


Visual Overview

Abstract
As part of the drug discovery and development process, it is important to understand the human metabolism of a candidate drug prior to clinical studies. Preclinical in vitro and in vivo experiments across species are conducted to build knowledge concerning human circulating metabolites in preparation for clinical studies; therefore, the quality of these experiments is critical. Within AstraZeneca, all metabolite identification (Met-ID) information is stored in a global database using ACDLabs software. In this study, the Met-ID information derived from in vitro and in vivo studies for 27 AstraZeneca drug candidates that underwent human absorption, distribution, metabolism, and excretion studies was extracted from the database. The retrospective analysis showed that 81% of human circulating metabolites were previously observed in preclinical in vitro and/or in vivo experiments. A detailed analysis was carried out to understand which human circulating metabolites were not captured in the preclinical experiments. Metabolites observed in human hepatocytes and rat plasma but not seen in circulation in humans (extraneous metabolites) were also investigated. The majority of human specific circulating metabolites derive from multistep biotransformation reactions that may not be observed in in vitro studies within the limited time frame in which cryopreserved hepatocytes are active. Factors leading to the formation of extraneous metabolites in preclinical studies seemed to be related to species differences with respect to transporter activity, secondary metabolism, and enzyme kinetics. This retrospective analysis assesses the predictive value of Met-ID experiments and improves our ability to discriminate between metabolites expected to circulate in humans and irrelevant metabolites seen in preclinical studies.
Introduction
A typical drug discovery and development campaign includes metabolite identification (Met-ID) studies performed on a large number of compounds with the aim of optimizing metabolic clearance, designing away from reactive metabolite formation, creating awareness of active metabolites, and, most importantly, building confidence with respect to which human circulating metabolites to expect in clinical studies (Isin et al., 2012; Stepan et al., 2013). This knowledge will support the planning and execution of appropriate safety studies (e.g., selection of toxicology animal species prior to clinical trials). Without a streamlined way of capturing this structural information, metabolite knowledge generated on the many hundreds of project compounds studied is easily lost, as is the ability for collective learning. To remedy this issue, we have established a companywide metabolite database with the aim of capturing information on metabolites generated at all stages of the drug discovery and development process from both in vitro and in vivo experiments carried out at all AstraZeneca sites.
Before taking drug candidates to the clinic to assess safety, tolerability, pharmacokinetic properties, and efficacy, it is essential to build confidence in the safety of the new chemical entity using a host of in silico as well as in vitro and in vivo preclinical models. In addition to the safety of the parent drug candidate, it is equally important to evaluate the safety of the expected human metabolites (Frederick and Obach, 2010; Nedderman et al., 2011). Prior to exposing human volunteers or patients to a drug candidate, Met-ID studies in vitro and in animal models constitute the basis as surrogates for safety testing of expected human metabolites (Luffer-Atlas, 2012). These approaches have the limitations inherent to the in vitro models (e.g., hepatocytes neglecting extrahepatic metabolism, viability of cells), as well as limitations owing to differences in biotransformation and drug disposition between the various species used in the safety studies, and they thus may be of questionable predictive value. From a biotransformation perspective, the output from these and other investigational studies is a starting point in building an understanding of the metabolic routes in humans and ultimately an understanding of the full disposition of the drug candidate of interest. It is also an important part of the preparations for the first clinical studies in which the drug candidate will be administered to humans for the first time (“first-in-human studies”). Although urine and plasma samples should both be collected for metabolite investigations in these first clinical studies, the focus is on metabolites in the systemic circulation for comparison with those metabolites circulating in the animal species used in the early preclinical safety studies. Plasma samples from these preclinical safety and the first human studies are analyzed for comparison of metabolite profiles and their relative exposure to each species (Gao and Obach, 2011; Ma and Chowdhury, 2011). Early identification of significantly different metabolite profiles between species may give time to proactively mitigate and preferably avoid resource-consuming problem-solving studies. Such studies may involve, but are definitely not limited to, synthesis of the metabolite(s) and dosing to animals to reach adequate exposure, or even identifying new animal species that will form the human metabolite to adequate exposure levels and repeating the in vivo toxicology program (Smith and Obach, 2006, 2009; Guengerich, 2009; Leclercq et al., 2009).
In this report, we describe an in-house database in which in vitro and in vivo animal and human biotransformation data on compounds from early as well as late-stage drug development projects have been captured and stored over a period of close to 1 decade. This database currently contains >12,500 biotransformation schemes for >5000 compounds. We present here our first investigations on how these data can form a knowledge base from which we can build an understanding of the quality of in vitro and preclinical systems for the prediction of circulating metabolites in humans.
Materials and Methods
Met-ID Studies.
Met-ID information was generated from the analysis of AstraZeneca proprietary compounds in in vitro incubations in liver microsomes (LMs) and hepatocytes from humans, rats, dogs, mice, and rabbits. Similarly, Met-ID information from reactive metabolite trapping experiments in human liver microsomes (HLMs) with glutathione, potassium cyanide, and methoxylamine and from Aroclor 1254–induced rat liver S9 fraction experiments were generated. Met-ID data from in vivo samples including plasma, urine, bile, and feces from dosed rats, dogs, mice, rabbits, cynomolgus monkeys, and marmosets and plasma, urine and feces from humans were likewise generated.
