.05. Concentration-dependent uptake of pitavastatin in HEK293 cells overexpressing NTCP (E), OATP1B1 (F), OATP1B3 (G), or OATP2B1 (H). Pitavastatin uptake followed Michaelis-Menten kinetics; the kinetic parameters are summarized in Table 2.
Kinetic parameters (mean ± standard deviation) of the transporter-mediated uptake of pitavastatin determined using either HEK293 cells (SLC transporters) or membrane vesicles (ABC transporters)
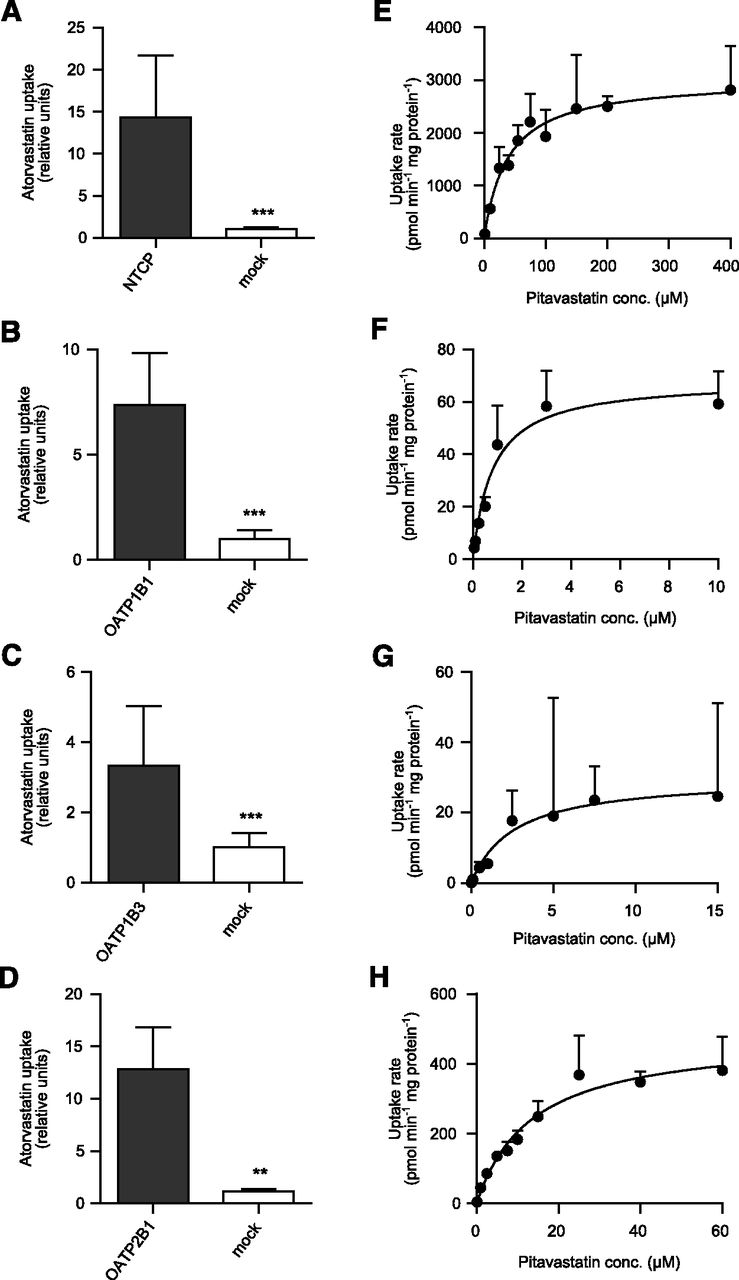
Each batch of membrane vesicles was characterized for transport activity using probe substrates (Fig. 3, A–C, D, and H). Transport of pitavastatin in the membrane vesicles was linear for at least 10 minutes (data not shown). Pitavastatin was identified as a substrate of BCRP, MDR1, and MRP3 (Fig. 3, E–G), but not of MRP2 and MRP4 (Fig. 3, D and H). The bile salt export pump (BSEP) was not included in our analysis since Hirano et al. were unable to detect any transport of pitavastatin by BSEP (Hirano et al., 2005a). The kinetic parameters describing the vesicular uptake mediated by efflux transporters are summarized in Table 2.

