


Abstract
Various in vitro preparations were compared with respect to their ability to mimic in vivo metabolism. For this purpose, S9-liver homogenate, microsomes, cryopreserved hepatocytes, cryopreserved liver slices and fresh liver, lung, kidney, and intestinal slices were incubated with three drugs in development, which are metabolized in vivo by a wide range of biotransformation pathways. Metabolites were identified and quantified with liquid chromatography-mass spectometry/UV from the in vitro incubations and compared with metabolite patterns in feces, urine, and bile of dosed rats. In vitro systems with intact liver cells produced the same metabolites as the rat in vivo and are a valuable tool to study drug metabolism. Phase I metabolites were almost all conjugated in intact cells, whereas S9-homogenate only conjugated by sulfation andN-acetylation. Microsomes and S9-homogenate are useful to study phase I metabolism but not for the prediction of in vivo metabolism. Extra-hepatic organ slices did not form any metabolites that were not produced by liver cells, but the relative amounts of the various metabolites differed considerably. Small intestinal slices were more active than liver slices in the formation of theN-glucuronide of compound C, which is the major metabolite in vivo. When the relative contribution of liver and small intestinal slices to the metabolism of this compound was taken into account, it appeared that the in vivo metabolite pattern could be well predicted. Results indicate that for adequate prediction of in vivo metabolism, fresh or cryopreserved liver slices or hepatocytes in combination with slices of the small intestines should be used.
In vitro research has been under growing interest for many disciplines within drug safety research but pre-eminently for the selection of animal species as models for human drug toxicity in late drug discovery and early development. In this phase, this is the only way to compare metabolism of a particular drug between animals and humans. From such a species comparison study, the animal species could be selected that forms the same major metabolites as humans do. A prerequisite for making an adequate selection is that the used in vitro system reliably predicts in vivo biotransformation of the drug (i.e., in vivo and in vitro patterns of the major metabolites should be comparable).
The in vitro interspecies comparison of drug metabolism approach in early drug development is normally very straightforward to serve speed. In our lab, normally a low and a high drug concentration are used, which are hopefully not too high such that toxicity occurs but high enough to produce detectable amounts of metabolite(s). In this phase, linearity in relation to drug concentration of the metabolite production is not studied, and other possible incubation variables are not optimized, like cell number used and incubation times. Only the major human metabolites are studied for their production by animal preparations.
As in vitro tool, both subcellular fractions and intact cells are intensively used. Subcellular fractions (i.e., S9-homogenates and microsomes) from various animal species and humans are commercially available and can be easily preserved for long periods of time. Disadvantages are the necessity of cofactor addition to facilitate metabolism and the lability (e.g., flavine monooxygenases) (Ekins et al., 1999), absence (e.g., cytosolar enzymes with microsomes) or inaccessibility for cofactor (e.g., glucuronyl-transferases) of some metabolic enzymes.
In contrast, in vitro preparations with intact cells (hepatocytes, tissue slices) possess the “complete” cellular machinery and have the ability for an integrated phase I and phase II metabolism of xenobiotics. A disadvantage of the use of hepatocytes is that isolation needs to be optimized for livers of every different animal species and involves collagenase digestion for disrupting cell-cell contacts. These problems are overcome when precision-cut tissue slices are used, which can be easily prepared from organs from various animal species, while the tissue architecture remains intact (Krumdieck et al., 1980). Several informative reviews discuss the applicability of liver slices in pharmaco-toxicological settings (Bach et al., 1996; Ekins, 1996b;Olinga et al., 1997b; Lerche-Langrand and Toutain, 2000). Recent studies have shown that extra-hepatic organ slices (lung, kidney, and intestinal slices) are almost as active as liver, metabolizing some drugs (Vickers, 1994; Vickers et al., 1995, 2001; de Kanter et al., 1999, 2002).
Long-term storage of slices and hepatocytes is more complicated than storage of subcellular fractions. Recently, we have developed a simple rapid freezing method for liver slices (de Kanter and Koster, 1995; de Kanter et al., 1998; de Graaf et al., 2000) and showed that post-thaw viability and phase I and II biotransformation activity of cryopreserved rat liver slices were maintained at least during 4 h after thawing. More complicated cryopreservation protocols exist for hepatocytes (Powis et al., 1987; Condouris et al., 1990; Diener et al., 1993). Cryopreserved hepatocytes are now commercially available.
In the present study, rat liver microsomes, S9-homogenate, cryopreserved hepatocytes, cryopreserved liver slices and fresh liver, lung, kidney, and intestinal slices are compared in their ability to predict in vivo metabolism. Furthermore, the metabolite patterns of precision-cut slices of liver, kidney, lung, and small intestine are combined on the basis of the relative contribution of these slices to the metabolism of the compound. Metabolites formed by the in vitro preparations are compared with those found in feces and urine of dosed rats. In addition, comparisons were made with the metabolite profile in bile, because some of the conjugates formed are deconjugated by microflora in the gut in vivo. The in vitro preparations are considered to be adequate predictors of in vivo metabolism if they meet two criteria. 1) They should produce the same metabolites as the intact animal and 2) they should produce these metabolites in approximately the same relative amounts as in vivo.
Three compounds from Solvay Pharmaceuticals Research, from here called compound A, B, and C were selected as model compounds, since these are metabolized in vivo via a wide range of metabolic routes. Metabolites were analyzed by LC1-MS/UV.
Materials and Methods
Materials.
Compound A, B, and C are products of Solvay Pharmaceuticals Research, UDPGA, 3-phosphoadenosine-5′-phosphosulfate, NADP+, NADPH, isocitrate, isocitrate dehydrogenase, phenylmethylsulfonyl fluoride, glycerol, glutathione, Acetyl CoA, Triton X-100, Krebs Henseleit buffer, insulin, gentamicin, and low melting agarose were obtained from Sigma-Aldrich (Axel, The Netherlands); William's medium E (WME), fetal calf serum (FCS) and phosphate-buffered saline were from Invitrogen (Breda, The Netherlands); DMSO (>99.9% pure; J. T. Baker, Deuenter, The Netherlands); hematoxylin and eosin were from Sigma-Aldrich (St. Louis, MO). d-Glucose, HPLC-water, and all other chemicals were from J. T. Baker, (Deventer, The Netherlands).
Preparation of S9-Homogenates and Microsomes of Rat Liver and Slices of Rat Liver, Lung, Kidney, and Small Intestine.
For preparation of S9-homogenates, microsomes and slices, male rats (Wistar, obtained from Harlan CPB, Horst, The Netherlands) were used. Rats had free access to food and water. They were anesthetized with 65% CO2, 35% O2 before the organs were removed.
