


Abstract
Characterization of the circulating metabolites for a new chemical entity in humans is essential for safety assessment, an understanding of their contributions to pharmacologic activities, and their potential involvement in drug-drug interactions. This review examines the abundance of metabolites relative to the total parent drug [metabolite-to-parent (M/P) ratio] from 125 drugs in relation to their structural and physicochemical characteristics, lipoidal permeability, protein binding, and fractional formation from parent (fm). Our analysis suggests that fm is the major determinant of total drug M/P ratio for amine, alcohol, N- and S-oxide, and carboxylic acid metabolites. Passage from the hepatocyte to systemic circulation does not appear to be limiting owing to the vast majority of metabolites formed being relatively lipid permeable. In some cases, active transport plays an important role in this process (e.g., carboxylic acid metabolites). Differences in total parent drug clearance and metabolite clearance are attenuated by the reduction in lipophilicity introduced by the metabolic step and resultant compensatory changes in unbound clearance and protein binding. A small subclass of these drugs (e.g., terfenadine) is unintentional prodrugs with very high parent drug clearance, resulting in very high M/P ratios. In contrast, arenol metabolites show a more complex relationship with fm due largely to the new metabolic routes (conjugation) available to the metabolite compared with the parent drug molecule. For these metabolites, a more thorough understanding of the elimination clearance of the metabolite is critical to discern the likelihood of whether the phenol will constitute a major circulating metabolite.
Introduction
An early understanding of the key metabolites for a new chemical entity is important in drug discovery and development. At the lead optimization stage, this can help to support medicinal chemistry efforts in designing more metabolically stable candidates via identification of soft spots in lead candidates that are rapidly cleared (Lin and Lu, 1997; Baillie, 2008). From a development perspective, characterization of metabolites provides a systematic understanding of the metabolic pathways of a drug and therefore the enzymes that are involved in their formation. This can aid in the proper design of drug-drug interaction (DDI) studies to evaluate the effect of inhibition or induction of the metabolic enzyme(s) contributing to the major pathway of clearance on systemic drug exposure. Assessment of circulating metabolites in humans also contributes to an overall understanding of their potential impact on the safety and efficacy of a drug. From a safety point of view, an effort toward gathering knowledge of the in vivo metabolic profile of a drug in humans has gained more importance since the publication of position papers on drug metabolites in safety testing as well as recently issued ICH (International Conference on Harmonisation) Guidance M3(R2) on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals (US FDA guidance; http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073246.pdf); (Davis-Bruno and Atrakchi, 2006; Nedderman et al., 2011). This guidance recommends that nonclinical safety evaluation is warranted if the exposure of a human circulating metabolite is observed at greater than 10% of the total drug-related exposure and at significantly greater levels in humans than the maximum exposure seen in the toxicity studies. Assessment of circulating metabolites in humans is also essential for a comprehensive understanding of their contributions to pharmacologic activities. Structural alteration of drugs via metabolism can sometimes lead to products that contribute to the therapeutic effect of the drug (on target activity) or cause an undesirable effect as a consequence of off-target activity (Fura, 2006). More recently, efforts have been directed at evaluating the potential involvement of metabolites in inhibitory DDI. Several clinically important drugs (especially clinically recognized P450 inhibitors) have been shown to have circulating metabolites that act as potent inhibitors of P450 enzymes and therefore contribute to in vivo DDIs (Isoherranen et al., 2009; Yeung et al., 2011). The most recent guidance by the European Medicines Agency (EMA) and United States Food and Drug Administration (FDA) also suggest evaluation of potential inhibitory effect of metabolites on the common drug metabolizing enzymes. This guidance recommends that circulating metabolite with an AUC of ≥25% relative to parent AUC (Draft FDA Guidance; http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf) and phase 1 metabolites with an AUC both larger than one fourth of the AUC of parent drug and larger than 10% of the drug-related exposure (EMA Guidance; http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf) should be investigated for their inhibitory potential. These recommendations necessitate characterization and quantification of the metabolites in plasma early on to allow a full assessment of their contributions to the pharmacological or toxicological effects and DDI potential.
In most cases, principal metabolites are identified by performing radiolabeled ADME studies where 14C or 3H analogs of the drugs are administered to humans (Roffey et al., 2007; Isin et al., 2012; Penner et al., 2012). The biologic matrices collected from this study are then analyzed to yield a quantitative profile of metabolites in circulation or in the excreta. While the abundance of metabolites in the excreta provides a means of assessing the clearance pathways and the fraction of the drug metabolized via a given pathway (fm), the exposure of metabolites in plasma obtained from this study serves as a starting point for prioritizing their further evaluation. The major metabolites are then synthesized and evaluated for pharmacological on-target and off-target interactions that may result in unpredicted toxicity or DDI. However, radiolabeled ADME studies in humans are expensive and resource intensive and hence not routinely feasible in early development. This can hinder the knowledge about relevant circulating plasma metabolites and hence their evaluation in a timely manner. Hence, until definitive data are obtained from human studies, one may need to rely on the in vitro systems for an assessment of potential metabolites that are generated. Human-derived reagents, such as the liver microsomes, hepatocytes, or S9 fractions, are the commonly used in vitro systems to study human drug metabolism and identify the principal metabolites, preferably prior to the human radiolabel ADME study (Dalvie et al., 2009). A comprehensive analysis of these in vitro systems by Dalvie et al. (2009) provide sufficient confidence in using these systems to reliably produce primary human metabolites. Although primary human metabolites can be predicted readily by in vitro systems, it is important to understand which metabolites formed in these systems are likely to circulate in vivo. This can help in prioritizing the synthesis of important metabolites for further characterization with respect to their pharmacological activity and/or their role as inhibitors or inducers of important drug metabolizing enzymes.
Smith and Dalvie (2012) recently attempted to analyze the influence of physicochemical properties on the metabolites that circulate. This review showed that metabolites with high lipoidal permeability will passively diffuse from liver to plasma and circulate (Smith and Dalvie, 2012). The aim of the current analysis was to extend this investigation and explore how the structures of the metabolite (e.g., amines, alcohols, etc.) and their associated physicochemical properties, as well as the fraction of dose converted to a given metabolite from the parent (fm) may influence their abundance in circulation.
We conducted a literature review to collect data on the plasma or serum area under the concentration time curve (AUC) values from 125 drugs and their metabolites after oral administration of single or multiple doses of the parent and then determine the relative abundance of metabolite to parent based on the ratio of plasma/serum AUC values (M/P ratio). Although several secondary and tertiary metabolites were detected circulating for several drugs, no attempt was made to classify and categorize these metabolites separately. In the vast majority of cases, the ratio of plasma or serum AUC of the metabolite to the corresponding parent was used, although in a few cases (as indicated by the asterisk in Tables 1–7), single time point concentrations were used because of paucity of the overall AUC data. In a few cases (clozapine, dextromethorphan, dothiepin, flecainide, and zolpiclone) where plasma concentration-time profiles were presented in the publication but the AUC values were not calculated, the data were captured using the software DigitizeIt version 1.5 (Eden Prairie, MN), and the AUC values were calculated by noncompartmental analysis using WinNonlin version 5.2 (Pharsight Corp., Cary, NC). The M/P ratio for these metabolites was then matched with the structure and physicochemical properties of the corresponding parent and metabolite pair to discern any possible relationship between the two. The physicochemical properties for the parent and metabolites were calculated using the ACD software version 12.01 (ACD Laboratories, Advanced Chemistry Development Inc., Toronto, ON, Canada), and the chemical structures of the parent and corresponding metabolites are shown in the supplemental file (Supplemental Tables 1–6). In addition, where data exist, information on the fm value was also estimated from the published human radiolabeled ADME studies. The fraction of dose for the metabolite of interest and its sequential metabolic products recovered in excreta (both urine and feces) was assumed to be representative of the in vivo fm value. An analysis was conducted in an attempt to identify parameter values for the physicochemical properties and fm that are associated with metabolites considered to have met the criteria for follow up DDI evaluations according to the regulatory guidance.
Theoretical Considerations of Determining In Vivo Metabolite Exposure
Theoretical models related to metabolite kinetics after oral or intravenous administration of the parent drug have been described extensively (Houston, 1982; Lutz et al., 2010) and will only be discussed briefly in this review. In vivo, a metabolite can display either a formation-rate limited or an elimination-rate limited kinetic. In the former case, the half-life of the terminal elimination phase of a metabolite plasma/serum concentration versus time curve will be equal to that of the parent drug, whereas in the latter case, the half-life of the terminal elimination phase of the metabolite will be greater than that of the parent. Most assessment of circulating concentrations of drugs and their metabolites are made without reference to protein binding, certainly historically and also at the initial profiling and identification stage of drug discovery and development. Regulatory guidelines reflect this in that the identification of “major” metabolite relies on total concentration compared with parent or drug-derived moieties, although there is recognition that certain metabolites may have very different binding characteristics than their parent. To understand the dynamics of metabolites, protein binding must be measured to derive unbound concentrations. This is reinforced by the tendency for a reduced lipophilicity for the metabolite, which may increase the fraction unbound and lowers unbound clearance, leading to disproportionate concentrations of unbound metabolite compared with unbound concentrations of parent. Unfortunately, protein binding data are lacking in many cases to provide more sophisticated analysis. In terms of the effects of changes in chemical structures and how they may affect the ratio of total drug to metabolite, the influence of protein binding may be attenuated. Most metabolic steps typically result in reduced lipophilicity, which will tend to decrease protein binding (or increase fraction unbound) and intrinsic metabolic clearance (unless the metabolic step introduces a route by which the parent cannot be metabolized), thereby attenuating the effect on the difference between parent and metabolite clearance (see Protein Binding). Similarly, the lowered lipophilicity will tend to decrease tissue or membrane affinity, which will be compensated for by the increased free fraction in plasma, thereby attenuating the effects on total volume of distribution. Thus, the ratio of total parent to metabolite is probably more sensitive to other factors as detailed below.