Accumulation of the probe substrate estradiol-17β-glucuronide (E17βG) in membrane vesicles overexpressing BCRP (A), MDR1 (B), or MRP3 (C) compared with passive uptake in control vesicles. ***P < 0.001; **P < 0.05. Concentration-dependent uptake of pitavastatin in membrane vesicles overexpressing BCRP (E), MDR1 (F), or MRP3 (G). Accumulation of 10 µM probe substrate or pitavastatin in inverted membrane vesicles expressing MRP2 (D) or MRP4 (H) compared with the passive uptake in control vesicles. Neither MRP2 nor MRP4 transported pitavastatin to a measurable extent.
Transporter Abundance in In Vitro Models and SCHHs.
The in vitro kinetic data from HEK293 cells and membrane vesicles presented here were used as input in our mechanistic model. Because the cell lines and membrane vesicles differ in transport protein abundance compared with hepatocytes in sandwich culture, we quantified this difference and used it as a scaling factor as described in Materials and Methods. The transporter abundances in the various in vitro systems are summarized in Table 3. Expression levels of the uptake transporters were 3- to 88-fold higher in the overexpressing HEK293 cells than in SCHHs. The efflux transporters displayed even larger abundance ratios, ranging from 24 to 2360, indicating a very high expression of transport proteins in the membrane vesicles in comparison with that in the hepatocytes.
Transporter abundance levels in the HEK293 cells (SLC transporters), inverted membrane vesicles (ABC transporters), and sandwich-cultured human hepatocytes (SCHHs)
Transporter expression was determined from abundance levels of surrogate peptides measured by targeted LC-MS/MS analysis (CV of the quantification methods <15%) (Balogh et al., 2012; Li et al., 2008b; Li et al., 2009; Qiu et al., 2013).
The transporters displayed an interindividual variability in hepatic abundance of 1.4- to 3.8-fold difference between the highest and lowest expressing SCHH batch (n = 4). OATP1B3 and BCRP displayed the highest interindividual variability in expression (>3-fold), whereas MDR1 expression was the most stable across batches (1.4-fold range in abundance).
Prediction of Time-Dependent Pitavastatin Disposition in SCHHs.
Pitavastatin disposition in four individual SCHH batches was simulated over a period of 60 minutes using a bottom-up modeling approach. The parameter values used as input for the simulations are summarized in Supplemental Table S1. The simulated curves were compared with experimental data (Fig. 4). The mechanistic model accurately predicted experimental results with an absolute average error of less than 2-fold in all experiments (Table 4).

Comparisons of simulations and experimental observations for pitavastatin disposition in four different batches of SCHHs: UU113 (A), UU115 (B), UU112 (C), and UU125 (D). Experimental data are presented as mean ± S.D. (n = 3). Solid lines represent the simulations. SCHHs (with or without disrupted bile canaliculi) were incubated with pitavastatin at a concentration of 0.1 µM (UU113), 0.6 µM (UU115), 1 µM (UU112), or 2.5 µM (UU125) for 1–45 minutes, after which intracellular (disrupted bile canaliculi) or combined intracellular and bile accumulation (intact bile canaliculi) was quantified by UPLC-MS/MS. During the experiments, medium samples were collected and analyzed for pitavastatin content.
Measures of predictive performance for simulations of pitavastatin disposition in four different batches of sandwich-cultured human hepatocytes (SCHHs)
Hepatic disposition was characterized by a rapid uptake during the first 20 minutes, followed by a steady state, indicating that the summed efflux rate (basolateral and canalicular) had increased to a level matching the uptake rate. Canalicular efflux of pitavastatin was low compared with the cellular uptake. This finding prevented us from getting reliable experimental data on the biliary accumulation owing to the small difference observed for the combined intracellular and bile accumulation and the intracellular accumulation. The highest biliary excretion index (BEI) observed was 9% at 10 minutes of incubation time for the batch incubated with the highest concentration of pitavastatin (UU125). This value is in the proximity of the BEI range of 12%–21% reported elsewhere (Abe et al., 2009). The BEI and intrinsic biliary clearance (CLbile) of the probe substrates taurocholate (BEI = 61%, CLbile=7.54 µl/min/mg protein) and rosuvastatin (BEI = 54%, CLbile=1.97 µl/min per milligram of protein) was also comparable to that observed by others (Bi et al., 2006; Abe et al., 2009). The excretion of substrates into the canalicular networks of our SCHHs was thus fully functional.