For preparation of liver S9-homogenates, freshly excised liver was homogenized on ice using a Potter-Elvehjem homogenator. Subsequently, homogenates were ultracentrifuged at 9000g for 20 min at 2 to 4°C. To the supernatant phenylmethylsulfonyl fluoride (final concentration 100 μM) was added. The S9-homogenate was stored at −80°C until use.
For preparation of microsomes, the S9-homogenate was subsequently centrifuged at 100,000g for 30 min (Williams and Wilson, 1978). The pellet was resuspended in KCl-Tris-EDTA buffer with a volume corresponding to that of 80% of the original volume of the liver. Then, the pellet was homogenized again using a Potter-Elvehem homogenizer (400 rpm). Finally, a volume of glycerol corresponding to 20% of the original volume of the liver was added to the microsomal suspension. The microsomes were frozen in liquid nitrogen and stored at −80°C until use.
Tissue cores (8 mm) from freshly excised kidneys and livers were prepared using an electrical drill (Metabo BSE 5010) with a tissue-coring tool (Alabama Research and Development, Munford, AL). For preparation of lung tissue cores, the lung was filled with 37°C 1.5% (w/v) low melting agarose with 0.9% (w/v) NaCl in distilled water and kept on ice for up to 1 h prior to coring. Cores from small intestines were prepared as follows: a 10 to 20 cm piece of the small intestine, starting 10 cm from the stomach was dissected, directly flushed with ice-cold University of Wisconsin medium, and subsequently filled with 37°C 3% (w/v) low-melting agarose with 0.9% (w/v) NaCl in distilled water. Both ends were tied, and the filled intestine was submerged in ice-cold University of Wisconsin medium for 5 to 10 min. After the agarose had solidified, the intestine was carefully cut into pieces of approximately 2 cm. Subsequently, the bottom of the sample holder of the tissue-slicer was sealed with a piece of parafilm and filled with 37°C agarose. A piece of the intestine was quickly submerged into the liquid agarose and the sample holder was then stored at 4°C for about 15 min until the agarose had solidified. The technique of intestinal slice preparation was obtained from Ruben de Kanter of the Groningen University Institute for Drug Exploration (publication reporting evaluation of viability of these slices is submitted).
Slices [10–14 mg for kidney and liver slices, 25–30 mg for lung slices, and 10–12 mg (1–2 mg without agarose) for small intestinal slices] were prepared subsequently using a Krumdieck tissue slicer (Alabama Research and Development), filled with oxygenated (95% O2, 5% CO2) ice-cold KH buffer and supplemented with NaHCO3 (25 mM) and CaCl2 (2.5 mM). After preparation, slices were washed and kept on ice in WME with 10% FCS until use.
Cryopreservation and Thawing of Slices and Hepatocytes.
Slices were cryopreserved according to the method developed by de Kanter and Koster (1995) and optimized by de Graaf et al. (2000). After storage in liquid nitrogen for 1 to 12 weeks, slices were thawed by placing the cryovials in a 37°C water bath. After visible ice had vanished, slices were washed shortly with WME, 10% FCS. Since in earlier studies (de Graaf et al., 2000) cryopreservation success has appeared to be variable to some extent, cryopreserved slices were selected based on viability. Only slices from livers with successful cryopreservation results (viability of at least 70% of fresh slices of the same liver after 4 h incubation and/or 50% after 24 h incubation) were selected. Viability was measured by estimating the percentage viable cells in a slice cross-section upon histomorphological examination as described previously (de Graaf et al., 2000).
Cryopreserved male Sprague-Dawly rat hepatocytes were obtained from Sanvertech (Heerhugowaard, The Netherlands). Hepatocytes were thawed according to recommendations of the supplier. Shortly, hepatocytes were quickly thawed by placing the cryovial in a 37°C water bath until visible ice vanished. Subsequently, the cryoprotectant concentration was lowered by dropwise addition of WME. Finally, the diluted cryoprotectant solution was removed from the hepatocytes after centrifugation, and the pellet was resuspended in WME supplemented with FCS (5%), 0.1 μM insulin, 50 mg/l gentamicin, andd-glucose (to a medium concentration of 25 mM).
Incubation of S9-Homogenate, Microsomes, Hepatocytes, and Tissue Slices.
The incubation conditions, especially the incubation times, are chosen such that the amounts of metabolites formed are maximal to make sure that the amounts formed are sufficient for analysis. It is assumed that loss of viability would affect the total rate of parent drug metabolism, not the relative production of metabolites.
A volume of S9-homogenate or of microsomes containing 1 mg of protein was incubated with 175, 110, and 104 μM compound A, B, or C, respectively. To the S9-homogenate UDPGA (final concentration 200 μM), 3-phosphoadenosine-5′-phosphosulfate (final concentration 100 μM), and acetyl CoA (final concentration 0.25 mM) were added to a total volume of 900 μl. Subsequently the incubation mixtures were placed in a 37°C water bath for 10 min under air. The reaction was started subsequently by adding 100-μl NADPH-regenerating system (containing NADP+, NADH, isocitrate, isocitrate dehydrogenase, and MgCl2 to a final concentration of 0.57, 0.57, 6.4, and 23.4 mM, respectively). After 2 h, the incubation was terminated by freezing the samples in acetone-CO2 ice. After this incubation time, these preparations are almost inactive.
Fresh and cryopreserved slices were incubated with 175 or 110 μM compound A and B, respectively, or 21 μM compound C during 24 h at 37°C, while floating in 5 ml of medium in a 25-ml Erlenmeyer flask (1 slice/Erlenmeyer), placed in a shaking waterbath (110 strikes/min), under humid carbogen (95% O2, 5% CO2). This incubation system has proven to maintain viability of fresh liver slices for at least 24 h (Olinga et al., 1997a). As incubation medium WME was used, supplemented with FCS (5%), 0.1 μM insulin, 50 mg/l gentamicin, andd-glucose (to a medium concentration of 25 mM).
Thawed hepatocytes were diluted with WME supplemented with FCS (5%), 0.1 μM insulin, 50 mg/l gentamicin, and d-glucose (to a medium concentration of 25 mM) to a concentration of approximately 1 · 106 living cells/ml. Subsequently, a 12-wells plate was filled with 1 ml of this solution per well. Hepatocytes were incubated for 6 h with 175 or 110 μM compound A or B or 21 μM compound C by placing the 12-wells plate in a shaking water bath (37°C) under humid carbogen atmosphere. According to the suppliers information, the cryopreserved hepatocytes have lost most of their activity after this incubation time.
In Vivo Studies with Compound A, B, and C.