The systemic exposure (total, bound plus free), as represented by the AUC of a metabolite (AUCm), will be dependent on the dose of the parent drug (D), the fraction of dose absorbed (after oral administration, Fa), the fraction of the absorbed drug dose that is converted to the metabolite (fm), the systemic availability of the metabolite (Fh,m), and the clearance of the metabolite (CLm), i.e., AUCm = (fm × Fh,m × Fa × D)/CLm (Houston, 1982). As illustrated in Fig. 1, the systemic availability of a metabolite can be conceptualized as the net fraction of the formed metabolite partitioning back into the circulation. Physiologically, this reflects the permeability rate of the metabolite across the sinusoid for passive diffusion (lipoidal permeability, fp) and the effect of sinusoidal efflux transporters (active transport) compared with the rate of further processing of the metabolite by metabolism within the hepatocyte or clearance directly from the hepatocyte or by canalicular transporters (biliary excretion). Lipoidal permeability decreases with decreasing lipophilicity and increasing polar surface area (tPSA). Unless the metabolite is a substrate for sinusoidal transporters, it is possible that its residence time in the liver will be increased by metabolism, favoring further processing and attenuating systemic exposure.

Scheme depicting potential routes of metabolite disposition.
In vivo, after oral dosing, the ratio of AUC for the metabolite-to-parent (M/P) ratio will be determined by fm × Fh,m × CLparent/CLm × Fh (where Fh is the fraction of absorbed parent drug dose that escape the liver) since dose (parent) and Fa are the same. For a drug cleared by metabolism to a single metabolite that diffuses completely from the hepatocyte to systemic circulation (e.g., a metabolite with high lipoidal permeability), this simplifies to CLparent/CLm × Fh, assuming linear processes. For a drug that undergoes metabolism to more than one metabolite and assuming complete diffusion of the metabolite(s) to systemic circulation, the M/P ratio is expressed as fm × CLparent/CLm × Fh, which indicates that the formation and elimination clearance (CLf and CLm, respectively) of the metabolite are major determinants of its M/P ratio. The differences between CLparent and CLm probably reflect actual chemical-structural change(s) as a result of the metabolism of a more labile functional group to a more stable functional group rather than simple reduction in lipophilicity (see above discussion on protein binding). These principles for highly diffusable metabolites have been established for some time (Levy et al., 1983) and provide a useful conceptual framework to guide the assessment and interpretation of our analysis regarding the potential relationship between structural and physicochemical properties of the metabolites and their abundance in circulation relative to the parent drug based on ratio of AUC.
Secondary or Primary Amines as Metabolites
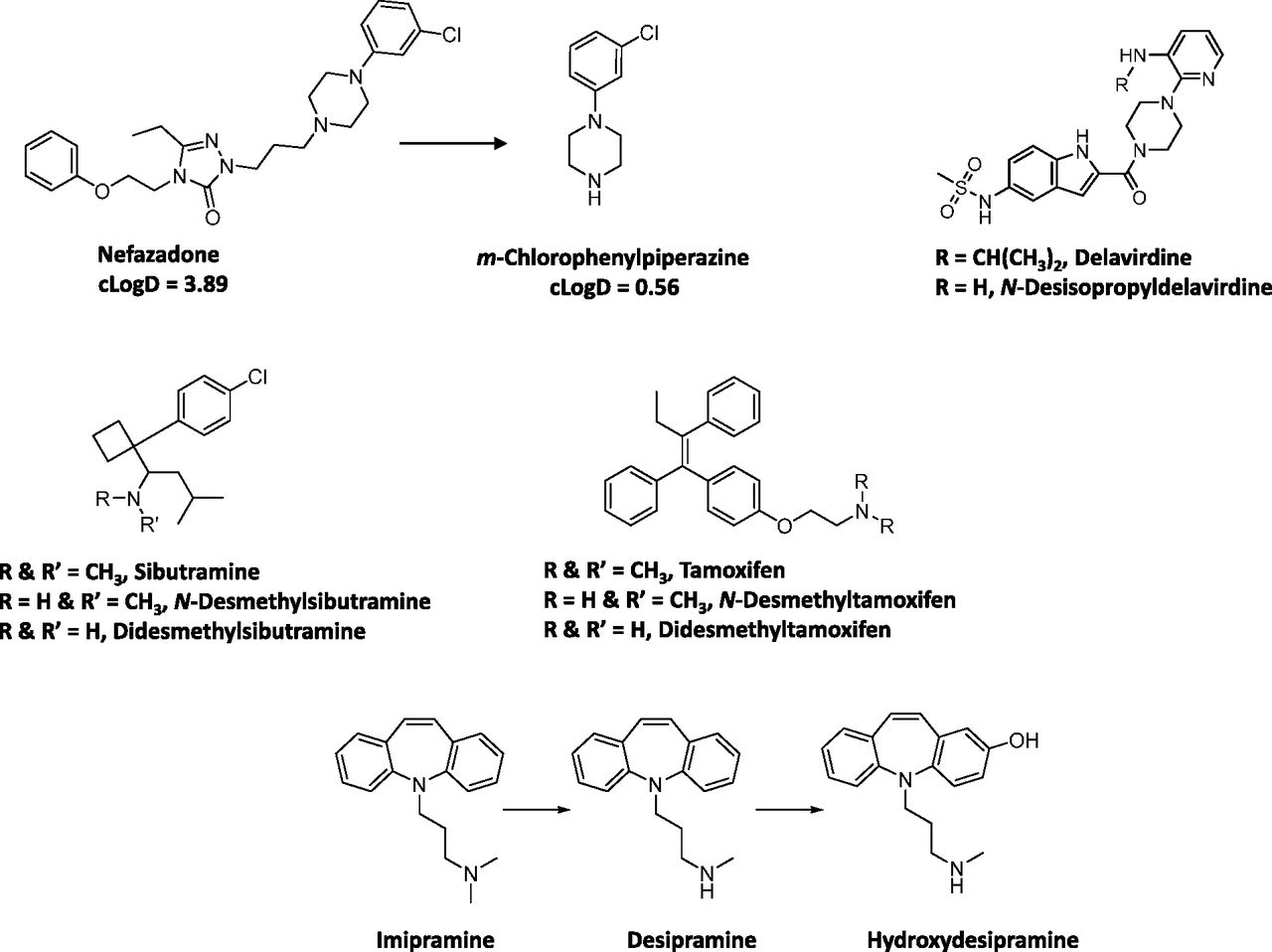
N-Dealkylation represents one of the most common biotransformation pathways for drugs containing secondary and tertiary amines. This oxidative pathway results in the formation of metabolites that contain primary or secondary amines or in some cases cleaved products as primary metabolites. In this dataset, 52 of a total of 125 drugs (42%) were N-dealkylated as part of the overall clearance process. Of these 52 drugs, 40 (77%) had an amine metabolite with an abundance, based on the M/P ratio, of >0.25 in human plasma (Table 1). This trend is not surprising since these metabolites retain lipophilicity, as represented by cLogD, which is within the range of −1 to 5 and is optimal for passive diffusion (Fig. 2). The physicochemical changes associated with N-dealkylation usually showed a small increase in tPSA of ∼9 Å and a reduction in cLogD of up to 1.0 log unit when simple alkyl substituents were cleaved (Table 1). However, when N-dealkylation reaction cleaved a molecule into smaller fragments, more pronounced changes in cLogD and tPSA were observed. For instance, cleavage of nefadozone to m-chlorophenylpiperazine (Fig. 3) was accompanied by a reduction in cLogD of ∼3 log units along with a decrease in tPSA of 36 Å. Interestingly, some drugs in this dataset were exceptions to this trend in that high abundance of amine metabolites were present in circulation despite their high polarity (i.e., cLogD < −1). For example, metabolites of ranolazine, tramadol, zolmitriptan, and sildenafil (the cleaved piperazine metabolite) displayed cLogD values ranging from −1.11 to −2.97 (Table 1). Furthermore, many cleaved metabolites such as 1-pyrimidinylpiperazine metabolite of buspirone and D617 or D620 metabolites of verapamil were also found circulating in human plasma with M/P ratios of >0.25 despite their high polarity. Although the reason for this is not known, one possibility for the high circulating concentrations of these metabolites relative to the parent could be attributed to transporter-mediated uptake into the blood. These hypotheses will require further experimental evaluation for confirmation.