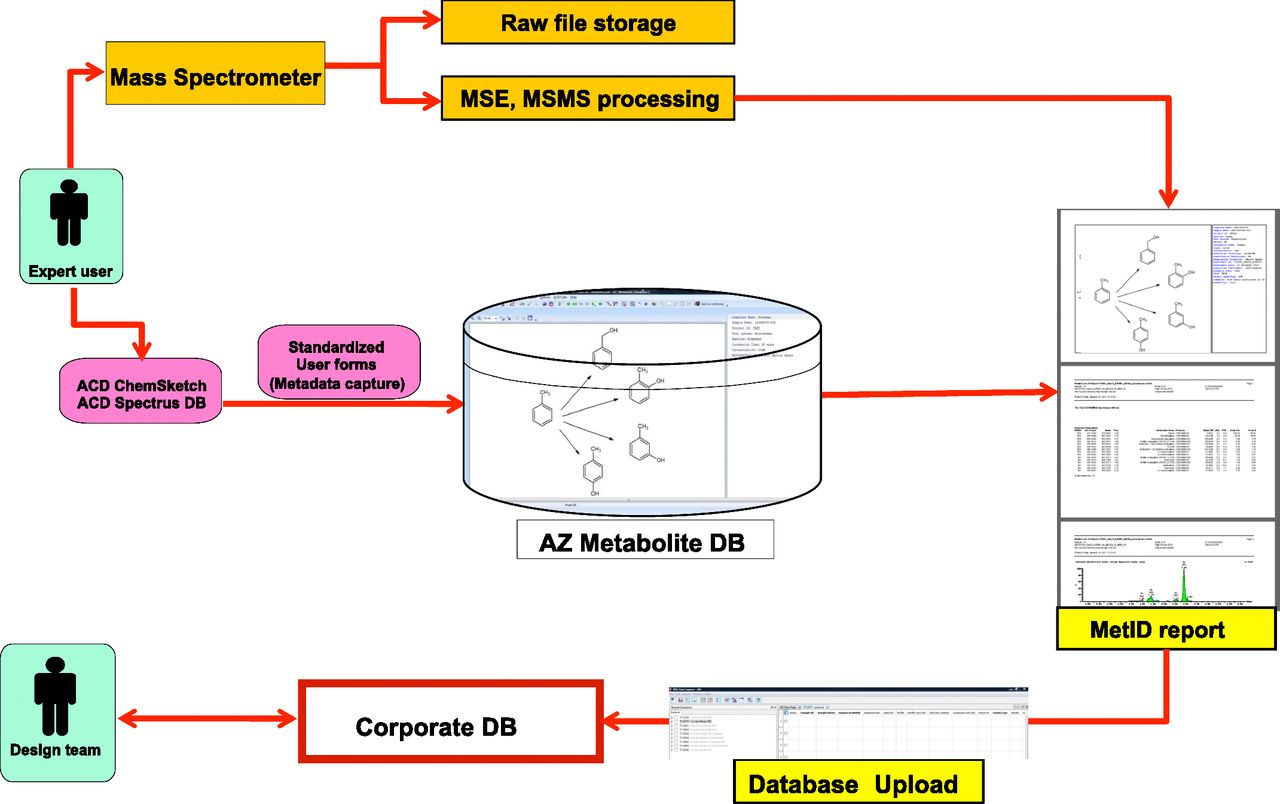
The analysis of in vitro and in vivo Met-ID experiments is typically performed by liquid chromatography coupled to tandem mass spectrometry on a Waters ultra high-performance liquid chromatograph system with a SYNAPT quadrupole time-of-flight mass spectrometer (Waters, Wilmslow, UK) or on ThermoFinnigan OrbitTrap instrumentation (Thermo Scientific, Waltham, MA) (Ekdahl et al., 2013). Tandem mass spectra of the parent compound and metabolites are acquired and analyzed using a combination of vendor software (e.g., MetaboLynx; Waters) and interpretation is carried out by biotransformation experts. Structures of both the parent and metabolites are drawn as a biotransformation scheme in ACDLabs Spectrus DB Enterprise software (Advanced Chemistry Development, Toronto, ON, Canada) with reaction arrows linking metabolites to the parent molecule. Metabolite numbering, quantification (by mass spectrometry response or radioactivity) of each metabolite, and biotransformation type are also captured at this stage. The scheme is then uploaded to the database, along with a standardized form capturing globally agreed experimental metadata fields (e.g., responsible scientist, test system, project code, date, etc.).
PDF reports of the new database record are also generated and shared with drug discovery teams via upload to the IBIS corporate drug discovery database (Fig. 1).

Biotransformation data capture and data flow showing publishing of Met-ID data into IBIS via the global metabolite database. AZ, AstraZeneca; DB, database; MSE, mass spectrometry at elevated collision energy; MSMS, tandem mass spectrometry.
ACD Database.
The ability of the software to draw and capture Markush structures (Markush, 1924; Cosgrove et al., 2012) to cover vagueness in structural assignments is a useful feature that facilitates reporting of metabolites in cases in which the site of metabolism cannot be narrowed down to a single atom (Yerin and Peirson, 2012). The common platform nature of the database allowed biotransformation scientists globally across the AstraZeneca organization to capture metabolite structural information in the form of biotransformation schemes and experimental metadata.
The ACD database currently contains >12,500 schemes and approximately 50,000 metabolic reactions on proprietary AstraZeneca compounds, of which only a small percentage are in the public domain.
AstraZeneca Metabolite Database Search.
The AstraZeneca metabolite database can be searched in two ways: by text or by structure searching. Experimental metadata (e.g., incubation time, compound and project identifiers, and responsible scientist) are captured as text fields alongside the biotransformation scheme. These are accessible within the database by a simple text-search function.