In vitro data were compared with data from various in vivo studies that were performed in the past. In vivo metabolism data of compound A were from one male Wistar rat, orally dosed with 273 μmol/kg14C-compound A. In vivo data about the metabolism of compound B were from one male Sprague-Dawley (SD) rat that was dosed orally with 11 μmol/kg 14C-compound B. In addition, one liver of a male Wistar rat was isolated and perfused with 172 μmol/kg 14C-compound B, and bile was collected. In vivo data of compound C were derived from two SD rats dosed with 1.6 μmol/kg of 14C-compound C. In addition, for compound C, an in vivo bile excretion study was performed. For this purpose, a male SD rat was dosed 4 times with 31 μmol/kg (1 time per 12 h) 14C-compound C.
Because both SD and Wistar rats were used for the various in vivo and in vitro studies, a study was done to assess possible differences in metabolism between the two strains. For this purpose, two Wistar rats were dosed with 110 μmol/kg compound B or 12 μmol/kg14C-compound C. Metabolite patterns were compared with those of SD rats.
Analysis by HPLC and LC-MS.
Metabolites of compound A, B, and C were determined (after sonication of the slices in their incubation medium) in the homogenate, after de-proteinization with methanol. For analysis by HPLC and LC-MS (both Hewlett Packard, Palo Alto, CA) an Inertsil ODS-3 column (150 × 2.1, 5 μm) was used with an OPTI-GUARD 1 mm C18 as precolumn. As mobile phase, a gradient over 40 min of 100% 3 g/l aqueous ammonium acetate to 100% methanol with a flow of 0.2 ml/min was used. UV-detection occurred at 320 nm for compound A and at 236 and 254 nm for compound B and C, respectively. The mass spectrum was taken at m/z 100 to 800 by electron spray ionization using ICL/ICIS software (Novatia, LLC, Princeton, NJ) for data acquisition and processing.
Quantification of Metabolites.
Peaks occurring in the UV signal were identified by MS, by searching for expected MH+ signals of possible metabolites. UV peak areas (% of total of metabolite peak areas) were used to evaluate the relative abundance of the metabolites in the chromatograms. In some cases, two different metabolites were identified by MS representing one single UV-peak. In this case the relative amounts of the metabolites involved are estimated using their MS peak area, taking differences in sensitivity for the MS into account as observed between conjugated and unconjugated metabolites: from other chromatograms the UV signal/MS signal ratio was determined and used to calculate the UV signal belonging to the MS signal.
Criteria for Judgment.
We considered the in vitro systems to be adequate for the prediction of in vivo metabolism if they met two criteria. 1) They should produce the same metabolites as the intact animal and 2) they should produce these metabolites in approximately the same relative amount as in vivo. To evaluate the latter, only metabolites that were formed in considerable amounts in vivo (i.e., with a peak area of at least 10% of the major metabolite) were taken into account. Of these metabolites, the percentage that they made up of the total amount of metabolites was calculated. The in vitro/in vivo ratio of this percentage should lie between 0.5 and 2. For example, if a metabolite made up 15% of the total amount of metabolites in vivo and 10% in vitro, the in vitro/in vivo ratio was 0.66; so in this case the in vitro system was considered to adequately predict the relative in vivo amount of the metabolite. The margins of 0.5 and 2 were chosen arbitrarily but reflect the intuitive way in vitro data are perceived as reasonably predictive for the in vivo situation.
Up-Scaling.
The combined in vitro metabolite profile of the different organ slices and liver slices was calculated as follows. First, the metabolism of the different slices was scaled up to the whole organ. For this purpose, the ratio of the organ weight and the slice weight was multiplied with the percentage of parent compound that was metabolized by the slices of that particular organ. Subsequently, these organ contributions were summed and normalized to 1. Thus, the relative contribution of each organ to the metabolism of the drug was obtained and used as a scaling factor. Then, for each organ, the relative abundance of the metabolites formed were multiplied with the scaling factor of that organ (giving the predicted relative contribution of that organ in the formation of the metabolites). Finally, for each of the metabolites, the relative contribution of each organ was summed to give the in vivo metabolite pattern.
For example, both liver and kidney slices metabolize compound X. For liver slices, a scaling factor of 0.4 is calculated (so, 40% of the in vivo metabolites is predicted to be made by the liver) and for kidney slices this factor is 0.6. Liver slices make metabolite 1 (10%), metabolite 2 (30%), and metabolite 3 (60%). Kidney slices make, respectively, 40, 20, and 40% of metabolite 1, 2, and 3. The combined metabolite pattern is calculated as follows: metabolite 1, 0.4 · 10 + 0.6 · 40 = 28%; metabolite 2, 0.4 · 30 + 0.6 · 20 = 24%; and metabolite 3, 0.4 · 60 + 0.6 · 40 = 48%.
Results
Viability of Cryopreserved Liver Slices.
In previous studies we have noticed that viability of slices of different livers after cryopreservation varies considerably (de Graaf et al., 2000). For this reason, in the present study we decided to select slices from experiments with the best cryopreservation results for further use in the drug metabolism studies. The results are shown in Table 1. The viability of fresh liver slices did not vary much between the different livers with 67 to 78% of the cells in a slice histomorphologically intact after 4 h of culturing and 55 to 72% after 24 h. (Note that in all slices, 1 or 2 outer cell layers are always damaged because of the slicing procedure, so that, even in very good slices, normally no more then approximately 80% of the cells are intact). In contrast with fresh slices, the quality of cryopreserved slices varied considerably, with 27 to 65% of the cells intact after 4 h of culturing and 8 to 62% after 24 h. For this reason, slices of liver 1, 3, 4, and 6 were thawed to be used in the drug metabolism studies.
Viability as determined by studying histomorphology of fresh and cryopreserved liver slices after 4 or 24 h in culture
Comparison of Metabolism between Sprague-Dawley and Wistar Rats.
For logistic reasons, in the present study different rat strains (SD and Wistar) were used for the various in vitro and in vivo studies. A study was undertaken to study possible differences in metabolism between the strains regarding compound B and C. The relative abundance of the metabolites of these compounds appeared to be well comparable for SD and Wistar rats: almost all metabolites did not differ more than 20% between the species, and only some metabolites of compound C differed by about 50% (data not shown).
In Vivo Metabolism of Compound A, B, and C.
Of compound A, 75% of the radioactivity was excreted by the Wistar rat within 24 h, 12% in feces and 63% in urine. Ca. 69% of the radioactivity in urine originated from the parent compound as well as ca. 50% in the feces. Of compound B, 74% of the radioactivity was excreted by the SD rats within 48 h, 63% in feces and 11% in urine. In feces, 5% of the radioactivity was found to be the unchanged parent compound, in urine 25%. In bile, 3% of the radioactivity was represented by the parent compound B. The SD rats dosed with compound C excreted 95% of the dose within 72 h, mainly in feces. Of the total dose, 50% was found to be metabolized. In bile, all administered compound C was found to be metabolized.
In Vitro Metabolism of Compounds A, B, and C.