Physicochemical properties, abundance, and estimated fm of amine metabolites
Structures of drugs and their metabolites are provided in Supplemental Table 1. References provided in Supplemental Material.

Relationship between M/P ratio and cLog D values for amine metabolites. (A) All metabolites; (B) expanded view.

Structures of selected drugs that form amine metabolites.
Our analysis also identified some amine metabolites that circulated in relatively low abundance despite having optimal cLogD values of between −1 and 5 (Table 1). On the basis of the conceptual framework discussed in Theoretical Considerations of Determining In Vivo Metabolite Exposure, this was ascribed to either a low CLf and/or a high CLm for the metabolite. For instance, low circulating concentrations of N-desalkyl metabolites of azimilide (Riley et al., 2005), elzasonan (Kamel et al., 2010), imatinib (Gschwind et al., 2005), itraconazole (Isoherranen et al., 2004), olanzapine (Kassahun et al., 1997), propafenone (Kroemer et al., 1989), and repaglinide (van Heiningen et al., 1999) can be attributed to the relatively minor contribution of N-dealkylation to the overall clearance of these drugs. On the other hand, low circulating concentrations of m-chlorophenylpiperazine (Fig. 3) may be ascribed to a high metabolic rate of this metabolite to the corresponding hydroxylated chlorophenylpiperazine (i.e., high CLm of this metabolite) (von Moltke et al., 1999). Ranitidine represents an interesting example in that its N-desmethyl metabolite showed both high polarity (cLogD of −2.22) and low fm (0.01) for this metabolic pathway (Table 1). Thus, it is not surprising that the abundance of this metabolite in circulation is negligible.
The differences in the M/P ratio of N-desisopropyldelavirdine (Fig. 3) after single and multiple dose administration of delavirdine are also informative to our analysis. After oral administration of a single dose, the M/P ratio of N-desisopropyldelavirdine to parent drug was ∼1.1 (Morse et al., 1997). However, after multiple dosing, this ratio was decreased to ∼0.12 to 0.15 (Borin et al., 1997a,b). This disconnect in M/P ratios after single and multiple doses could possibly be attributed to decreased CYP3A-mediated formation of N-desisopropyl metabolite of delavirdine, leading to the lower concentration in the systemic circulation. In vitro, formation of the N-desisopropyldelavirdine is mediated by CYP3A (Voorman et al., 1998a,b). However, delavirdine has also been shown to inactivate CYP3A (Voorman et al., 1998b), which may account for the reduction in oral clearance of delavirdine and hence a decrease in formation of N-desisopropyldelavirdine with repeated dose administration.
Although most aliphatic tertiary amines can undergo secondary metabolism to primary amines via the corresponding secondary amines, only two drugs, namely sibutramine and tamoxifen, yielded primary amines (didesethylsibutramine and didesethyltamoxifen, respectively, Fig. 3) that were circulating in greater than 25% of the parent AUC (Kim et al., 2009; Yeung et al., 2011). Moreover, other drugs (Table 1) that theoretically could be converted to primary amines (based on their structures and lipophilicity) showed very little or no didesalkyl metabolites in circulation. For instance, the M/P ratios for the didesalkyl metabolite of amiodarone (McDonald et al., 2012) or citalopram (Herrlin et al., 2003) were less than 0.1 despite having similar cLogD values to that of the secondary amine metabolites. It is likely that this disconnect can be ascribed to a low fraction of the secondary amine being metabolized to the corresponding primary amine and is indicative of increased metabolic stability of the secondary amine with respect to the subsequent N-demethylation/N-dealkylation pathway compared with the parent tertiary amine. Alternatively, the structural change from a tertiary amine to a secondary amine may impart a different biotransformation pathway such as hydroxylation on the secondary amine (e.g., desipramine to hydroxydesipramine and nortriptyline to hydroxynortriptyline; Fig. 3) rather than formation of a primary amine.
Overall, this dataset suggests that the amine metabolites can be present in circulation that exceeds the threshold of 25% parent AUC over a wide range of cLogD values (Fig. 2). Thus, the fm or CLm appear to be the more important factors in determining the abundance of the amine metabolites in circulation. For those amine metabolites that have cLogD values below −1, they may still be present in circulation in abundance that exceeds the M/P ratio of 0.25, possibly related to transport-mediated processes. Hence, assessment of fm, CLm, cLogD, and transport properties will provide a useful guide to estimate the potential for the amine metabolites to circulate in abundance that may exceed the threshold of 0.25 relative to that for the parent AUC.
Alcohols and Ketones as Metabolites
In this dataset, oxygenation of compounds constitutes the second most common biotransformation process. The corresponding hydroxylated metabolites could be formed either by oxygen insertion into the alkyl or alicyclic and aromatic groups or via O-dealkylation of aliphatic or aromatic ethers. Analysis of the dataset for hydroxylation of aliphatic or alicyclic moieties revealed that of the 125 drugs examined, 40 drugs were shown to yield metabolites with this modification (Table 2), some of which were further oxidized to ketones (Table 3). Examination of physicochemical properties of these metabolites suggested that formation of an aliphatic or alicyclic alcohol was associated with an increase in tPSA of up to ∼20 Å and a reduction of cLogD by ∼1 to 2 log units, thus increasing their polarity as expected. Although a general trend of a decrease in the lipophilicity for these metabolites relative to the parent drug was observed, the vast majority of these metabolites retained favorable lipophilicity (cLogD values between −1 and 5) for diffusion into the systemic circulation.
Physicochemical properties, abundance, and estimated fm of amine metabolites
Structures of drugs and their metabolites are provided in Supplemental Table 2. References provided in Supplemental Material.
Physicochemical properties, abundance and estimated fm of ketone metabolites
Structures of drugs and their metabolites are provided in Supplemental Table 2B. References provided in Supplemental Material.
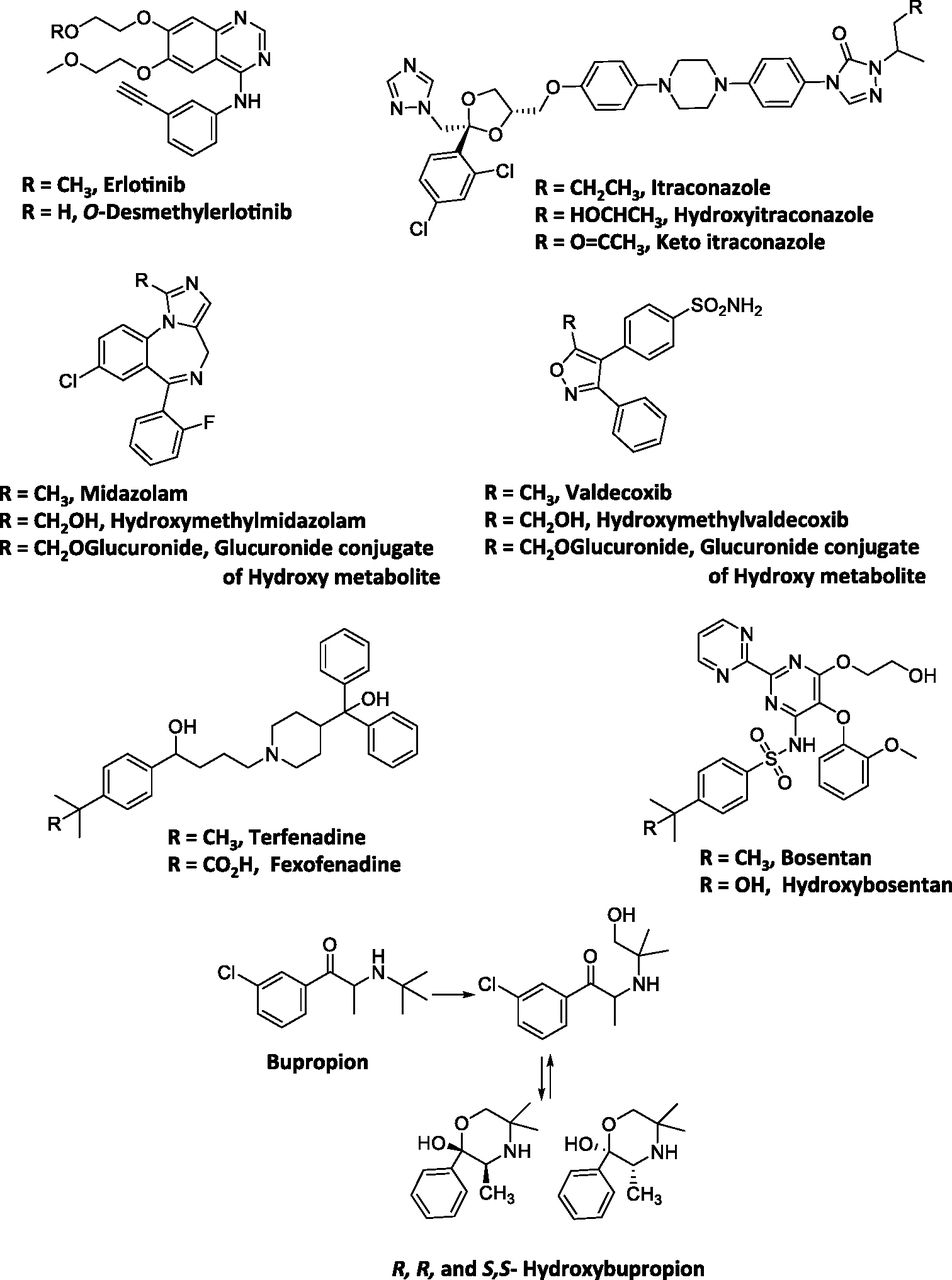
Most of the hydroxylated metabolites in this dataset were found circulating as free aglycones rather than their glucuronide or sulfate conjugates. This was possibly attributed to high pKa values of the hydroxyl groups, which were in the range of ∼12 to 16 (Table 2). Chemically, high pKa value could render conjugation of the hydroxyl group with the glucuronosyl moiety from uridine 5′-diphospho-glucuronic acid or the sulfate group from 3′-phosphoadenosine 5′-phosphosulfate unfavorable (if the alkoxy anion RO− is considered the reactive species in the conjugation reaction), with a net effect of reducing CLm of alcohol metabolites via conjugation reactions. The optimal cLogD and pKa properties impart favorable ADME characteristics to these metabolites and therefore enable their entry into the systemic circulation as free aglycones and maintain a sufficiently high exposure (>25% parent AUC). The contribution of the metabolic pathway also constituted an important parameter in determining if the metabolite circulated in the system in high concentrations, as in the case of amine metabolites. For instance, in the case of erlotinib (Fig. 4), its O-desmethyl metabolite was found in low levels in systemic circulation despite its high pKa (13.9) and high cLogD (2.41). Assessment of the contribution of this metabolic pathway revealed that O-demethylation is a relatively minor route of metabolism for this drug and only accounts for 13% of the dose in humans (Ling et al., 2006). Hence, low CLf most likely accounted for the low abundance of this metabolite (OSI-420, 5% of the parent) in human plasma (Frohna et al., 2006).