Prediction of Transporter Contribution to Hepatic Uptake and Efflux in SCHHs.
By characterizing the uptake/efflux mediated by each individual transporter, we were able to predict their contribution to the disposition in SCHHs over time (Supplemental Figs. S2 and S3). OATP1B1 was identified as the major uptake transporter, with a contribution of 87% in batch UU113, 79% in batch UU115, 58% in batch UU112, and 70% in batch UU125 at steady state. NTCP and OATP1B3 were the second most important transporters (Supplemental Fig. S2). NTCP played a particularly large role in batch UU112, with more than 20% contribution to the active uptake. Transporter contributions to hepatic uptake did not vary much over time as a result of the small changes in medium concentration and the low substrate concentration in relation to transporter Km values.
Of the canalicular transporters, BCRP was found to be more important than MDR1 for the biliary efflux. Over time, however, its contribution decreased as a result of increased intracellular accumulation of pitavastatin causing a saturation of the transporter. Hence, MDR1 became more involved in the canalicular transport over time (Supplemental Fig. S3). The saturation was, as expected, more evident in the experiments with higher starting concentration, resulting in higher accumulation of pitavastatin intracellularly.
Sensitivity Analysis.
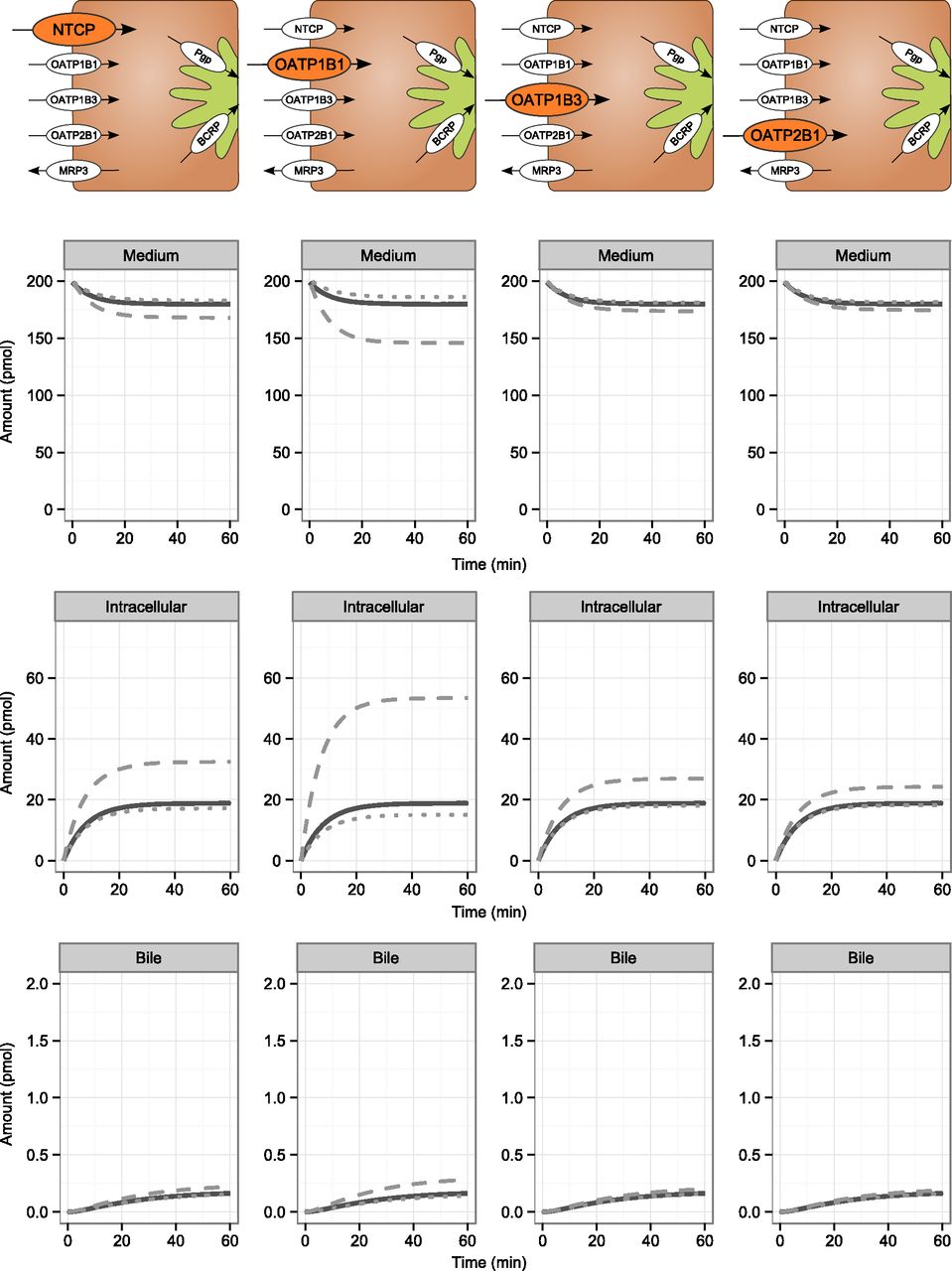
The impact of altered transporter activities on medium, intracellular, and bile concentration-time profiles were investigated by increasing and decreasing the maximal transport rate of each transporter 10-fold while keeping the other parameters fixed. Although increasing or decreasing the activity of OATP1B3 and OATP2B1 changed their contributions to hepatic uptake several-fold, it had a low impact on the time profiles (Fig. 5). Altered OATP1B1 activity, on the other hand, influenced medium, intracellular, and bile profiles (Fig. 5) to a larger extent. The influence of NTCP activity on the time profiles was greatest for batch UU112, in which NTCP was predicted to contribute the most to hepatic uptake of the four batches studied. However, the impact of altered NTCP activity was not nearly as large as that of OATP1B1 (Fig. 5).

Impact of altered uptake transporter activities on the simulated time profiles of medium, intracellular, and bile accumulation of pitavastatin for SCHH batch UU112. The maximal transport rate of each transporter was varied over a 0.1-fold (dotted line) to 10-fold (dashed line) range of the experimental value. The reference simulation is represented by the solid line.