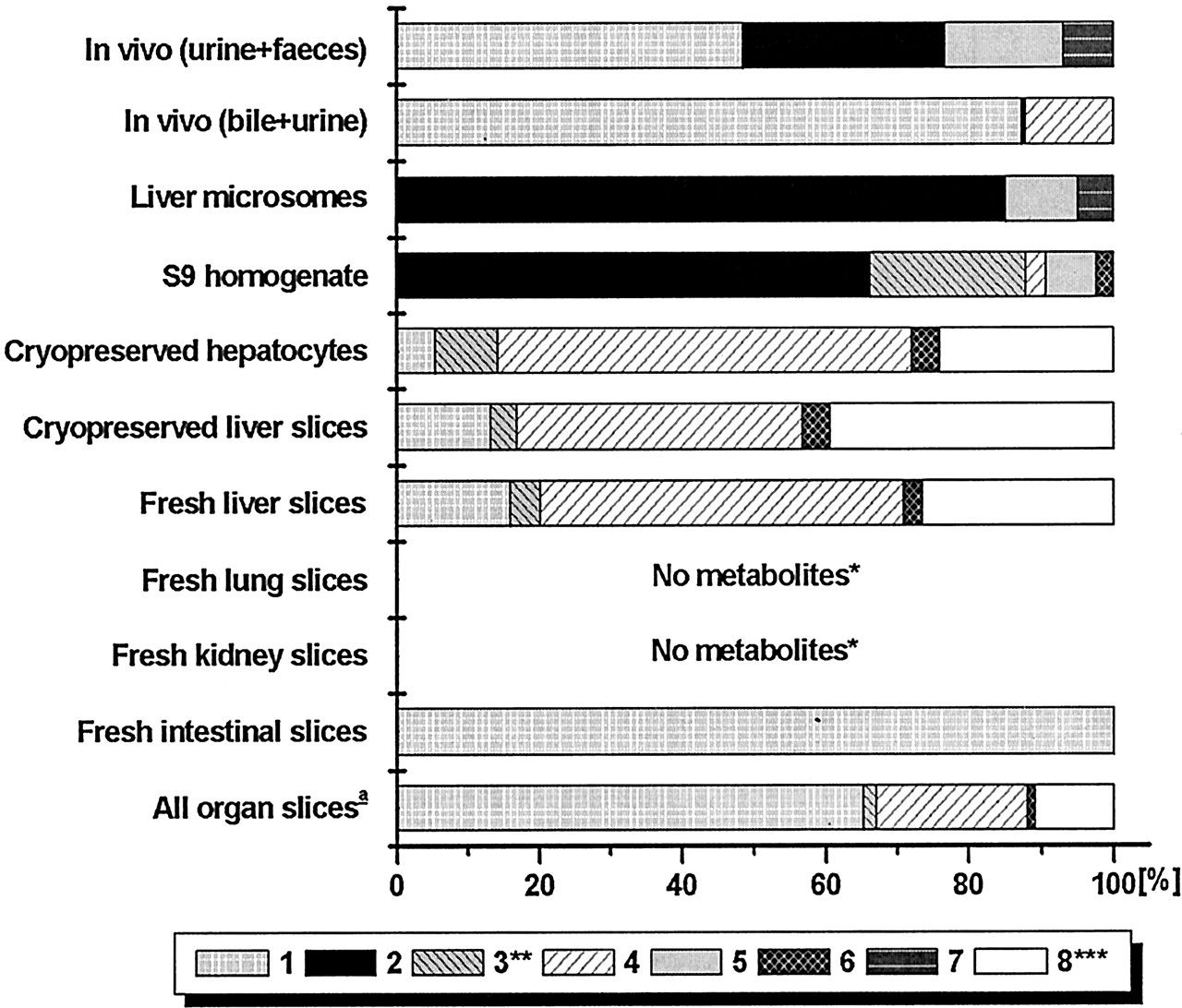
The various preparations used varied in their activity of metabolism of the three compounds (Table 2). All three compounds were most actively metabolized by intact liver cell preparations, whereas intestinal slices also metabolized compounds A and C. Kidney slices had some activity but lung slices hardly produced metabolites.
Relative activities of the in vitro systems in metabolism of compound A, B, and C
In Vivo and in Vitro Metabolite Patterns of Compound A.
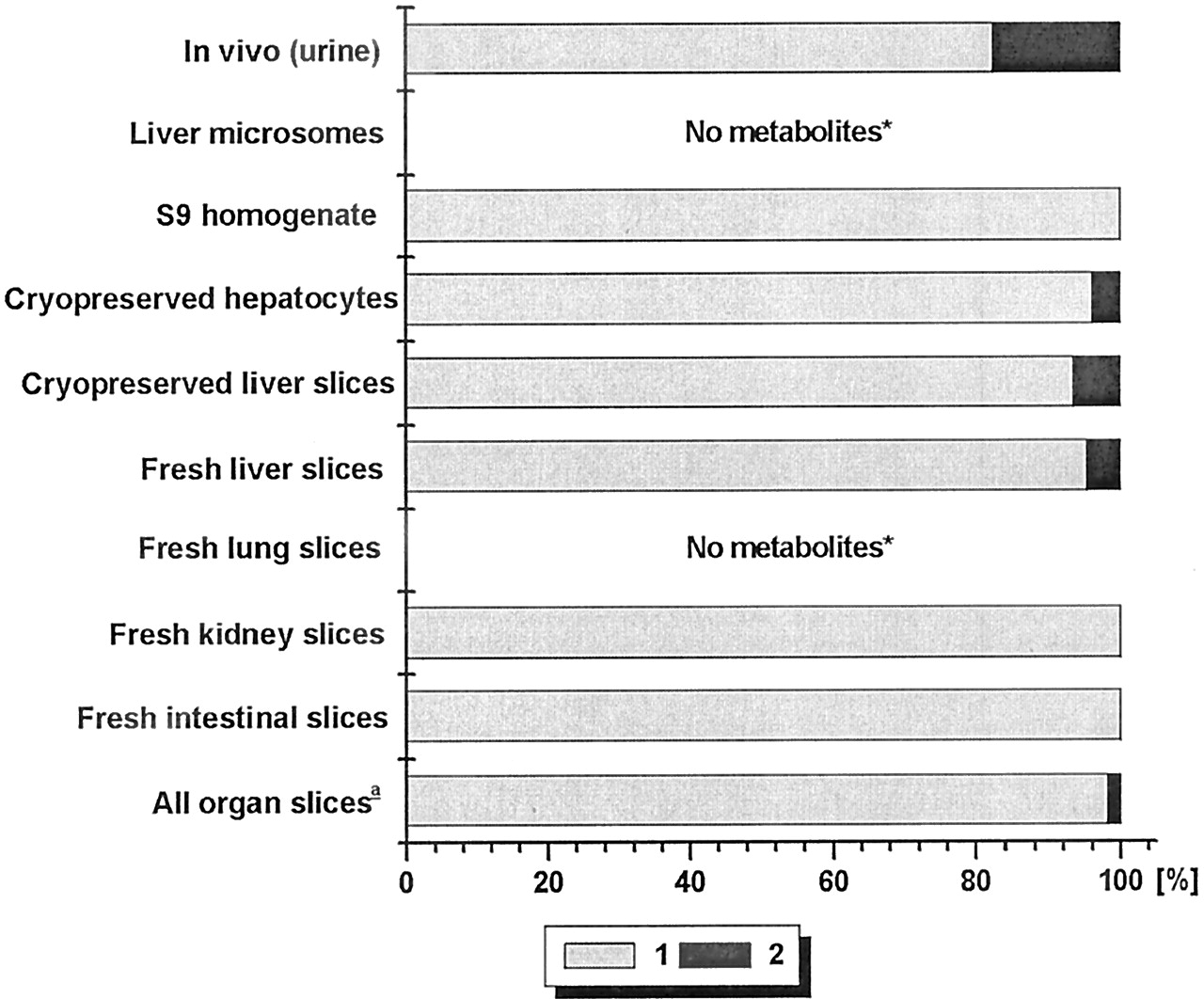
The major routes of biotransformation of compound A are given in Fig.1. As shown in Fig.2, by far the most important metabolite in the rat in vivo is the product of N-acetylation (metabolite 1). Another, still unknown metabolite was found in vivo (2). All in vitro preparations with intact cells (organ slices and hepatocytes) produced the major N-acetyl metabolite but lung slices only in negligible amounts. Metabolite 2 was only formed in vitro by the intact cell systems derived from the liver. Of the subcellular systems, S9-homogenate did acetylate compound A, whereas microsomes did not, because they lack the enzyme required for this reaction. Of the slices from the various organs, liver slices were the most active in the metabolism of compound A (Table 2). There is not much difference between the amounts of metabolites formed by fresh or cryopreserved slices. Cryopreserved hepatocytes were only incubated for 6 h with compound A but metabolized the same amount of this compound as liver slices did in 24 h.

Identified metabolic pathways of compound A.
A1, molecular fragment attached to the part that is acetylated. (1)-(2), metabolites referred to in the text.

Relative amounts of identified metabolites of compound A formed by various test systems.
1, acetylated A; 2, unknown metabolite. For all test systems >75% of the metabolites were identified. Slices were from three livers; Microsomes from two livers; S9-homogenate from two livers; hepatocytes from one batch. In vivo data were from one rat. ★, <1% of the mother compound was metabolized. a, relative amounts of metabolites are calculated as described underMaterials and Methods, with data in Table 3