Structures of selected drugs that form alcohol metabolites.
Secondary Alcohols as Metabolites
Drugs that yielded secondary alcohols as metabolites such as flutamide, itraconazole, metoprolol (5α-hydroxylation), pioglitazone, taranabant, or buspirone also circulated as the aglycones in humans (Table 2). Although this observation could be explained on the basis of their high pKa and cLogD values, an additional consideration to this observation could be that these metabolites are readily oxidized to their corresponding ketones, as exemplified by pioglitazone or itraconazole for which the ketone metabolite was also primarily detected in plasma (Table 3) (Eckland and Danhof, 2000; Budde et al., 2003). Although this was not observed in all cases, the possible reductase and dehydrogenase catalyzed redox cycle between the alcohol → ketone → alcohol (Oppermann and Maser, 2000) cannot be ruled out for these drugs. Taken together, these factors may result in reducing CLm for the secondary alcohols, enabling these metabolites to circulate as free aglycones.
Primary Alcohols as Metabolites
It is well recognized that many drugs or metabolites containing a primary alcohol tend generally to undergo further oxidation to a carboxylic acid. Consequently, the alcohol metabolite may appear in low abundance, if at all, in circulation with the carboxylic acid as a major component in plasma (see Carboxylic Acid Metabolites below). For example terfenadine (Fig. 4) is converted to the t-butyl alcohol metabolite by CYP3A4 followed by subsequent oxidation to the corresponding carboxylic acid (fexofenadine), which is the primary metabolite found in human plasma (Abernethy et al., 2001). Other drugs that exhibit similar behavior include celecoxib, metoprolol, and montelukast. However, there are exceptions where metabolites could circulate as alcohols instead of the corresponding carboxylic acids. For instance, oxidation of the tertiary butyl functionality of bupropion yields hydroxybupropion (Hesse et al., 2000). The plasma exposure of this hydroxylated metabolite, unlike those of metoprolol, montelukast, and terfenadine, is 7 to 41 times higher than that of parent. It can be postulated that the lack of subsequent oxidation to the carboxylic acid may be related to the spatial arrangement of the hydroxy group relative to the carbonyl group in the molecule. Intramolecular cyclization of the hydroxyl metabolite leads to the formation of the corresponding cyclic R,R-hydroxybupropion and S,S-hydroxybupropion isomers (Fig. 4) and possibly reduces its chance of undergoing further oxidation to the carboxylic acid. Additionally, the CLm of the hydroxyl metabolite via the carboxylic acid pathway is probably low relative to its CLf.
Midazolam and valdecoxib (Fig. 4) represent an example of drugs in which the hydroxylated metabolite is primarily detected as glucuronide conjugate and not as an aglycone or carboxylic acid. Glucuronide conjugate of 1′-hydroxymidazolam was the major metabolite excreted in urine and circulating in plasma even though the plasma AUC of 1′-hydroxymidazolam was ∼0.48 of parent after oral administration of midazolam (Heizmann and Ziegler, 1981; Eap et al., 2004). Similarly, although valdecoxib was oxidized to a hydroxymethyl metabolite (Yuan et al., 2002), the abundance of this metabolite relative to parent in plasma was low (∼0.1) (Sarapa et al., 2005). Results from a human ADME study indicated that this metabolite appears to favor glucuronidation as evidenced by a recovery of 23% of dose in urine as the glucuronide conjugate of this metabolite (Yuan et al., 2002). Interestingly, in both cases, the methyl group undergoing hydroxylation is attached to 5-membered heterocyclic rings (the isoxazole in the case of valdecoxib and imidazole in the case of midazolam). It is possible that the heterocyclic rings may influence the propensity for these alcohol metabolites to undergo glucuronidation. In fact the pKa values of the hydroxymethyl metabolites suggest that the hydroxyl groups are relatively more acidic (13.09 and 13.59, respectively) compared with the primary alcohols formed via oxidation of other drugs (Table 2). Chemically, decreased pKa could potentially increase the susceptibility of these metabolites to undergo further conjugation reaction relative to other hydroxymethyl derivatives. Additionally, potential intramolecular interactions of the hydroxyl group with the nitrogen or oxygen atom in the imidazole or isoxazole rings of the two drugs could possibly increase the propensity of conjugation by rendering the hydroxyl group more nucleophilic. One caveat involves the limitation of our current understanding of the interaction between alcohol metabolites and the conjugation enzymes such as uridine diphosphate glucuronosyltransferase to allow a priori prediction of how well the metabolites with this structural motif may undergo conjugation (i.e., assessment of CLm). This parameter still requires further in vitro experimentation to ascertain, such as the approach used by Lutz and Isoherranen (2012).
Bosentan represents another example of the exception to the trend that a hydroxyl metabolite on the primary carbon will undergo further oxidation to a carboxylic acid. In humans, the major metabolic pathway for bosentan involves oxidation of the t-butyl group to yield the hydroxyl metabolite Ro 48-5033. This pathway constitutes at least 35% of the overall biotransformation for bosentan (Weber et al., 1999). However, after oral administration, plasma AUC of t-butyl hydroxybosentan to parent was only ∼0.13, and the corresponding carboxylic acid was not reported. On the basis of the pharmacokinetic framework described in Theoretical Considerations of Determining In Vivo Metabolite Exposure, it can be postulated that the elimination clearance of this metabolite (by pathways other than further oxidation to carboxylic acid) is more likely favored relative to its formation. Bosentan is a substrate for efflux transporter such as bile salt export pump (Hartman et al., 2010). It may be hypothesized that despite the structural modification, the t-butyl hydroxyl metabolite of bosentan may still retain the affinity to the efflux transporters as with the parent so that biliary elimination rather than further oxidation is favored. If this hypothesis holds true, it will illustrate the importance of understanding the key factors (e.g., structural motifs, physicochemical properties) that affect the affinity of a metabolite to interact with transporters versus metabolic enzyme(s), which will determine the elimination characteristics of the metabolite. Overall, the findings from these empirical observations can be taken into consideration when formulating a rational strategy to anticipate and prioritize which alcohol metabolite(s) should be considered for synthesis and quantitation early on to address the potential drug metabolites in safety testing or DDI issues.
Arenol Metabolites
Metabolites formed via hydroxylation of aromatic rings or O-dealkylation of aromatic ethers (arenols) represented very different characteristics compared with their alcoholic counterparts. Thirty-six drugs in this dataset revealed arenol formation as a part of their biotransformation pathway (Table 4). The physicochemical properties of the arenol metabolites were moderately changed compared with the parent drug, with increase in tPSA by ∼11 to 20 Å and decrease in cLogD by ∼0.5 to 1 log unit associated with these metabolic steps (Table 4). Although the cLogD values of arenols, which typically ranged from −0.6 to 4, showed considerable overlap with those for the alcohol metabolites, the pKa value of the hydroxyl group in these metabolites ranged from ∼4 to 11 (Tables 2 and 3) and were more acidic compared with the alcohol functionality (pKa of 12–16). As seen in Table 4, in contrast to alcohol metabolites, many of the arenol metabolites circulated as glucuronide or sulfate conjugates in humans. Chemically, lower pKa of arenols would make these metabolites more ionizable to yield a phenoxy anion and hence favor the nucleophilic addition to uridine 5′-diphospho-glucuronic acid or 3′-phosphoadenosine 5′-phosphosulfate. Thus, it is not surprising that these metabolites are susceptible to further conjugation and circulated either as glucuronide (e.g., apremilast, atomoxetine, chlorpromazine, codeine to morphine, desloratadine, desipramine, dextromethorphan, duloxetine, efavirenz, imipramine, loxapine, and propranolol) or sulfate (e.g., apixaban, pantoprazole, and rosiglitazone) conjugates (Table 4). Although in vivo data are lacking, in vitro data suggest that 7-OH S-warfarin likely undergoes extensive glucuronide conjugation as well (Bratton et al., 2012).