Exact, similarity, or substructure searches can be performed and are accessed via the ChemSketch drawing module. Parent or metabolite structures or substructures are drawn in ChemSketch or simply imported from the database and suitably modified. The desired structure is selected and searched against the entire database.
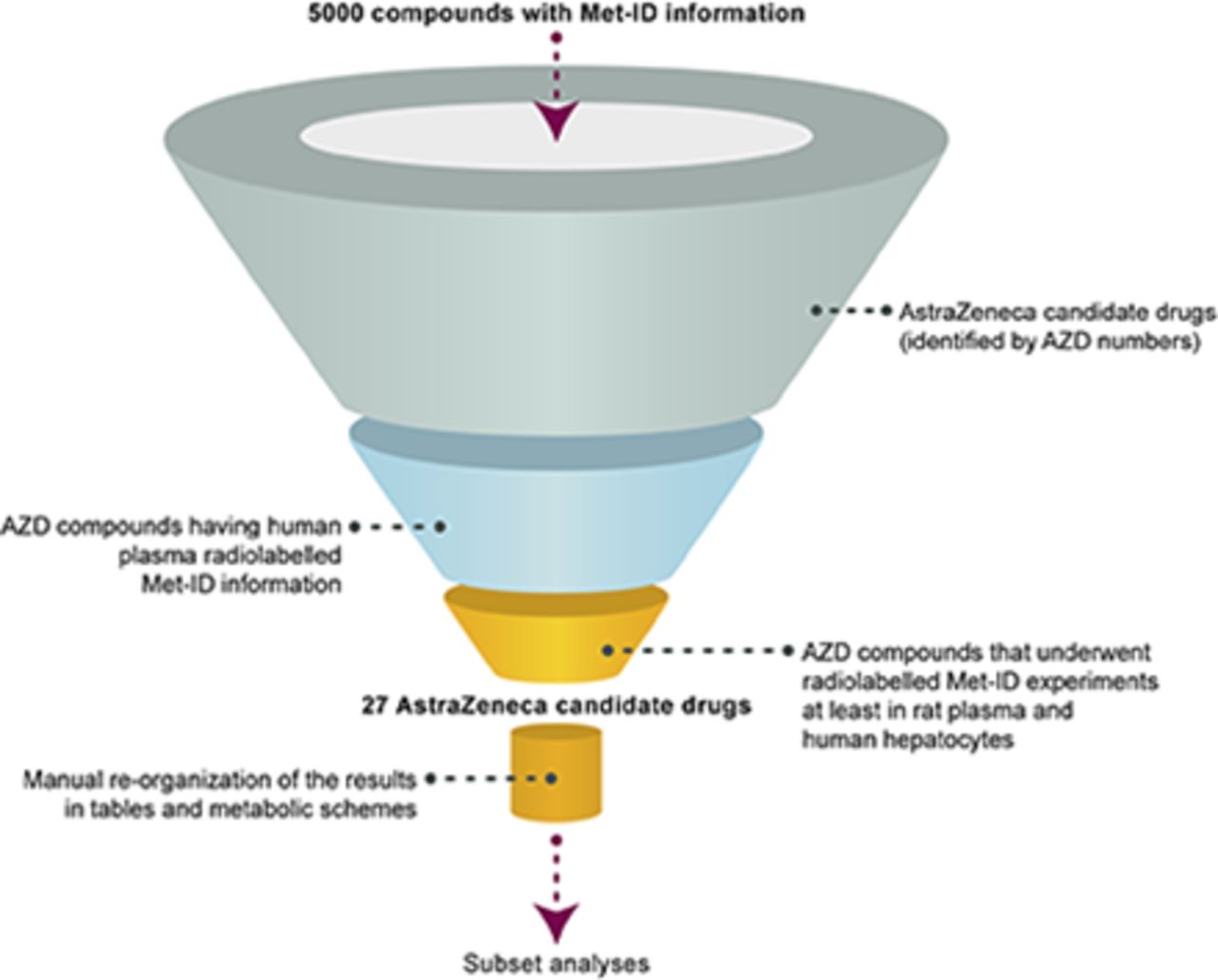
For the purposes of this work, Met-ID information on AstraZeneca compounds that underwent radiolabeled studies in humans and for which plasma metabolite profiles were available was extracted using the text-search option. The list was refined further by carrying out text searching for compounds having human hepatocytes and rat plasma Met-ID information (Fig. 2). Subsequently, the substructure search option was used to determine in which matrices each metabolite was observed.

A graphic illustration of the filters applied to the search to obtain the subset analyzed. The first three filters were applied using the text-search option, whereas the manual reorganization part was done together with a substructure search to identify the matrices in which the same metabolites were observed.
Results
Database Extraction.
As a result of the database extraction effort, a list of 27 candidate drugs that underwent radiolabeled cross-species in vitro, preclinical in vivo, and human absorption, distribution, metabolism, and excretion (ADME) studies was compiled. For each compound, the database provided a list of records corresponding to the various biotransformation reactions reported to occur in different matrices (e.g., hepatocytes, LMs, induced rat liver S9 fraction, urine, plasma, feces, bile) of different species (e.g., humans, rats, dogs, monkeys, mice, rabbits). An example of an ACD database record containing Met-ID information obtained from the analysis of male rat plasma is given in Supplemental Fig. 1. Prior to the descriptive and structural analysis, a unique metabolite scheme, including all of the metabolites observed during these Met-ID studies, was built on the extracted data for each of the 27 compounds. One such summary scheme is shown in Fig. 3. Moreover, tables listing all metabolites versus matrices were created for each compound to facilitate the various analyses; as an example, the table created for AZD1 is given in Supplemental Table 1.