Clearly, only intact cellular systems derived from liver made both important in vivo metabolites of compound A. Therefore, to calculate a combined metabolite pattern taking extra-hepatic metabolism into account to predict the in vivo metabolite profile of compound A, data of liver slices and kidney and small intestinal slices were used. The scaling factors for the organ slices are shown in Table3. Both metabolites are predicted, but the amount of metabolite 2 is underestimated (Fig. 2). This is also seen when the ratios of the relative amounts of metabolites formed in vitro and in vivo are compared (Table 4); for all intact liver cell preparations, the ratio for this metabolite is below 0.5 and, when all organ slices are combined, this is even lower. However, the major metabolite, N-acetyl A, was predicted correctly; all ratios were close to 1.
Calculation of scaling factors for compound A
Semi-quantitative prediction of in vivo metabolism of compound A, B, and C by various in vitro systems
In Vivo and in Vitro Metabolite Patterns of Compound B.
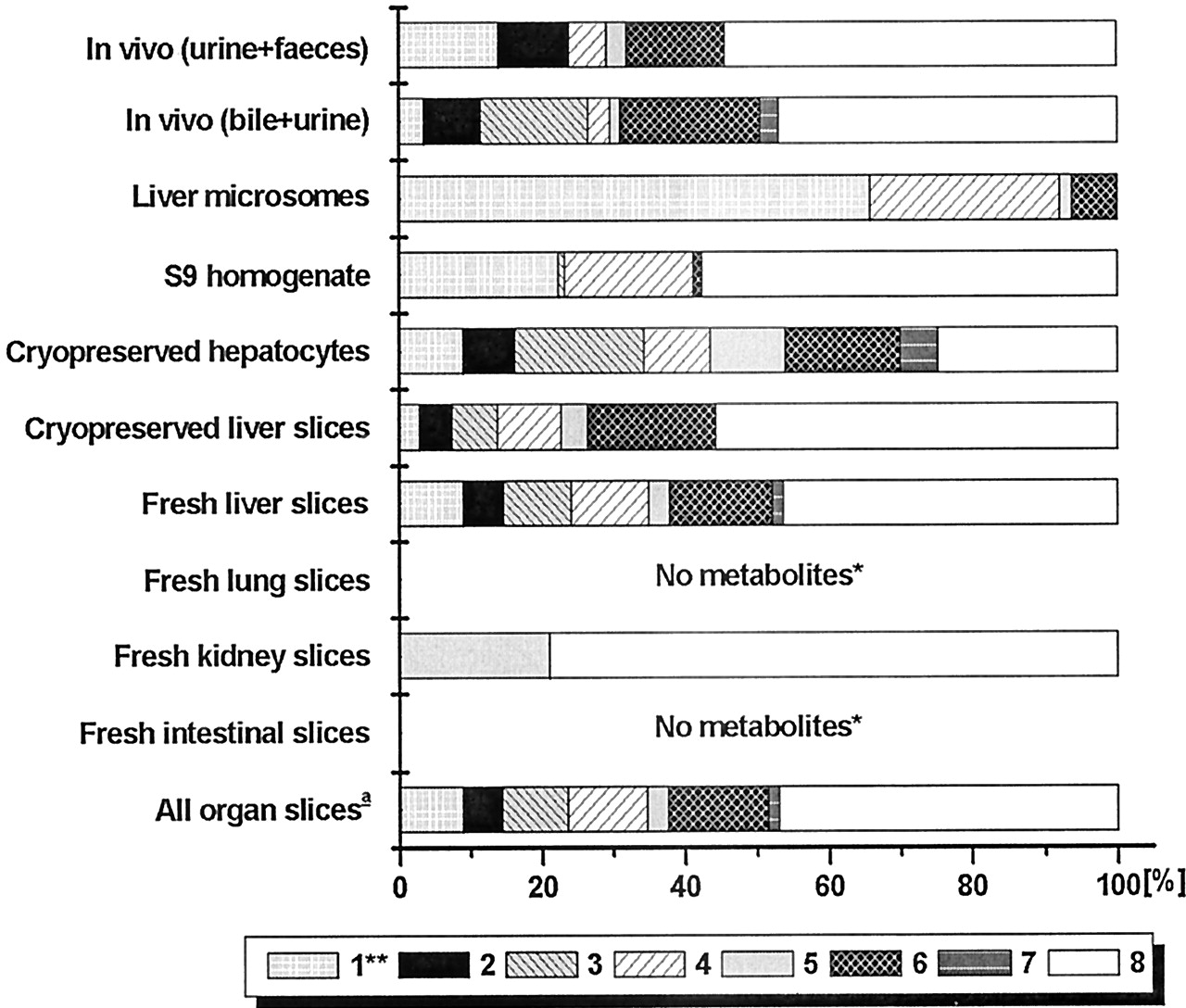
Compound B is metabolized in vivo via a wide range of metabolic pathways (Fig. 3), the major metabolite being the sulfate conjugate, 8. This metabolites was produced in considerable amounts by fresh and cryopreserved liver slices, cryopreserved hepatocytes, S9-homogenate, and in small amount by kidney slices (Fig. 4). Only intact cell systems derived from liver formed all metabolites of compound B that are produced by the rat in vivo. Some conjugates (i.e., 3 and 7) produced in vitro were only found in bile and not in other excreta. Microsomes only formed phase I metabolites of compound B (i.e., 1, 4, 5, and 6). S9-homogenate produced all phase I metabolites except for 4, and it produced both sulfate conjugates (3 and 8) but not the glucuronide conjugates (2 and 7). These results show that only the intact-cell systems can be used for up-scaling to predict in vivo metabolite pattern.