Physicochemical properties, abundance and estimated fm of arenol metabolites
Structures of drugs and their metabolites are provided in Supplemental Table 3. References provided in Supplemental Material.
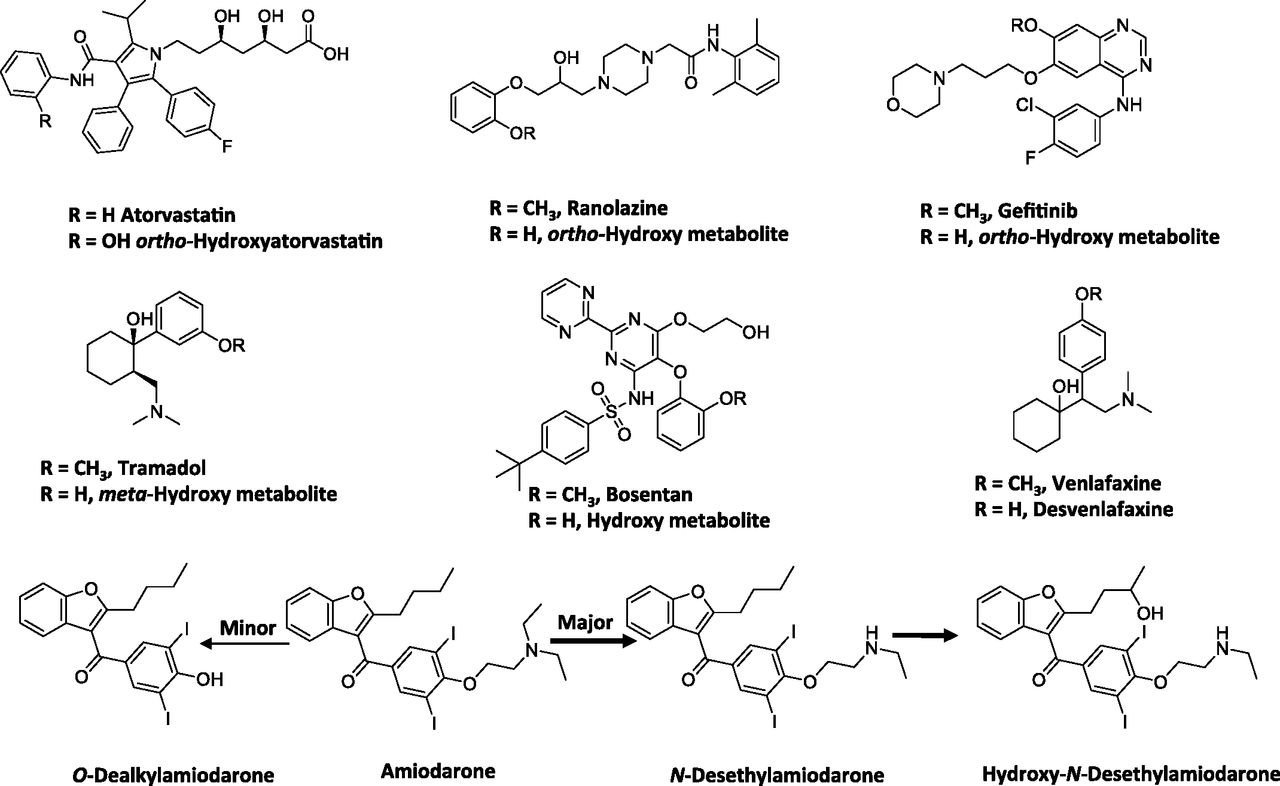
Empirically, our meta-analysis suggested that conjugates of para-hydroxylated arenols were major circulating metabolites rather than the aglycones, while the ortho- and possibly meta-hydroxylated metabolites were primarily found as aglycones in circulation. For instance, the hydroxylated metabolites of atorvastatin, ranolazine, gefitinib, and tramadol (Fig. 5) primarily circulated as the aglycones with an M/P ratio of 0.25 or greater (Table 4). One reason for this could be limited formation of the conjugates of ortho-hydroxyarenols. It is also possible that the position of the hydroxy group on the aromatic ring may influence the efficiency of conjugation. Alternatively, the substrate affinity of the ortho-hydroxylated metabolites for various efflux transporters (e.g., multidrug resistance protein 2 versus multidrug resistance protein 3) may alter the disposition of these metabolites (i.e., being transported to the bile versus to the systemic circulation). The validity of these hypotheses will need to be confirmed with further research.

Structures of selected drugs that form arenol metabolites.
Some examples from this dataset illustrate the utility of the pharmacokinetic framework discussed in Theoretical Considerations of Determining In Vivo Metabolite Exposure in rationalizing the relative abundance of the arenol metabolite to parent. For instance, the ratio of O-desalkylamiodarone to amiodarone is low (0.1) (McDonald et al., 2012), suggesting that the CLf is low relative to the CLm of this metabolite. It can be postulated that since N-deethylation and 3′-hydroxy monodeethylation (Fig. 5) constitutes the major metabolic pathways rather than O-dealkylation of amiodarone, the low ratio may possibly reflect a low CLf for this metabolite. Another example may include the O-desmethyl metabolite for bosentan (Ro 47-8634) (Fig. 5), which also showed low abundance relative to the parent in plasma (∼0.05) (van Giersbergen et al., 2002). The relatively low fraction of dose attributable to the O-demethylation pathway in the excreta for this drug (∼6.4% of dose) is consistent with this interpretation (Weber et al., 1999).
Another drug that displayed an exception to the general trend was venlafaxine. O-Dealkylation of venlafaxine resulted in the formation of desvenlafaxine (Fig. 5) (Howell et al., 1993). The systemic exposure of this metabolite was four- to sixfold higher than that of parent in CYP2D6 extensive metabolizers (EM) (Patat et al., 1998; Hynninen et al., 2008). Furthermore, although a significant portion of the dose (26%) was excreted in urine as the glucuronide conjugate of desvenlafaxine, 29% of the dose was excreted as the aglycone (Howell et al., 1993). Also, after oral administration of desvenlafaxine, renal clearance constitutes a major clearance mechanism for the parent (DeMaio et al., 2011). This example illustrates that a change in physicochemical properties associated with the metabolite may alter its clearance mechanism, in this case, to renal elimination. This can result in a significant impact on the elimination clearance of the metabolite and thus affect the anticipated AUC ratio of metabolite to parent. Overall this analysis suggested the complexity of predicting the exact nature of the metabolites that undergo aromatic hydroxylation or O-dealkylation and emphasized the importance of understanding the formation and elimination (e.g., conjugation) characteristics of the metabolites. It also highlighted the importance of obtaining good estimates of both CLf and CLm to yield a more accurate assessment of the AUC ratio of metabolite (aglycone) to parent.
S- and N-Oxides Metabolites
S-Oxides
Twelve drugs in this dataset were metabolized to the corresponding sulfoxide and/or sulfone metabolites (Table 5). Formation of a sulfoxide via oxidation of the sulfur moiety lowered the cLogD by ∼1 to 2 log units and increased tPSA by ∼11 Å relative to the parent drug. Despite this, most of the metabolic products from sulfoxidation retained sufficient lipophilicity, as reflected by the cLogD values in the range of −1 to 3 (Table 5). This favorable physicochemical property probably enabled these entities to reach systemic circulation, because most drugs (6 of 8) that were converted to the sulfoxide metabolite had an M/P ratio exceeding 0.25. On the other hand, relatively low amounts of the circulating sulfoxide metabolite of montelukast and ranitidine were detected in human plasma. Montelukast is primarily metabolized in humans via hydroxylation and subsequent conversion to the carboxylic acid, whereas S-oxidation constitutes a minor pathway of metabolism for this drug. Low levels of the montelukast sulfoxide in human plasma despite the favorable cLogD and tPSA values (1.74 and 107, respectively) were therefore likely attributed to a low CLf, whereas the sulfoxide metabolite of ranitidine exhibited both low fm (0.01) and high polarity (cLogD of −3.43).
Physicochemical properties, abundance and estimated fm of N- and S-oxide metabolites
Structures of drugs and their metabolites are provided in Supplemental Table 4. References provided in Supplemental Material.