Canalicular transport activities of BCRP and MDR1 had minimal effects on medium and intracellular profiles. They were instead more important for the accumulation in the bile compartment (Fig. 6). The basolateral transporter MRP3 did not influence any of the time profiles, indicating a very minor involvement of active basolateral efflux in pitavastatin disposition. In all sensitivity analyses, the increase in transport activity was associated with larger changes in time profiles than was a decrease. This finding reflects the overlapping function of the transporters in the uptake and efflux of pitavastatin.

Impact of altered efflux transporter activities on the simulated time profiles of medium, intracellular, and bile accumulation of pitavastatin for SCHH batch UU112. The maximal transport rate of each transporter was varied over a 0.1-fold (dotted line) to 10-fold (dashed line) range of the experimental value. The reference simulation is represented by the solid line.
To analyze the sensitivity of the model to parameters other than transport activities, Kflux and fraction unbound were varied 10-fold above and below their initial values (Supplemental Fig. S4). The Kflux parameter did not influence the medium and intracellular profiles but had a small impact on the biliary profile. The fraction unbound, on the other hand, greatly influenced medium and intracellular accumulation of pitavastatin.
Discussion
In this study, we have shown that experiments using in vitro systems, in which each transporter is studied in isolation, can be used to simulate drug disposition in more complex systems such as SCHHs. The simulations were performed on an individual level by including the interindividual variability in transporter abundances.
The bottom-up approach of modeling used herein is less frequently used than the top-down approach, in which parameters describing the disposition are estimated by fitting the model to in vitro/in vivo observations. Although top-down modeling can reveal important information about the drug being modeled, such as the rate-limiting step of the elimination, it normally has limited use for drugs, where observations from hepatocyte or in vivo experiments are lacking. The bottom-up approach, on the other hand, is readily applicable to early drug development. The input data on which it depends are kinetic determinations of transport processes in well-defined in vitro systems and knowledge of the transporter abundance in the in vitro system in relation to the more complex system being modeled. This gives the advantage of being able to estimate the kinetics of the average individual and, more importantly, of the more extreme individuals, who are the ones at risk of toxicity or lack of efficacy, by including interindividual variability in transporter abundances in the model.