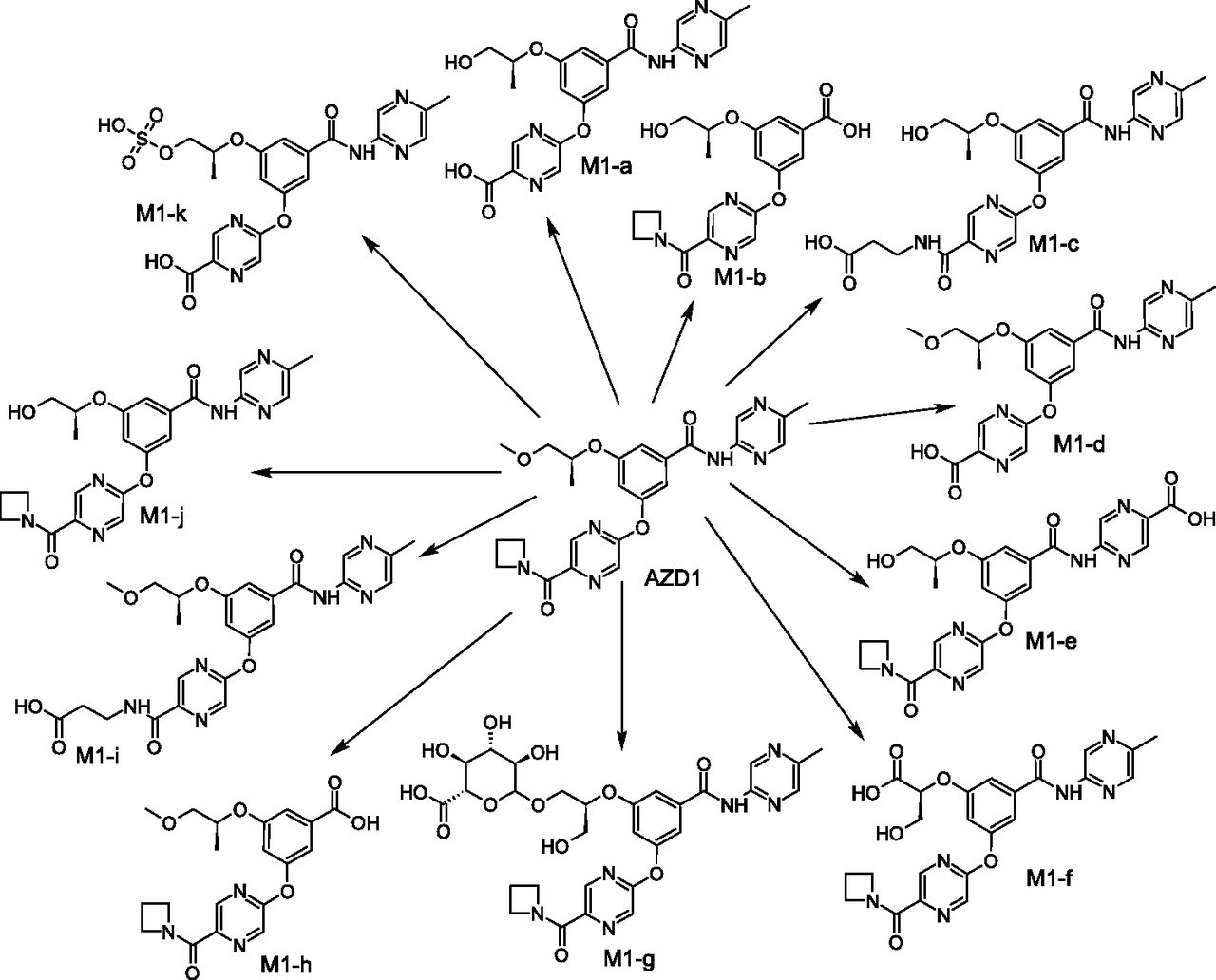
Comprehensive metabolic scheme for compound AZD1. The manually constructed scheme includes all of the metabolites observed in all of the matrices on which Met-ID experiments were performed.
Descriptive Analysis.
The aim of the first part of the work was to evaluate the quality of our in vitro and in vivo preclinical experiments in terms of human metabolite prediction (Fig. 4). All 27 AstraZeneca compounds underwent Met-ID studies in human and rat hepatocytes and in vivo in rats and humans using radiolabeled compound. Depending on the drug project, some compounds were also tested either in vitro and/or in vivo in other species (e.g., cynomolgus monkeys, marmosets, mice, or dogs). However, because these species were not uniformly used as a test system for the whole subset of candidate drugs reported in this work, these data were included only in the descriptive analysis.

Definition of the terms used in this article to describe the experiments performed at different stages of the drug discovery and development process.
From the Met-ID studies on the 27 AstraZeneca candidate drugs, a total of 564 metabolite structures were identified, including those with Markush labeling. Of the 564 metabolite structures, 234 were observed in human in vivo and/or in vitro samples and 144 of these were found in systemic circulation (i.e., observed in plasma). Of the 144 human circulating metabolites, 117 (81%) were observed before the human ADME study in at least one other test system. The actual number of previously observed human circulating metabolites varied, however, between candidate drugs (Fig. 5). In 16 of the 27 cases, it would have been possible to predict all human circulating metabolites based on Met-ID experiments prior to beginning first-in-human studies.

Bar chart comparison of the number of human circulating metabolites (in red) with the ones previously observed in preclinical studies (in blue).