Drugs containing a sulfoxide moiety can generally undergo further oxidation to a sulfone, which can also circulate as the major metabolite. For instance, flosequinan, omeprazole, lansoprazole in CYP2C19 poor metabolizers (PM), and thioridazine were converted to their corresponding sulfone metabolites and had M/P ratios >0.25 (Table 5). This was ascribed to their favorable cLog D values, because in most cases the conversion of a sulfoxide to a sulfone metabolite increased its cLogD by up to ∼1 log unit and decreased tPSA by ∼6 Å (Table 5). The only outlier to this trend was pantoprazole, which showed an M/P ratio <0.25. Although the reason for this is not known, the discrepancy could also be ascribed to relatively less contribution of this metabolic pathway toward clearance of this drug, because the major pathway appears to be O-dealkylation of the methoxy ether to the corresponding arenol metabolite. Of interest, in this analysis, for the parent drugs that contain sulfide in its structure, oxidative metabolism yields sulfoxide as the major circulating metabolite (e.g., albendazole, axitinib, chlorpromazine, dothiepin, thioridazine). However, the corresponding sulfone tends to be either not present as major circulating metabolite or is present in abundance considerably lower than the corresponding sulfoxide (e.g., thioridazine). Similar to the amines (see Secondary or Primary Amines as Metabolites), this may also be attributed to the low CLf for this sequential oxidation step.
N-Oxides
In this analysis, 11 drugs were converted to aliphatic or aromatic N-oxides (Table 5). Three drugs showed the formation of aromatic N-oxide, two of which are pyridine N-oxide (roflumilast and regorafenib) and one is a pyrimidine N-oxide (voriconazole). The remaining eight drugs formed aliphatic or cyclic N-oxide. Changes in physicochemical properties from N-oxidation are of a similar order to S-oxidation to a sulfoxide with an increase of tPSA of 14 Å and a lowering of lipophilicity of ∼0.5 to 2 log units. All except ranitidine N-oxide and zolmitriptan N-oxide showed a cLogD value of greater than zero (Table 5), which conferred a favorable lipophilic property to enable these metabolites to reach the systemic circulation provided that a sufficient amount was formed. A notable feature of N-oxide metabolites is their apparent rapid interconversion with parent, leading to formation of both parent drug and other primary and secondary metabolites. Interconversion after administration of the metabolite to humans and animals has been shown for chlorpromazine (Jaworski et al., 1990), clozapine (Chang et al., 1998), and voriconazole (Roffey et al., 2003). The interconversion will tend to lower the M/P ratio of this type of metabolite.
Carboxylic Acid Metabolites
Eleven drugs in this dataset were oxidized to a carboxylic acid metabolite via various metabolic reactions (Table 5). Seven of these drugs, including celecoxib, losartan, metoprolol (O-demethylation), montelukast, quetiapine, N-dealkyl product of ranolazine (CVT-2534), and terfenadine, underwent oxidation of either the parent drug or the intermediate alcohol metabolite to the corresponding carboxylic acid (see Alcohols and Ketones as Metabolites). As observed from the M/P ratios, in all cases, the carboxylic acid was the major circulating metabolic product possibly because of its small volume of distribution and high plasma protein binding (Obach et al., 2008). The data from these seven drugs/metabolites were consistent with this hypothesis in that the AUC of the carboxylic acid metabolite was considerably higher compared with their corresponding alcohol metabolite. Another factor that may also influence the high M/P ratio for the carboxylic acid metabolites is related to the new functional group being introduced to the metabolite, which is metabolically more stable than the parent (i.e., reduced CLm). This is illustrated by the example of losartan. A comparison of the pharmacokinetics of losartan to its carboxylic acid metabolite indicates that the volume of distribution (∼0.45 l/kg) and plasma clearance (∼9 ml/min/kg) of losartan was greater than that of carboxylosartan (plasma clearance ∼0.7 ml/min/kg and volume of distribution ∼0.14 l/kg) (Christ, 1995; Lo et al., 1995). Additionally, the renal clearance (∼1 ml/min/kg) of losartan accounted for 11% of its total plasma clearance, whereas the renal clearance of carboxylosartan (∼0.4 ml/min/kg) accounted for 55% of its total plasma clearance. The free fraction for the metabolite (∼0.0035) showed an approximately fourfold decrease compared with the parent (∼0.0135), suggesting that the effect of the additional acidic function in the metabolite has little effect on unbound volume of distribution or renal clearance. However, there was a sevenfold reduction (593 ml/min/kg for losartan and 86 ml/min/kg for carboxylosartan) in unbound nonrenal clearance, which can be postulated to be due mainly to the oxidation of the more labile hydroxyl group in losartan to a substantially more metabolically stable carboxyl function. In addition to losartan, metabolic conversion of terfenadine to fexofenadine likely represents another example in support of this postulate.
In this dataset, 8 of 11 carboxylic acid metabolites showed negative cLogD values ranging from −0.06 (carboxylosartan) to −3.4 (zolmitriptan indoleacetic acid), suggesting that these metabolites have high polarity (Table 6). However, 7 out of 8 of these metabolites had an M/P ratio of 0.25 or higher, with repaglinide carboxylic acid (Fig. 6) as the only exception. Although the mechanism(s) by which passage of these polar carboxylic acid metabolites into systemic circulation is not well understood, one possible explanation may involve transporter-mediated processes. For instance, fexofenadine is a substrate for hMRP3 (human multidrug resistance protein 3) (Matsushima et al., 2008), which mediates the sinusoidal efflux of a variety of organic anions. This transporter therefore may play a role in mediating the efflux of fexofenadine from the liver to the systemic circulation, as demonstrated in the Mrp3(−/−) mouse model (Matsushima et al., 2008). Moreover, the example from the carboxylic acid metabolite of repaglinide illustrates the potential complexity of these active processes. In humans, the M/P ratio is low (0.05) despite a large fraction of the repaglinide dose being metabolized to this metabolite as evidenced by the recovery of ∼60% of the dose as this metabolite in feces (presumably via biliary excretion since the formation of this metabolite involves oxidative metabolic process) and ∼2% of the dose in urine (van Heiningen et al., 1999; Honkalammi et al., 2011). This suggests that the CLm for this metabolite probably is considerably higher relative to its CLf. Thus, it is possible that the structural motif, physicochemical, and/or ADME properties for repaglinide carboxylic acid metabolite favor an interaction with the transporters involved in biliary excretion, whereas those for the other 7 carboxylic acid metabolites may have properties that favor an interaction with transporters that enables the passage of these metabolites into the systemic circulation. Interestingly, repaglinide and its carboxylic acid metabolite also showed one of the largest physicochemical changes among the parent/metabolites reviewed in this analysis, with an increase in tPSA from ∼80 to 125 Å and a ∼4 log unit reduction in cLogD to approximately −2 (Table 6). It is possible that another contributing factor to the low plasma concentration of repaglinide carboxylic acid metabolite may be attributed to low fp, which, in this case, may become limiting for partitioning from the hepatocytes to systemic circulation. As our knowledge in transporter-mediated processes in relation to structural and physicochemical properties is relatively nascent, further research into this area is needed to elucidate these mechanisms.
Physicochemical properties, abundance and estimated fm of carboxylic acid metabolites
Structures of drugs and their metabolites are provided in Supplemental Table 5. References provided in Supplemental Material.

Scheme showing conversion of repaglinide to repaglinide carboxylic acid and methadone to EDDP metabolite.
Novel Metabolites
We included in Table 7 novel metabolites that are formed in a “unique” manner, although in fact they are still formed mainly via oxidations and reductions. In most cases, the changes in physicochemical properties of metabolites are relatively small compared with the parent drug (Table 7). Most metabolites exhibited cLogD values within the range of −1 to 5, which conferred favorable lipophilic characteristics for their passage from the liver to the systemic circulation. For most of these metabolites, unless the structural modifications render the CLm to be substantially different than CLparent, the M/P ratios will mostly be determined by CLf. The exception to this was methadone. Conversion of methadone to 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP) metabolite (Fig. 6) showed a reduction in cLogD value by almost 6 log units from 2.29 to −3.51, and the tPSA of this metabolite is also very small (∼3 Å). Thus low M/P ratio of this metabolite (∼0.16) could be ascribed, at least in part, to the high polarity and hence low lipoidal permeability for diffusion into the systemic circulation.
Physicochemical properties, abundance and estimated fm of novel metabolites
Structures of drugs and their metabolites are provided in Supplemental Table 6. References provided in Supplemental Material.
Theoretical Considerations: Which Factors Appear Dominant?
Lipoidal Permeability
The liver is the principal site of metabolism and receives approximately 25% of the cardiac output. This equates to a blood flow of around 0.8 ml/min/g in human and 3-4 ml/min/g in rat. The capillaries supplying the hepatocytes are sinusoidal with a broken basement membrane and very leaky endothelial openings of 100–300 nm in diameter (Balaz, 2009). Drug and metabolite molecules are free to access or leave the surface of the hepatocyte without regard to physicochemistry. The membrane permeability of the hepatocyte to drugs and their metabolites has been explored in a series of studies by Chou et al. (1995). The authors’ analysis of the data concludes that the lower limit of PS is 0.3–0.5 ml/min/g liver for compounds with logD values below −3. Permeability then increases dramatically with increases of logD from −3 to 1.5; at still greater logD values, a plateau rate of PS of 200–300 ml/min/g liver is reached. In our dataset, we have ascribed a lower cLogD value of −1 as a threshold at or above which free passive diffusion of drug and metabolites across the membrane is likely to occur and will not be a limiting factor in determining the presence of circulating metabolites. Around this value PS exceeds liver blood flow, suggesting blood perfusion rate limitations (rather than membrane permeability limitations) on influx and efflux of metabolites.
