


Abstract
The main route of elimination of vildagliptin, which is an inhibitor of dipeptidyl peptidase-4 (DPP-4), in humans is cyano group hydrolysis to produce a carboxylic acid metabolite M20.7. Our in vitro study previously demonstrated that DPP-4 itself greatly contributed to the hydrolysis of vildagliptin in mouse, rat, and human livers. To investigate whether hepatic DPP-4 contributes to the hydrolysis of vildagliptin in vivo, in the present study, we conducted in vivo pharmacokinetics studies of vildagliptin in mice coadministered with vildagliptin and sitagliptin, which is another DPP-4 inhibitor, and also in streptozotocin (STZ)-induced diabetic mice. The area under the plasma concentration-time curve (AUC) value of M20.7 in mice coadministered with vildagliptin and sitagliptin was significantly lower than that in mice administered vildagliptin alone (P < 0.01). Although plasma DPP-4 expression level was increased 1.9-fold, hepatic DPP-4 activity was decreased in STZ-induced diabetic mice. The AUC values of M20.7 in STZ-induced diabetic mice were lower than those in control mice (P < 0.01). Additionally, the AUC values of M20.7 significantly positively correlated with hepatic DPP-4 activities in the individual mice (Rs = 0.943, P < 0.05). These findings indicated that DPP-4 greatly contributed to the hydrolysis of vildagliptin in vivo and that not plasma, but hepatic DPP-4 controlled pharmacokinetics of vildagliptin. Furthermore, enzyme assays of 23 individual human liver samples showed that there was a 3.6-fold interindividual variability in vildagliptin-hydrolyzing activities. Predetermination of the interindividual variability of hepatic vildagliptin-hydrolyzing activity might be useful for the prediction of blood vildagliptin concentrations in vivo.
Introduction
Vildagliptin (LAF237) is an inhibitor of dipeptidyl peptidase-4 (DPP-4; EC 3.4.14.5, also known as CD26) for the treatment of type 2 diabetes mellitus (T2DM) (Villhauer et al., 2003). Because DPP-4 inhibitors, also called incretin enhancers, improve glucose control with a low risk of hypoglycemia, they are attracting attention among therapeutic agents for T2DM (Scheen, 2010; Deacon, 2011). To date, at least 11 DPP-4 inhibitors have been approved in the world (Deacon and Lebovitz, 2016). Twice-daily administration is suggested for vildagliptin due to its shorter half-life, whereas most other DPP-4 inhibitors allow single oral administration per day for management of T2DM (Deacon, 2011). Vildagliptin is largely metabolized before excretion, whereas most of the approved DPP-4 inhibitors are excreted into urine without undergoing metabolism (He et al., 2009a). This allows us to use vildagliptin on patients with renal impairment (Lukashevich et al., 2011).
The main route of elimination of vildagliptin in humans is cyano group hydrolysis to produce a carboxylic acid metabolite M20.7 (LAY151), which is pharmacologically inactive (He et al., 2009a) (Fig. 1). It has been reported that the parental vildagliptin and its main metabolite M20.7 account for the majority of vildagliptin-related materials in human plasma (approximately 26% and 56%, respectively) and that the liver has an M20.7 formation activity (He, et al., 2007, 2009a). Cytochrome P450s (CYPs) and nitrilase-like proteins did not exhibit the formation of M20.7 (He et al., 2009a; Asakura et al., 2014). Although the main metabolic enzyme responsible for vildagliptin cyano group hydrolysis in humans was unknown, our in vitro study previously demonstrated that DPP-4, which is the pharmacological target of vildagliptin, greatly contributed to the cyano group hydrolysis of vildagliptin in human liver (Asakura et al., 2015). Minor metabolites, which resulted from amide bond hydrolysis (M15.3), glucuronidation (M20.2), and oxidation on the pyrrolidine moiety of vildagliptin (M20.9 and M21.6), have been identified in blood and urine (He et al., 2009a) (Fig. 1). The main circulating metabolite of vildagliptin in humans, mice, rats, and dogs is M20.7 (He et al., 2009b), whereas that in rabbits and monkeys is M15.3 and M20.2, respectively (Table 1). In humans, the formation of M20.7 is the main clearance pathway of vildagliptin (Table 1). In contrast, the main clearance pathway of vildagliptin is the renal excretion of parental vildagliptin in mice (Table 1). Thus, the main clearance pathway of vildagliptin is slightly different among species. Because the main circulating metabolite of vildagliptin in mice is M20.7 as observed in humans (Table 1), it was suggested that mice could be one of the useful model animals for the in vivo pharmacokinetics studies of vildagliptin and its main metabolite M20.7.