A descriptive analysis was performed to assess the frequency at which human excreted metabolites (i.e., observed in urine and/or feces samples) were observed in preclinical in vitro or in vivo studies prior to beginning first-in-human studies. The analysis showed that 73% of the excreted human metabolites (157 of 216) were observed in at least one study type prior to the human ADME studies.
An evaluation of the predictive value of the different in vitro and in vivo systems used preclinically was carried out for human circulating metabolites. The Venn diagram in Fig. 6 shows that by performing in vitro experiments only (all studied test species and systems), 46% (33% human hepatocytes only) of human circulating metabolites were observed. On the other hand, by performing in vivo studies only (all studied test species and matrices), 77% of circulating metabolites in humans were observed.

This analysis includes all of the in vitro data available (hepatocytes and/or microsomes and/or S9 fractions from humans, rats, dogs, monkeys, rabbits, mice, and common marmosets) and in vivo data (plasma, urine, feces, and bile from humans, rats, dogs, monkeys, mice, and marmosets) for the 27 compounds. The numbers indicate the quantity of observed metabolites.
Human Circulating Metabolites Not Observed in Preclinical Studies.
For the 27 candidate drugs studied, only 27 circulating human metabolites (of 144) were seen for the first time in humans, whereas the remainder were previously observed in in vitro and/or in vivo preclinical studies (Fig. 6). The biotransformation routes leading to these 27 previously unobserved metabolites were scrutinized to understand the underlying reasons for this lack of predictability. An analysis of the metabolic schemes indicates that the metabolites missed were primarily derived from multistep reactions, and evidence for the pathways leading to these metabolites had already been captured in in vitro experiments by the identification of intermediate or precursor metabolites. In several cases, these multistep pathways resulted in the formation of a conjugated metabolite but examples of oxidative multistep biotransformations were also identified. The initial enzymatic O-dealkylation of AZD24 is followed by phosphate hydrolysis leading to M24-a (Fig. 7), which was observed in human hepatocytes as well as in rat plasma and urine. However, metabolite M24-b, derived from the sulphonation of the intermediate phenol (M24-a), was observed for the first time in human plasma.

Multistep biotransformation of AZD24 observed in human plasma resulting in the formation of the previously unobserved sulphonated metabolite M24-b.
Another example of a previously unobserved human circulating metabolite involves AZD8 (Guo et al., 2015). A hydroxylated metabolite M8-a (Fig. 8) was observed in human plasma and urine, mouse and rat plasma, and human hepatocytes. The metabolite M8-b derives from the reaction of the drug (and/or M8-b) with endogenous CO2 to form a carbamic acid intermediary metabolite, which is subsequently conjugated with glucuronic acid. This resulting N-carbamoyl glucuronide M8-b was captured in human plasma only and was hence unobserved prior to human studies (Schaefer, 2006).

M8-b shows a previously unobserved human metabolite deriving from multistep metabolism of AZD8.
Only one compound (AZD10) had a metabolite found for the first time in human plasma, in which no precursors were captured in in vitro systems. As shown in Fig. 9, the hydrolytic ring opening of the candidate drug AZD10 gives the previously unobserved metabolite M10-a.

Metabolism of a methylpyrazinone moiety leading to a previously unobserved human circulating metabolite.
Extraneous Metabolites.
The analysis of the 27 AstraZeneca compounds indicates that 58% of the metabolites observed during the preclinical Met-ID studies (in vitro and/or in vivo) are not relevant to what is observed subsequently in human circulation. In other words, during preclinical Met-ID work, 330 of the metabolites formed and characterized are not found in circulation in humans.
A deeper structural analysis of the metabolites observed in human hepatocytes and rat plasma, but not identified in human plasma, was performed and the results are shown in Table 1. Human hepatocytes and rat plasma were studied since these matrices were considered the most relevant to the retrospective analysis objective of this work. In particular, human hepatocytes allowed an in vivo/in vitro intraspecies comparison to be made and rat plasma enabled the cross-species comparison. The rat was chosen over other species because it was used in radiolabeled Met-ID studies for all of the subset of compounds analyzed.
Number of human circulating metabolites and extraneous metabolites observed in the different matrices
The extraneous metabolites are subdivided into three classes: not observed but evidence of the same pathways captured in human plasma, not observed in plasma but seen as excreted, and true misleading metabolites. The first two are the extraneous metabolites that are actually precursors or intermediates to the final metabolites captured in human plasma. The last class comprises metabolites irrelevant to the human circulating metabolites.
Structural Analysis of Extraneous Metabolites Captured in Human Hepatocytes.
Several metabolites observed in human cryopreserved hepatocytes but not in human plasma appear to be further metabolized in humans and hence these extraneous metabolites are intermediates or precursors of what is observed in in vivo studies. An example is depicted in Fig. 10 where M7-a is a metabolite observed in in vitro systems only (human and rat hepatocytes). The metabolites formed via further metabolism of M7-a (N-oxidation to M7-b, oxidation to carboxylic acid M7-c, glucuronidation to M7-d) are not observed in hepatocytes but are captured in human urine (M7-b, M7-c, M7-d) and human plasma (M7-b).

Example of a metabolite (M7-a) observed in human hepatocytes but not observed in humans in vivo. In the latter, products of further metabolism of M7-a were captured.