We also considered the abundance of circulating concentrations of metabolite relative to the parent drug from the viewpoint of fm and fp. The term fp was thought to be important inasmuch as a low permeability rate from the hepatocyte would attenuate appearance in the circulation if further metabolism or canalicular biliary excretion occurs. It appears that whether considered from in vitro or in vivo excretion data, fm is a much more dominant feature in determining the circulating M/P ratios relative to fp. This is further substantiated by an additional analysis showing that there was no apparent relationship between M/P ratio and change in cLogD or tPSA values from parent drug to its corresponding metabolites (data not shown). This partly reflects the observation that most metabolites still possess favorable lipoidal permeability characteristics because of a limited change in lipophilicity and tPSA. As illustrated by the drugs evaluated in this dataset, in most cases the metabolic steps associated with circulating metabolites lower lipophilicity (cLogD) by up to ∼2 log units and raise tPSA by up to ∼20 Å. Although these metabolites typically have lower lipoidal permeability than their parent drugs, the changes are unlikely to be significant enough to render the permeability rate from the hepatocytes be a limiting factor in restricting the passage of metabolites from the liver to the systemic circulation. It is also reasonable to assume that lowered lipophilicity will result in lowered rates of metabolism and canalicular biliary excretion, because the binding sites of most drug metabolizing enzymes and transporters are hydrophobic in nature. Thus, it can be conceived that the net effect of these changes associated with lower lipophilicity for the metabolites would render fm to be a more dominant determinant relative to fp.
Metabolite-to-Parent Ratio in Systemic Circulation for High Clearance Drugs
In most cases, in vivo data are lacking for CLm so that the ratio of CLparent/CLm typically cannot directly be calculated. As discussed previously (see Theoretical Considerations of Determining In Vivo Metabolite Exposure), CLm is an important parameter in determining the M/P ratio; thus, it represents an opportunity for further investigations to develop better predictive models for this parameter. Some of the drugs examined in this dataset are very rapidly and extensively metabolized in humans and act virtually as prodrugs. These include flutamide (oral clearance value of >600 ml/min/kg), buspirone (intravenous clearance value of 28 ml/min/kg and 4% bioavailability), and terfenadine (oral clearance value of >800 ml/min/kg) (Anjum et al., 1999; Mahmood and Sahajwalla, 1999; Abernethy et al., 2001). These drugs all have very high M/P ratios in systemic circulation. Although characterization of the in vivo CLf and CLm values for these metabolites are incomplete, the high M/P ratio after oral administration of the parent drug suggests that the fraction of the parent drug escaping the liver is low and/or that the CLf far exceeds the CLm for these metabolites (see Theoretical Considerations of Determining In Vivo Metabolite Exposure). Such drugs probably will not be a major factor in current drug discovery programs because of the emphasis on bioavailability and metabolic stability in compound design.
Protein Binding
In Theoretical Considerations of Determining In Vivo Metabolite Exposure, it was postulated that overall the reduction in lipophilicity seen in most cases of metabolism would generally result in a higher free fraction and a lower intrinsic (unbound) metabolic clearance, thus attenuating the effects on total drug clearance versus total metabolite clearance. Data are available for several drugs that undergo different metabolic routes to their corresponding metabolites to illustrate this hypothesis (Table 8). In all cases, metabolism lowered lipophilicity and increased free fraction. For those metabolites that are cleared predominantly by metabolism (5/6, desmethylclozapine being predominantly renal), their total clearances are within 3-fold of that for the parent even though their unbound metabolic or hepatobiliary (nonrenal) clearance differences can be much larger. This attenuation of differences in clearance is exemplified by darifenacin and its hydroxyl metabolite. The total clearance of the parent and metabolite is similar, but there is a 10-fold difference in unbound clearance and fraction unbound. Similar trends are observed for chlorpromazine, diltiazem, and tolterodine (Table 8). These examples illustrate that the reduction in lipophilicity seen with metabolism does not have pronounced effects on the ratio of CLparent/CLm.
Although the effect of reduction in lipophilicity generally tends to attenuate the influence on total drug clearance versus total metabolite clearance, an exception to this has been observed and is related to the new functional group being introduced into the metabolite, which is itself highly labile to a metabolic route unavailable to the parent. In this case, the metabolite may display a higher clearance compared with the parent. This exception is illustrated by the metabolic conversion of propranolol to hydroxypropranolol, which is cleared predominantly by conjugation of the newly introduced phenolic group. The clearance of this metabolite is higher than its parent (Table 8), consistent with its very low concentration after systemic administration and its presence being observed only after the rapid first-pass effects seen for parent after oral administration. This reinforces the observation that in addition to protein binding, additional factors such as the new structural motif(s) introduced into the metabolite should be taken into consideration when determining the potential influence of structural-chemical changes on the CLm of the metabolite.
Pivotal Role of fm
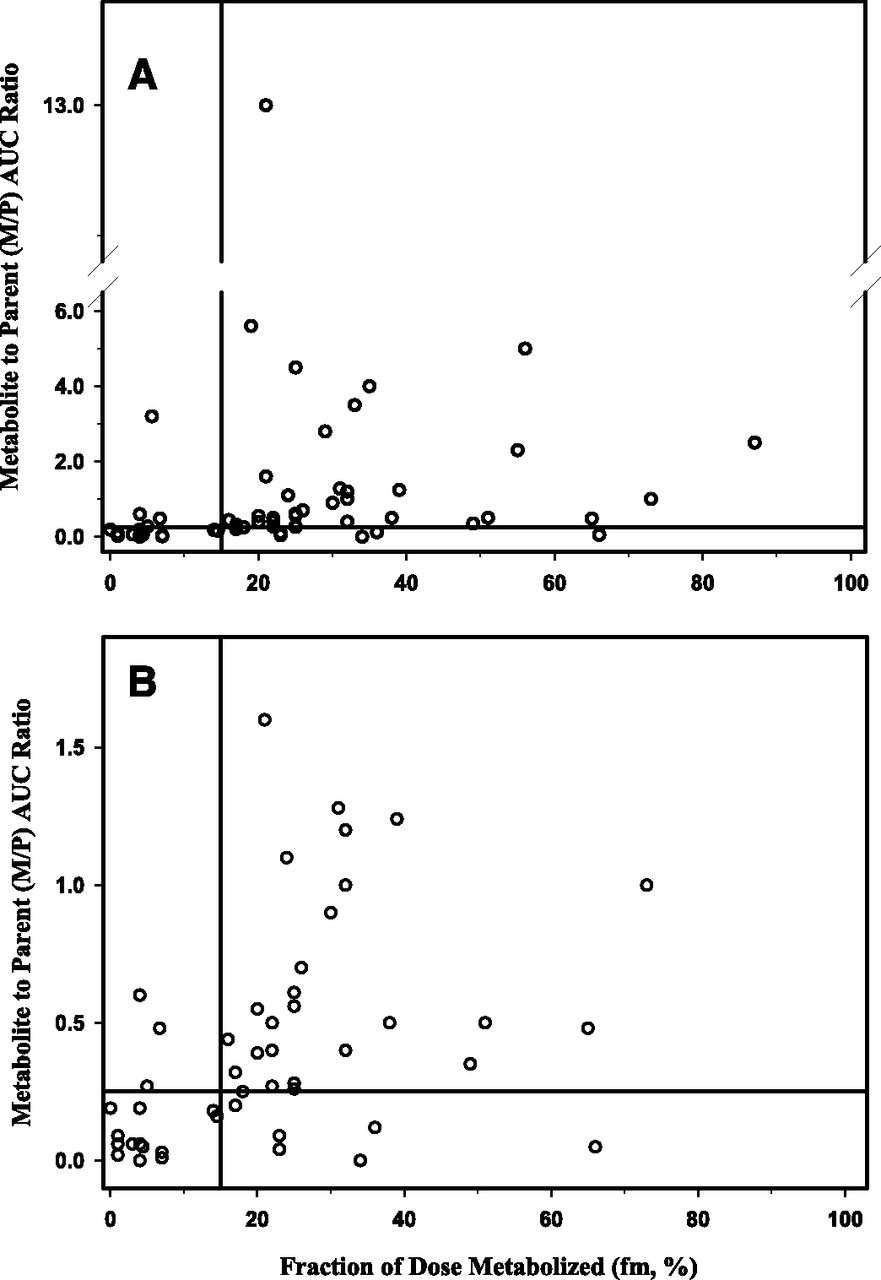
Given the pivotal role that fm plays in determining the AUC of a given metabolite (see Theoretical Considerations of Determining In Vivo Metabolite Exposure) and hence its relative abundance in relation to the parent drug, an additional analysis was conducted to ascertain if there is a threshold value that can be identified to be associated with a M/P ratio of >25%. The data (M/P ratios and fm values) used in this analysis are listed in Tables 1–6. The fm values were estimated from data obtained in human ADME studies with radiolabeled drugs. The fraction of dose for the metabolite of interest and its sequential metabolic products recovered in excreta (both urine and feces) were assumed to be representative of the in vivo fm value. Data were available for a total of 60 metabolites in this dataset, and the results are presented in Fig. 7. Visual inspection of this data suggests that fm value of 0.15 appears to be a reasonable threshold value to distinguish the metabolites that may circulate with abundance exceeding the threshold of 25% of parent AUC compared with those that do not. By using this threshold fm value, four metabolites have been identified as “false negative” in that while the fm values are below 0.15, the M/P ratios exceed the threshold of 0.25. These four metabolites are cyclized indole products of elzasonan, 2-hydroxycyclopentyl ruxolitinib, zolmitriptan N-oxide, and N-desmethylzolmitriptan. The reason(s) for this is not readily apparent, but it could be postulated that underestimation of fm [possibly related to incomplete collection of radioactive dose and/or characterization of the metabolite profiles for elzasonan (79% of dose recovered) and potential back-conversion of N-oxide to parent in feces for zolmitriptan] may have contributed to this discordance.