The bottom-up approach also has the advantage of revealing the contribution of each transporter to the hepatic disposition. Transporter contributions have implications for the clinical importance of reduced function genetic variants and drug-drug interactions. The hepatic uptake of pitavastatin was mainly dependent on OATP1B1, with a contribution of 57%–87% in the four batches of SCHHs. This result is in agreement with the contribution reported in published studies, ranging from 32% to 98% of total active uptake (Hirano et al., 2004; Williamson et al., 2013; Kunze et al., 2014). The high variability in OATP1B1 contribution may be a result of genetic variations (Niemi et al., 2011) but could also be a result of hepatocyte isolation and culture (Ulvestad et al., 2011; Vildhede et al., 2015). The importance of OATP1B1 for pitavastatin disposition is supported by clinical studies where associations between OATP1B1 reduced function genetic variants and increased pitavastatin plasma concentrations are reported (Ieiri et al., 2007; Oh et al., 2013).
Besides OATP1B1, NTCP has been suggested as an important transporter in pitavastatin disposition, with a 29% contribution to the active hepatic uptake (Bi et al., 2013). In our four SCHH simulations, NTCP contributed to 6%, 9%, 9%, and 22% of total active uptake, indicating that it can be an important transporter in some hepatocyte batches, depending on the relative transporter abundance levels.
Of the canalicular transporters, BCRP was the most important, mediating more than 75% of the biliary efflux in all SCHH batches. Studies in Bcrp1-deficient mice and Mrp2-deficient rats suggest a major role of Bcrp1 and a minor role of Mrp2 in the disposition of pitavastatin, respectively (Fujino et al., 2002; Hirano et al., 2005b). Whereas a similar transporter contribution pattern in humans is possible, species differences cannot be excluded.
Different culture conditions and substrate concentrations can result in variable biliary excretion. For quantitative extrapolation to the in vivo situation, it is therefore important to use standardized protocols that have been optimized for such purpose (Lee et al., 2010b). In vitro-in vivo extrapolation was not the scope of this study. Instead, our study serves as a proof-of-concept for the applicability of the bottom-up modeling approach undertaken. A limitation of our study was the low biliary efflux in relation to the high uptake of pitavastatin in our sandwich cultures, resulting in high relative measurement error for the derived bile concentrations. This experimental limitation complicated comparisons of predicted and observed biliary accumulation, and we could therefore not verify our predictions of the biliary accumulation. Follow-up experiments using a substrate with higher biliary accumulation could improve the confidence in our predictions of the bile accumulation by allowing for such comparisons. Nevertheless, our simulations generally agreed well with experimental observations with an exception for the combined intracellular and bile accumulation for batch UU112. The lack of dose proportionality for this batch is, however, physiologically unlikely, given that the expression of transporters is comparable to the other batches. Moreover, to recover the intracellular and bile accumulation observed for UU112, the uptake transporter capacities would need to be increased several-fold, resulting in an overprediction of the intracellular accumulation (simulations not shown). We therefore hypothesize that the high intracellular and bile accumulation observed for UU112 is an experimental error (e.g., owing to inefficient washing in the experiment in presence of divalent cations).
The mechanistic model presented herein relies on the following assumptions: 1) functional activity is directly related to transporter abundance and is not limited by other factors such as cofactor concentrations; 2), transporters are independent of each other; 3) all transporters quantified are active; 4) pitavastatin is not bound to any considerable extent in the medium compartment; 5) intracellular metabolism is negligible; 6) pitavastatin is not distributed back into the cells from the biliary compartment; and 7) the dynamic pulsing of bile compartments can be described as a linear release of accumulated drug.
A linear correlation between transporter function and expression has been reported for both uptake and efflux transporters in cell models and membrane vesicles (Miliotis et al., 2011; Kumar et al., 2015). This finding supports our first assumption of a functional dependence on transport protein abundance. The transporters may also be considered to operate independently of each other during the short time frame of the experiments. Regulatory changes in transport activities, such as after induction of transport protein expression, will occur over longer time periods than those applied here. Further, changes in protein expression from culturing the hepatocytes are taken into account by the protein abundance-dependent scaling factor.