Identified metabolic pathways of compound B.
The molecular fragments attached to the part that is metabolized are indicated as B1 through B7. (1)-(8), metabolites referred to in the text.
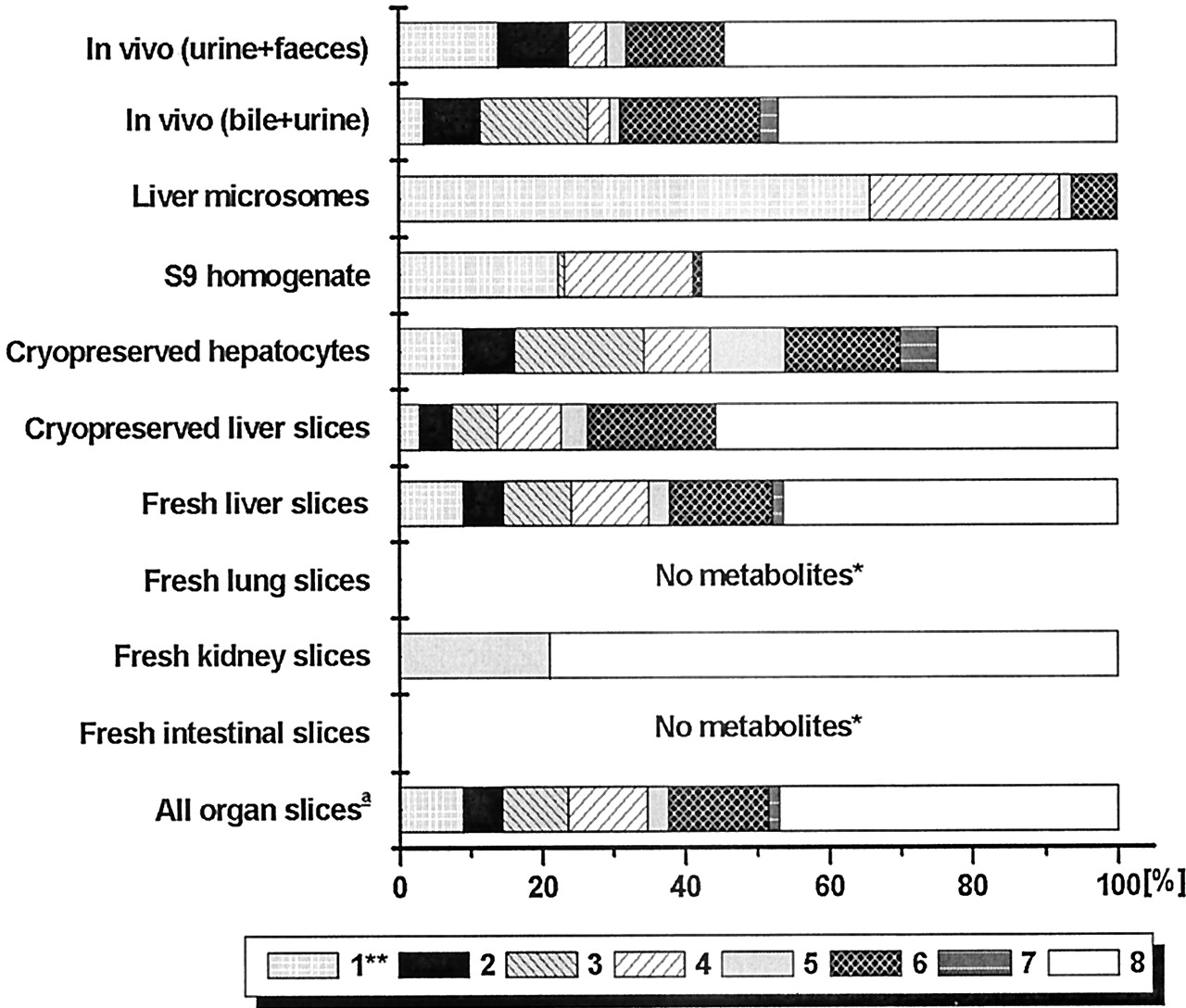
Relative amounts of identified metabolites of compound B.
1, hydroxy-B; 2, [hydroxy-B]-O-glucuronide; 3, [hydroxy-B]-O-sulfate; 4 and 5, fragments afterN-dealkylation; 6, B-carboxylic acid; 7, B-glucuronide; 8, B-sulfate. For all test systems >75% of the metabolites were identified. Slices were from three livers, microsomes from two livers, S9-homogenate from two livers, hepatocytes from 1 batch. In vivo data were from one rat; bile was from one rat. ★, <1% of the mother compound was metabolized. ★★, in some in vitro incubations a second hydroxyl metabolite was found in small amounts.a, metabolite pattern is calculated as described under Materials and Methods, with data in Table 5.