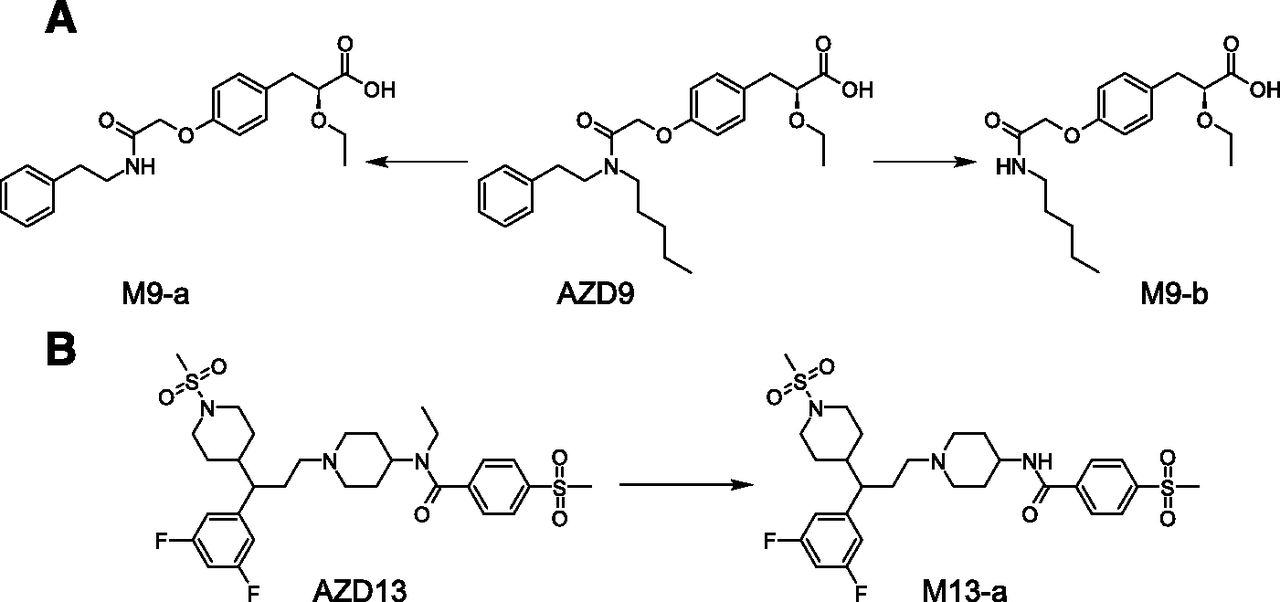
Interestingly, in several examples, metabolites seen in human hepatocytes but not in human plasma (or urine) are formed via amide dealkylation of tertiary amides. Examples for this biotransformation pathway are the secondary amide metabolites M9-a, M9-b derived from AZ9 (Fig. 11A), and M13-a derived from AZD13 (Fig. 11B), which were formed in human hepatocytes but were not found in human plasma or urine.

Examples of metabolites deriving from amide N-dealkylation found in human hepatocytes but not in human plasma.
Structural Analysis of Extraneous Metabolites Captured in Rat Plasma.
Metabolites captured in rat plasma but not observed in human plasma were investigated to understand whether certain biotransformations are species specific.
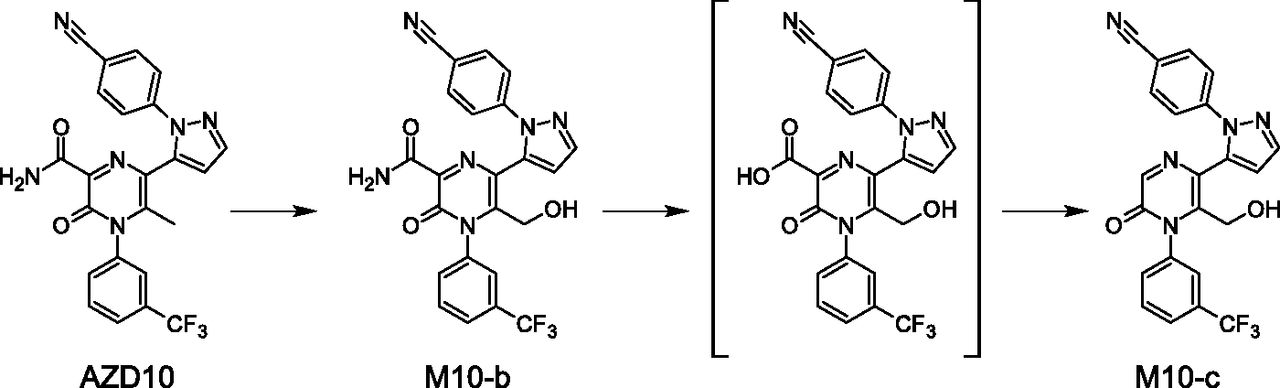
Of the metabolites seen in rat plasma but not in human plasma, a relatively large number are organic anions. However, although they are not observed in plasma, some of these metabolites are found in human urine. The majority of metabolites captured in rat plasma, but not circulating in humans, were formed via biotransformations occurring on alkyl substituents of heterocyclic aromatic rings. No evidence for these pathways or further metabolism was found in humans in vitro or in vivo. An example is the C-methyl-hydroxylation of the 2-methyl-pyrazine ring of AZD10, leading to the formation of M10-b (Fig. 12). M10-c is a metabolite derived from further metabolism of M10-b. The proposed pathways involve the hydrolysis of the amide to carboxylic acid and a decarboxylation of the carboxylic acid to form M10-c. Neither M10-b nor M10-c was seen in human matrices.

Metabolism occurring on a pyrazinone moiety leading to metabolites observed in rat plasma but not in human plasma.