Relationship of metabolite to parent AUC ratio as a function of fm. (A) All metabolites; (B) Expanded view.
When the M/P ratio for the aglycone of the arenol metabolites were used in this analysis, there were 14 “false positives” identified (i.e., fm of >0.15 but M/P of <0.25). Among these 14 “false positives,” the most common structures are the arenol metabolites where the circulating level of the aglycone is negligible. However, when the M/P values for the conjugates of these arenol metabolites were plotted instead (Fig. 7), the number of “false positive” is reduced to 6. Three of these six metabolites appear to be conjugated (5-hydroxyelzasonan, O-desalkyl flecainide, and 5-methylhydroxy valdecoxib) and excreted; however, the conjugate was not reported to be present in plasma. Two additional metabolites (t-butyl hydroxybosentan and repaglinide carboxylic acid) may also display similar properties in that once formed, these metabolites likely favors excretion into the bile rather than being transported to the systemic circulation. N-desmethylzopiclone represents a marginal case in that the fm value is estimated to be 0.17 while the M/P ratio is 0.2.
By use of the threshold fm value of 0.15, the false-negative and the false-positive rates (when the M/P ratios of the conjugates of the arenol metabolites are considered) are 10% (4 out of 40 metabolites) and 30% (6 out of 20 metabolites), respectively, which represent a reasonable trade-off since further reduction in the false-negative rate will increase the false-positive rate considerably. Moreover, this suggested threshold value may be further refined as more data become available in the future.
Special Considerations
Genetic Polymorphisms
Genetic polymorphisms of the drug metabolizing enzymes play an important role in influencing the disposition of many drugs and metabolites. In this analysis, a few examples illustrate the important influence of genetic polymorphisms on the abundance of major circulating metabolites. The first set of examples involves the differences in the abundance of circulating metabolites between EM and PM of CYP2C19 and CYP2D6. In the former case, the M/P ratio of lansoprazole sulfone in CYP2C19 PM (0.88) was considerably higher than those in the CYP2C19 homozygous EM and heterozygous EM (<0.1). Another example that showed a similar pattern was atomoxetine, which is metabolized by CYP2D6 (aromatic 4-hydroxylation, major pathway in CYP2D6 EM) and CYP2C19 (N-demethylation) (Sauer et al., 2005). In CYP2D6 EM subjects, 4-OH atomoxetine glucuronide constitutes the major circulating moiety, while the plasma AUC ratios of N-desmethyl atomoxetine and 4-hydroxyatomoxetine to parent were low (<0.1) (Sauer et al., 2003). Moreover, in CYP2D6 PM, the AUC ratio of N-desmethyl atomoxetine to parent drug was 0.33 to 0.42 (Sauer et al., 2003; FDA Clinical Pharmacology Review: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21-411_Strattera_biopharmr_P2.pdf), thus constituting a moiety that needs to be considered for assessment for potential DDI under the current EMA and draft FDA Guidance. These examples highlight the need to evaluate the abundance of circulating metabolites in relation to the genotype and phenotype of the metabolizing enzymes, if genetic polymorphisms are known to exist for the particular enzyme of interest.
Another set of examples that illustrate the importance of gene dose on the abundance of the circulating metabolites include nortriptyline and codeine. In humans, nortriptyline undergoes CYP2D6-mediated hydroxylation to form the cyclic E-10-hydroxynortriptyline (major form of this metabolite), while the formation of Z-10-hydroxynortriptyline (minor form of this metabolite) is not related to CYP2D6 polymorphism. Dalen and co-workers (1998) demonstrated that the ratio of AUC of 10-hydroxynortriptyline to parent increased with the number of functional CYP2D6 genes, ranging from 0.36 in subjects with no functional CYP2D6 genes to 13 in subjects with 13 functional CYP2D6 genes. Likewise, in humans, codeine undergoes CYP2D6-mediated demethylation to form morphine, which is glucuronidated to morphine 3-glucuronide and morphine 6-glucuronide. In subjects who are CYP2D6 PM, ratio of plasma AUC for morphine to codeine was minimal (0.008), whereas this ratio was higher in subjects who are CYP2D6 EM (0.06) and CYP2D6 ultrarapid metabolizers (0.08) (Kirchheiner et al., 2007). Furthermore, in these same subjects, the systemic exposure (plasma AUC) of morphine 3-glucuronide and morphine 6-glucuronide (pharmacologically active) in CYP2D6 EM and ultrarapid metabolizers were substantially higher than in CYP2D6 PM. Thus, gene dose can exert a significant influence on the systemic exposure to the circulating metabolites and should be taken into consideration when attempting to project the abundance of circulating metabolites.
Caveats to This Analysis
This analysis is intended to assess general trends regarding a possible relationship of structural motif and the associated physicochemical properties of the metabolites to their relative abundance in systemic circulation. Attempts to rationalize some of the observed trends were made based on existing known biologic and/or chemistry principles. However, there are some significant knowledge gaps in certain areas where mechanistic insights are lacking to fully understand the observed results. Plausible hypotheses for further testing are postulated for subsequent confirmation when feasible. Not surprisingly, results of this analysis point to the need for 1) further understanding of the scaling for conjugation reactions (e.g., glucuronidation and sulfation) to provide a more accurate estimate of CLm for the alcohol and arenol metabolites to better predict the ratio of AUC of metabolite to parent, 2) an integrated approach to combine metabolism and transport data for quantitative prediction, particularly the transport processes in the liver that mediate biliary excretion versus passage into systemic circulation, and 3) a more complete understanding of the interplay between physicochemical properties for passive processes with spatial arrangement of the compound for its affinity to enzymatic/active transport processes.
Another caveat to our results involves the complexity of the metabolic pathways. In this dataset, the vast majority of the circulating metabolites is formed from primary biotransformation pathways. However, in humans, there are examples where the major circulating metabolites are derived from secondary or tertiary biotransformation steps, e.g., torceptrapib (Dalvie et al., 2008). Since the ability for the in vitro systems to predict these secondary or tertiary metabolites is poor (Dalvie et al., 2009), there is still the necessity to confirm the major circulating metabolites in a human in vivo ADME study with radiolabeled drug. In addition, this analysis only addressed the relative abundance of the metabolite to parent, and there was no attempt to assess structure-physicochemical properties in relation to pharmacologic activity. Clearly, in drug discovery and development programs, an understanding of both aspects is essential.
Summary
To our knowledge, this is one of the first attempts to assess the relative abundance of circulating metabolites using an integrated approach of principles of metabolite kinetics along with structure-physicochemical properties of the parent/metabolites. The analysis was conducted in a dataset comprising 125 drugs with their circulating metabolites. The major factors identified in this analysis that will favor a given circulating metabolite to exceed the threshold of 25% of parent appears be a fm value of >15%, a cLogD value of greater than −1, and a structural motif that minimizes the CLm relative to its CLf. Potential hypotheses are proposed where exceptions to these general trends are observed, which provides opportunities for further scientific investigations to refine these recommendations.
Authorship Contributions:
Participated in research design: Loi, Smith, Dalvie.
Performed data analysis: Loi, Smith, Dalvie.
Wrote or contributed to the writing of the manuscript: Loi, Smith, Dalvie.
Footnotes
- Received November 20, 2012.
- Accepted March 1, 2013.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ADME
- absorption, distribution, metabolism, excretion
- AUC
- area-under-the concentration time curve
- AUCm
- area-under-the concentration time curve for a metabolite
- CLf
- formation clearance of metabolite
- CLm
- elimination clearance of metabolite
- CLparent
- clearance of parent drug
- CVT-2534
- ranolazine
- P450
- cytochrome P450
- DDI
- drug-drug interactions
- EM
- extensive metabolizer
- EMA
- European Medicines Agency
- Fa
- fraction of dose absorbed after oral administration
- FDA
- Food and Drug Administration
- Fh,m
- systemic availability of the metabolite
- fm
- fraction of dose converted to a given metabolite from the parent
- fp
- lipoidal permeability
- M/P
- metabolite-to-parent
- PM
- poor metabolizer
- Ro 47-8634
- bosentan
- tPSA
- topological polar surface area
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}