In vitro metabolite patterns are compared with those in bile + urine. This is a more adequate comparison than that with feces + urine since in feces, some metabolites are found to be deconjugated (Fig. 4). Slices from kidney, lung, and the small intestines metabolized small amounts of compound B and, therefore, the combined metabolite pattern based on metabolism by all organ slices (scaling factors given in Table5) resembles that of liver slices (Fig.4). Since cryopreserved hepatocytes and fresh and cryopreserved liver slices all give a similar metabolite profile, these preparations all predict in vivo metabolite patterns of compound B well, with in vitro/in vivo ratios all being between 0.5 and 2 (Table 4).
Calculation of scaling factors for compound B
In Vivo and in Vitro Metabolism of Compound C.
Compound C was metabolized in vivo to four major metabolites, notably an N-glucuronide (metabolite 1 in Fig.5) and three hydroxylated metabolites excreted in feces in unconjugated form (2, 5 and 7). In bile, theN-glucuronide was prominent, but also the glucuronidated hydroxy metabolite (4) was seen. The sulfate conjugate (3) was detected with the MS in bile, but the amount was too small to be quantified by HPLC/UV.

Identified metabolic pathways of compound C.
The molecular fragments attached to the part that are metabolized are indicated as C1 through C5. (1)-(8), metabolites referred to in the text.
The N-glucuronide was only one of the minor metabolites in in vitro systems with intact cells derived from liver (Fig. 6; Table 6). Subcellular fractions did not make this glucuronide conjugate at all. However, theN-glucuronide was formed in considerable amounts by slices of the small intestine. In fact, intestinal slices (1–2 mg of tissue) produced approximately 3 times more of this metabolite than liver slices (10–15 mg of tissue). A number of metabolites were formed by all systems with intact cells from liver but not found in in vivo (3, 6, and 8; Fig. 6). A considerable amount of a metabolite with the same molecular weight as the parent drug was found in vitro but not in vivo. Presumably, this metabolite was a C-N-oxide that was converted to the original drug by the mass spectrometer. Unconjugated hydroxy metabolites were not detected in (liver) slices or hepatocyte incubations. With microsomes and S9-homogenate, however, substantial amounts of these metabolites were found. Fresh liver slices and hepatocytes converted the same amount of compound C during incubation (Table 3). Cryopreserved liver slices and intestinal slices were a little less active. Negligible amounts of metabolites were found after incubation of compound C with lung and kidney slices.
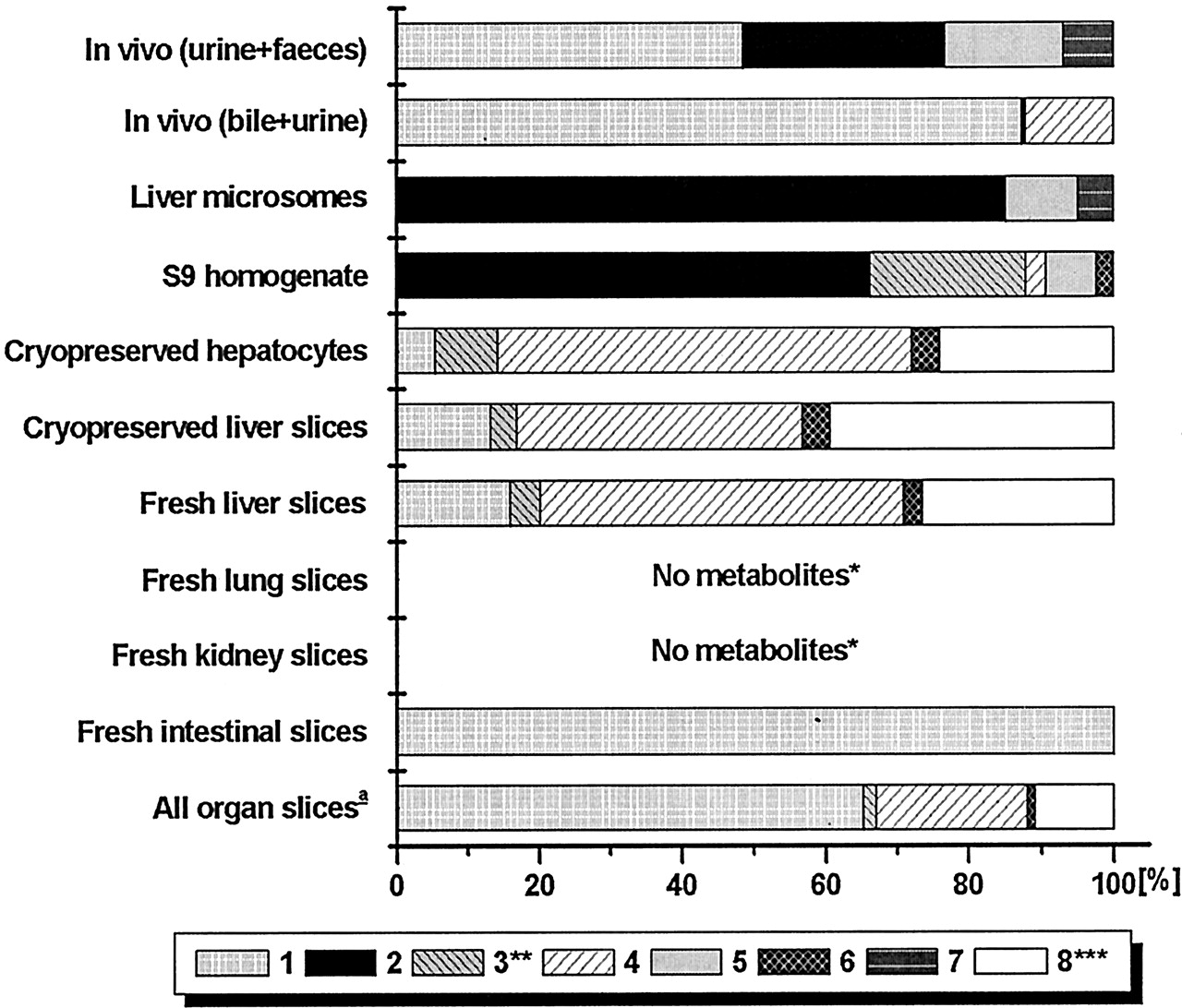
Relative amount of identified metabolites of compound C formed by various test systems.
1, [C]-N-glucuronide; 2, 3-hydroxy-C; 3, [4-hydroxy-C]-O-sulfate; 4, [4-hydroxy-C]-O-glucuronide; 5, 3-hydroxy-C; 6, [3-hydroxy-C]-O-sulfate; 7, dihydroxy-C; 8, C-N-oxide. For all test systems >75% of the metabolites were identified. Slices were from three livers; Microsomes were from two livers, S9-homogenate from two livers, hepatocytes from one batch. In vivo data were from two rats, bile from one rat. ★, <1% of the parent compound was metabolized. ★★, a hydroxy-C and [hydroxy-C]-O-sulfate were found at the same retention time because the sulfate was split off by the mass spectrometer. ★★★, a metabolite with the same mass as the parent was found, presumably because C-N-oxide was converted to the original drug by the mass spectrometer. a, metabolite pattern is calculated as described under Materials and Methods, with data in Table 6.