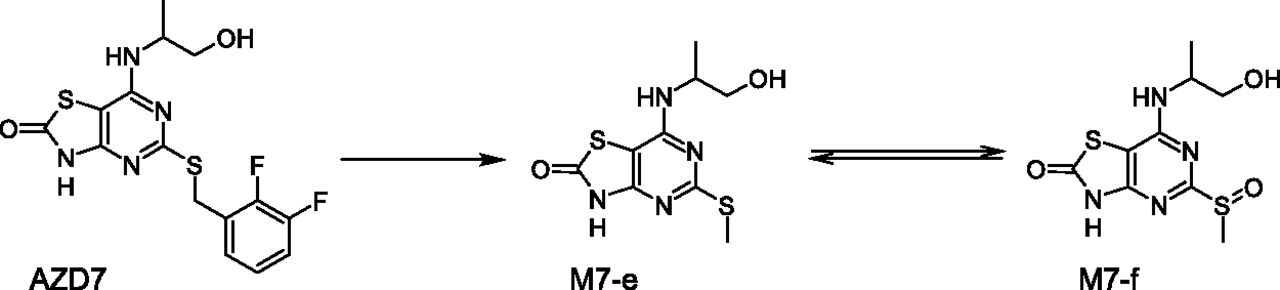
Some of the extraneous metabolites reported to be present in rat plasma suggest that biotransformations differ between species. An example for a reversible metabolic pathway as seen in rat and human plasma is shown in Fig. 13. M7-e derived from compound AZD7 is observed in vivo in rats but not in humans. On the other hand, M7-f is observed in human plasma and has been reported to form in rat hepatocytes but is not observed in rat plasma.

AZD7 underwent metabolism showing the redox reversibility between rats and humans.
Discussion
The AstraZeneca metabolite database provides a central repository for all metabolite structural information on AstraZeneca compounds. This database is a structure- and text-searchable resource that allows global sharing of metabolite data across AstraZeneca sites, therapeutic areas, and projects and ensures that metabolite knowledge accumulated over many years is permanently accessible. The database contains Met-ID data generated across the entire value chain from early discovery to product life cycle management. The majority of the data are from compounds synthesized during the discovery phase with human liver microsomal or hepatocyte Met-ID data; however, a subset of development compounds contains a more comprehensive package of data, including Met-ID from human ADME studies.
The AstraZeneca metabolite database differs from commercially available drug metabolite databases (BIOVIA Metabolite Database; BIOVIA, San Diego, CA) since it is built on studies of proprietary drugs and drug-like compounds combined with strictly defined experimental protocols and analytical methodologies.
The descriptive analysis of 27 AstraZeneca candidate drugs showed that in vitro and in vivo preclinical experiments were able to generate 81% of the human circulating metabolites and 73% of human excreted metabolites captured in the human ADME studies. In vitro experiments in human hepatocytes alone were able to predict 33% of human circulating metabolites. Inclusion of data from other in vitro systems in addition to human hepatocytes increases the predictability to 46%. This number is comparable to the outcome of an analysis by Anderson et al. (2009), who showed that in vitro data predicted the major human circulating metabolites for 41% of the studied drug candidates. In another study, human hepatocytes were reported to predict complete circulating metabolite profiles for 65% of the studied compounds (Dalvie et al., 2009). This observation is not surprising considering the caveats associated with in vitro systems (see below and De Graaf et al., 2002; Dalvie et al., 2009), particularly the limited incubation times as a result of the decaying metabolic activity, which leads to the inability of predicting secondary metabolites (Dalvie et al., 2008). Micropatterned hepatocyte coculture systems are reported to have considerably improved ability to predict major human metabolites over more conventional in vitro human systems such as microsomes and hepatocytes (Wang et al., 2010). That these types of systems can show enhanced metabolic capability over primary hepatocyte suspensions was demonstrated by the incubation of a set of 27 drug compounds for 7 days in a micropatterned coculture of hepatocytes. It was found that of the circulating human metabolites exceeding 10% of total circulating drug-related material, 75% were found in the in vitro coculture system compared with 53% found in human hepatocyte suspensions. Various other in vitro approaches to maintain metabolic capability for extended periods of time have recently been explored, including three-dimensional cell cultures such as spheroids (Godoy et al., 2013), liver bioreactor systems (Darnell et al., 2012), and stem cell–derived hepatocytes (Ulvestad et al., 2013), as well as different technical approaches such as the relay method (Ballard et al., 2014). Although these approaches show promise that further improved in vitro systems will predict a greater number of human metabolites (particularly secondary or tertiary metabolites), it remains uncertain whether the metabolites would be captured as excreted or in circulation in humans.
In vitro experiments as a whole (i.e., human hepatocytes and LMs; rat hepatocytes, LMs, and liver S9 fractions; and dog, mouse, rabbit, monkey, marmoset LMs and hepatocytes) gave very little information in addition to what was observed in vivo. Less than 4% of human circulating metabolites were unique to in vitro experiments. On the basis of this analysis, nonhuman in vivo systems may be perceived as a better system than human hepatocytes for predicting human circulating metabolites. However, the issue around extraneous metabolites (see below) observed in these in vivo systems must be taken into account for a proper comparison.
An analysis of human circulating metabolites not captured in preclinical experiments/studies was carried out and the results provided a plausible explanation for why some metabolites were seen for the first time in human circulation. Interestingly, only one metabolite (Fig. 9) was observed for the first time in humans and no reaction type or molecular motif consistently leading to a unique human metabolite was observed. Six of the metabolites classified as previously unseen are vague Markush structures. Because of the broadness of the Markush labeling, identification of the exact biotransformation that occurred is not possible; therefore, uncertainty remains regarding their elucidation.