Transporter abundance levels were quantified in crude membrane fractions. Because canalicular efflux proteins are known to undergo recycling from intracellular membrane pools (Wakabayashi et al., 2006), a fraction of the measured protein levels in SCHHs may reflect transport proteins localized in intracellular compartments rather than in the plasma membrane, where they are functionally active. Assuming all transporters are active, although in fact some are localized intracellularly, would cause the model to overpredict the canalicular efflux; however, pitavastatin efflux was (if anything) underpredicted, which may indicate that SCHHs depend less on recycling of canalicular transporters than their in vivo counterpart. The underprediction could also reflect an involvement of MRP2 in the canalicular efflux. Whereas we did not detect any MRP2-mediated transport of pitavastatin in our membrane vesicles, results from other in vitro studies suggest that MRP2 may transport pitavastatin (Hirano et al., 2005b; Ellis et al., 2013). In addition, genetic variants of MRP2 have been associated with altered pitavastatin exposure in healthy volunteers (Oh et al., 2013). In contrast to the canalicular transporters, the OATP transporters have been suggested not to undergo recycling from intracellular pools (Roma et al., 2008).
The assumption that all quantified transporters are functionally active requires that the transport proteins are oriented correctly in the membrane, which is not the case for all transport proteins present in the membrane vesicle preparations given that only a fraction of the vesicles are oriented inside-out. To take this into account, we corrected our protein abundance measurements by using an estimated fraction of inside-out vesicles of one-third (Keppler et al., 1998); however, the proportion of inverted vesicles varies from batch to batch depending on preparation techniques. We therefore performed additional simulations to investigate the sensitivity of our predictions to the finverted parameter. The parameter affected bile accumulation to a limited extent; hence, our assumption did not affect the overall results and conclusions. For substrates whose hepatic disposition is more dependent on canalicular efflux, on the other hand, this parameter could have a significant impact on the results. In such cases, finverted should be determined for each vesicular batch that is used (Loe et al., 1996; Volk and Schneider, 2003).
Pitavastatin is metabolized to a limited extent (Elsby et al., 2012). Data presented in this manuscript also indicate that the assumption of a very low nonspecific binding is valid since the measured amount of pitavastatin in the medium samples matched the amount of drug added in the experiments. Negligible passive diffusion across the canalicular membrane is a common assumption when modeling hepatic disposition; the rationale for this assumption is the stiffness of the apical membrane with its high content of sphingolipids and cholesterol (Zegers and Hoekstra, 1998). The description of the dynamic pulsing of the bile canalicular compartments as a linear function is a simplification of reality; however, this approach has successfully been used to describe rosuvastatin disposition in sandwich cultures in a previous study (Pfeifer et al., 2013) and was hence applied herein.
In summary, this is the first study to model hepatic uptake and efflux on the detailed level of individual transporters using a bottom-up approach. With the knowledge of transporter contributions derived from the model, changes in disposition resulting from genetic variants or drug-drug interactions are expected to be predicted with higher precision. Our modeling approach may be expanded to include more processes, such as metabolism, and can also be applied for a more deconvoluted parameterization of PBPK models.
Acknowledgments
The authors thank Dr. Ed LeCluyse for sharing expertise in hepatocyte isolations and sandwich cultures; Drs. Johan Palm, Marie Brännström, and Constanze Hilgendorf at AstraZeneca for providing the HEK vesicular batches used throughout the study; and Drs. Agneta Norén, Frans Duraj, and Jozef Urdzik for assisting with human liver tissue material for the hepatocyte isolations.
Authorship Contributions
Participated in research design: Vildhede, Mateus, Artursson, Kjellsson. Artursson was in charge of the overall conception and design of the study.
Conducted experiments: Vildhede, Mateus, Khan, Lai.
Performed data analysis: Vildhede, Kjellsson.
Wrote or contributed to the writing of the manuscript: Vildhede, Mateus, Khan, Lai, Karlgren, Artursson, Kjellsson.
Footnotes
- Received August 12, 2015.
- Accepted February 1, 2016.
This work was supported by the Swedish Research Council [grant approval no. 2822]; the Lars Hierta Memorial Foundation; and O. E. and Edla Johansson’s Scientific Foundation.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- BCRP
- breast cancer resistance protein
- BEI
- biliary excretion index
- CL
- clearance
- DDI
- drug-drug interaction
- DPBS
- Dulbecco’s phosphate-buffered saline HBSS, Hank’s balanced salt solution
- HEK
- human embryonic kidney
- MDR
- multidrug resistance protein
- MRP
- multidrug resistance-associated protein
- NTCP
- sodium taurocholate cotransporting polypeptide
- OATP
- organic anion transporting polypeptide
- SCHH
- sandwich-cultured human hepatocyte
- UPLC-MS/MS
- ultra-high performance liquid chromatography-tandem mass spectrometry
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}