Calculation of scaling factors for compound C
Since only the intact cell preparations predicted all major in vivo metabolites, the data from these preparations were used for up-scaling. These preparations produced only conjugated hydroxyl metabolites (that were found to be deconjugated in feces) and, therefore, comparison was made with the combined bile and urine data. The results in Table 4 show that the liver preparations underestimate the N-glucuronide and overestimate the glucuronidated hydroxyl metabolite. When the relatively large contribution of intestinal slices to theN-glucuronidation of compound C is taken into account in the calculation of the combined metabolite pattern of the various organ slices, the obtained pattern closely resembles in vivo metabolism (Table 4, last column; scaling factors are given in Table 7).
Discussion
In the present study, we compared in vitro metabolism by preparations from rat liver with increasing structural organization and by slices from other metabolizing organs (rat lung, kidney, intestines) with in vivo biotransformation. It appeared that there are substantial differences in metabolism, both qualitatively and quantitatively, between the different systems.
Microsomes and S9-homogenate produced considerable amounts of phase I metabolites of the drugs tested. However, as microsomes possess only UDP-glucuronyltransferases inaccessible for its cofactor UDPGA and no other phase II enzymes, this preparation did not produce the phase II metabolites formed in vivo from compounds A, B, and C. S9-homogenate formed only the sulfate and acetyl conjugates since cytosolic enzymes, phase II enzymes as acetyl-transferases, and sulfo-transferases are present, but, like in microsomes, glucuronyl-transferases cannot contribute to drug metabolism. With compound C, small amounts of an O-glucuronide metabolite were found with S9-homogenate nevertheless. In contrast with the other compounds, compound C was dissolved in DMSO (medium concentration 1%). Possibly, DMSO disturbed the endoplasmic reticulum membrane, making it more permeable and activating the glucuronyl transferases. These results clearly show that intact cell preparations are required to reliably predict the metabolites formed in vivo.
In general, fresh and cryopreserved slices and cryopreserved hepatocytes produced the various metabolites in approximately the same relative amounts. Liver slices, containing approximately the same amount of hepatocytes per slice as were used for the hepatocyte incubations, however, needed a 24-h incubation to produce the same absolute amount of metabolites as hepatocytes in 6 h. The lower drug-metabolizing activity of liver slices, which is also shown byEkins et al. (1995) may be caused by slow drug transport into the center of the slice, limiting its metabolism.
Our results indicate that cryopreserved liver slices are as good as in vitro tool for studying drug metabolism as fresh liver slices. However, selection of slices by viability is in our opinion necessary, because the success of cryopreservation differs between livers (de Graaf et al., 2000). In previous studies, we noticed that cryopreserved liver slices with a low viability did not only produce smaller amounts of metabolites, but also the relative amounts of the metabolites changed (unpublished observations). Particularly conjugation activity decreases in unsuccessfully cryopreserved slices, possibly by cofactor loss, whereas phase I biotransformation is preserved longer (Maas et al., 2000; Vanhulle et al., 2001). The selection of slices on the base of their histomorphological appearance after thawing and culturing, as done in the present study, appeared to be useful.
Slices from extra-hepatic origin produced metabolites that were also formed by liver slices or hepatocytes, but the relative amount in which the various metabolites were formed differed considerably. With compound C, the major metabolite in vivo was theN-glucuronide, whereas in liver slices and hepatocytes, this was the O-glucuronide. Similar results were reported bySandker et al. (1994) with Org 3770 and by Pahernik et al. (1995) with pimobendan. It was suggested that the N-glucuronide was mainly formed extra-hepatically (Sandker et al., 1994). In the present study, small intestinal slices appeared to be very active producers of the N-glucuronide conjugate of compound C in comparison to liver slices, indicating that the intestines indeed are important in the in vivo metabolism of compound C. Differences in relative activities of various biotransformation pathways between liver, lung, and kidney slices were also reported with ethoxycoumarin and testosterone by de Kanter et al. (1999, 2002). These results indicate that for a reliable prediction of in vivo metabolism of some compounds, the use of both hepatic and extra-hepatical slices is required.
The up-scaling procedure we applied combines the contribution of each of the organs by a scaling factor based on the amount of metabolism per slice of the various organs, the slice weights and the organ weights. This procedure assumes that the relative rates of metabolism of the various organs is the same in vitro and in vivo, as well as the relative amounts of metabolites formed by an organ. These assumptions are only valid when the degree of saturation of metabolism is similar in vitro and in vivo. This factor was not examined however in the present study. Nevertheless, in vivo metabolism was found to be well predicted by either intact liver cells and/or the combined organ slices. The predicted relative in vivo amounts of metabolites were in most cases within the set limits of 0.5 and 2 of the amounts that were actually formed in vivo. So, this approach could be useful in a prospective study, predicting metabolite patterns of various animal species and humans for proper selection of an animal model for human drug toxicity studies.
In conclusion, in vitro systems with intact cells (slices and hepatocytes) from liver produced the same metabolites of the three compounds studied as the rat in vivo and for that reason seem to be a valuable tool to predict drug metabolism in vivo. Deconjugation of biliary conjugates in feces should be taken into account when comparing in vitro metabolite patterns with those in feces and urine. Since phase I metabolites were found in only small amounts with intact cells, microsomes and S9-homogenate can be adequately used to study phase I metabolism but are unsuitable to predict in vivo metabolite patterns. Extra-hepatic organ slices produced metabolites that were also formed by liver cells, but the relative amount of the various metabolites differed considerably. For compounds that are metabolized byN-glucuronidation, the small intestine may be an important metabolizing organ and the amount of N-glucuronides formed may be underestimated when only liver cell preparations are used. Therefore, for the prediction of the relative amounts of metabolites formed in vivo, the use of intestinal slices in combination with liver slices may be required.
Acknowledgments
The authors thank Dr. Bas Blaauboer and Prof. Dr. Willem Seinen (Institute for Risk Assessment Sciences, Division of Toxicology, Utrecht University) for critically reading the manuscript and valuable discussion.
Footnotes
-
This project is supported by a grant of from the Dutch Platform for Alternatives for Animal Experimentation.
- Abbreviations used are::
- LC
- liquid chromatography
- MS
- mass spectometry
- UDPGA
- UDP-glucuronic acid
- WME
- Williams medium E
- FCS
- fetal calf serum
- DMSO
- dimethylsulfoxide
- HPLC
- high-performance liquid chromatography
- SD
- Sprague-Dawley
- Received July 25, 2001.
- Accepted June 26, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}