In 13 of the 27 cases of previously unobserved human circulating metabolites, evidence of biotransformation pathways seen in human plasma is observed during in vitro cross-species experiments. Therefore, because of the limited viability of cryopreserved hepatocytes, the metabolism found in vivo in humans is more extensive and the final metabolite is thus not captured in the in vitro systems. Therefore, the predictability of human hepatocytes increases to 87% if metabolic pathways instead of specific human circulating metabolites are considered, which is similar to that reported by Dalvie et al. (2009) with respect to metabolic pathways. When LMs, hepatocytes, microsomes, and S9 fractions of other species (i.e., rat, mouse, dog, rabbit) are also included, the predictability of in vitro systems goes up to 92% with regard to biotransformation pathways. This assessment emphasizes the importance of the critical evaluation of observed metabolites by biotransformation scientists in an attempt to predict and later elucidate complete metabolic pathways in humans. Other examples (4 of 27) of previously unseen metabolites suggest that species specificity with respect to glucuronidation may play a role as to why human circulating metabolites are not seen in other species. Since conjugated metabolites are generally considered to be nontoxic, less concern has been expressed by the U.S. Food and Drug Administration over their presence in the circulation (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079266.pdf).
The number of human circulating metabolites predicted from human hepatocytes is similar to the number predicted from rat plasma. The number of extraneous metabolites in the two matrices is also comparable (Table 1). However, structural analysis reveals that extraneous metabolites found in human hepatocytes often tend to be a precursor or an intermediate of a human circulating metabolite (5 metabolites) or are seen as excreted metabolites (15 metabolites). This latter figure could be greater (20 metabolites) if metabolites were considered that could be excreted via bile and for which metabolite profile in feces were not analyzed. This could be the case for metabolites deriving from amide dealkylation and for glucuronides and other polar metabolites that, once formed in the liver, may be directly excreted via the bile or rapidly excreted via the urine without accumulation in plasma to detectable concentrations (Loi et al., 2013).
Overall, these analyses showed that in vitro experiments in human hepatocytes generate a few misleading metabolites owing to specific reactions taking place in human in vitro systems, and only five metabolites appear to be irrelevant to human in vivo metabolites. One of these five irrelevant metabolites underwent formylation biotransformation, and this might be the result of an artifact of the in vitro experiment itself (i.e., high concentration of formic acid). The other four metabolites found uniquely in human hepatocytes are metabolites of an AstraZeneca marketed drug and might reflect more extensive Met-ID work done on this drug.
When metabolites found in human hepatocytes are interpreted with the aim of predicting human circulating metabolites, two pieces of evidence must be considered: 1) the metabolites may be further metabolized in vivo in humans, and 2) finding metabolites in hepatocytes does not indicate whether they will enter the circulation.
On the other hand, extraneous metabolites in rat plasma are generally irrelevant to metabolites seen in human plasma (25 metabolites) or excreted metabolites (8 metabolites), suggesting that the prediction of human metabolites from in vivo studies may be misleading.
Misleading metabolites in rat plasma are mainly organic anions, which are formed in human in vitro systems but are not further metabolized in humans compared with rats. Therefore, whether a metabolite will be found in the circulation may depend not only on its formation per se but also on the species-specific expression of drug transporters such as the organic anion transporters (OATs). Species differences with respect to location on the apical or basolateral membranes of some human OATs and rat Oats (OAT2) are reported in literature (Burckhardt, 2012). OATs such as Oat5 have been demonstrated in rat kidney but not in humans, and their role is either to uptake xenobiotics from the primary urine into the cells or to release organic anions into the lumen.
A common trend was observed during the analysis of extraneous metabolites in rat plasma. In three cases, biotransformations occurring on substituents of heterocyclic aromatic rings led to metabolites that were observed in rats only. Different substrate specificity between otherwise similar drug-metabolizing enzymes in rats and humans might provide explanations for these observations. Therefore, in contrast with what was observed for extraneous metabolites found in human hepatocytes, extraneous metabolites found in rat plasma are not related to human metabolites.
Summary
The value of a corporate metabolite database was highlighted through the analysis of all metabolites generated during discovery and development for a set of 27 candidate drugs. The analysis showed that human hepatocytes alone were only able to predict 33% of the human circulating metabolites. However, the majority of metabolites missed were further transformations of the metabolites seen in hepatocytes. If instead the criterion was the indication of metabolic pathways, the predictability of human hepatocytes to human plasma increased to 87%. This indicates that the advances now being made in human hepatic in vitro systems, with improved viability and longer incubation times, should considerably improve our ability to predict which metabolites humans will be systemically exposed to.
Rat plasma was similar to human hepatocytes in the number of correctly predicted human circulating metabolites. However, the considerably larger number of metabolites irrelevant to human metabolism seen in rat plasma makes this a much less useful tool than human hepatocytes.
Acknowledgments
The authors dedicate this article to Prof. Neal Castagnoli, Jr. for his contributions to the field of drug metabolism and AstraZeneca drug discovery and development projects over a span of more than 30 years.
Authorship Contributions
Participated in research design: Iegre, Hayes, Thompson, Weidolf, Isin.
Performed data analysis: Iegre, Hayes, Weidolf.
Wrote or contributed to the writing of the manuscript: Iegre, Hayes, Thompson, Weidolf, Isin.
Footnotes
- Received October 28, 2015.
- Accepted February 10, 2016.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ADME
- absorption, distribution, metabolism, and excretion
- HLM
- human liver microsome
- LM
- liver microsome
- Met-ID
- metabolite identification
- OAT
- organic anion transporter
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}