Metabolic pathways of vildagliptin in humans. The major metabolic pathway of vildagliptin in humans is DPP-4–mediated hydrolysis at the cyano group to produce a carboxylic acid metabolite M20.7 (He et al., 2009a; Asakura et al., 2015). Vildagliptin is also metabolized to form amide bond hydrolysis (M15.3), glucuronidation (M20.2), and oxidation on the pyrrolidine moiety of vildagliptin (M20.9 and M21.6). Reported amount of vildagliptin and metabolites in urine and feces is indicated as percentage of dose (He et al., 2009a).
Species differences of metabolite levels in plasma after oral administration
Australian public assessment report for vildagliptin, Department of Health and Ageing, Therapeutic Goods Administration, 2010 April. Available from: http://www.tga.gov.au/auspar/auspar-vildagliptin (accessed June 7, 2016).
DPP-4, a serine protease belonging to type II transmembrane glycoproteins, is ubiquitously expressed in various tissues (Mentlein, 1999; Gorrell et al., 2001). In addition to the integral membrane form, a soluble form of DPP-4 presents in blood (Durinx et al., 2000). The soluble form of DPP-4 also has dipeptidyl peptidase activity (Durinx et al., 2000). It has been reported that DPP-4 expression level in blood positively correlates with body mass index (BMI), the amount of visceral adipose tissue, and insulin resistance (Lamers et al., 2011; Sell et al., 2013). Furthermore, several reports describe that DPP-4 activity in blood is increased in type 1 diabetes mellitus (Varga et al., 2011; Osawa et al., 2016) and T2DM (Ryskjaer J et al., 2006; Sell et al., 2013). It has also been reported that the DPP-4 activity level in blood of streptozotocin (STZ)-induced type 1 diabetic mice is higher than that of normal mice (Das et al., 2014).
Although hepatic DPP-4 greatly contributes to the cyano group hydrolysis of vildagliptin in vitro (Asakura et al., 2015), not only hepatic DPP-4 but also DPP-4 in other tissues and blood might also be involved in the formation of M20.7 in vivo. If DPP-4 in blood is involved in the formation of M20.7 in vivo, the increase of DPP-4 activity level in blood according to obesity and diabetes status can induce the formation of M20.7 in patients treated with vildagliptin. However, it has not been reported whether the increase of DPP-4 activity level in blood can have an effect on the formation rate of M20.7 in vivo. In the present study, first we conducted in vivo pharmacokinetics studies using sitagliptin as a competitive inhibitor of the DPP-4–mediated cyano group hydrolysis of vildagliptin to investigate whether DPP-4 contributes to the cyano group hydrolysis of vildagliptin in vivo. Second, to investigate the effect of the increased DPP-4 activity level in blood on the blood concentrations of vildagliptin and M20.7, we conducted pharmacokinetics studies in STZ-induced diabetic mice. Finally, to evaluate the contribution rate of DPP-4 in liver, kidney, small intestine, and blood to cyano group hydrolysis of vildagliptin, we performed DPP-4 activity assays using a synthetic substrate H-glycyl-prolyl-7-amino-4-methylcoumarin (Gly-Pro-AMC) and hydrolysis assays of the cyano group of vildagliptin using tissue samples and blood.
Materials and Methods
Chemicals and Reagents.
Vildagliptin was synthesized in our laboratory using the standard technique (Asakura et al., 2014). Vildagliptin carboxylic acid metabolite (M20.7) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Sitagliptin was obtained from Carbosynth (Berkshire, UK). STZ was purchased from Wako (Osaka, Japan). The 7-amino-4-methylcoumarin (AMC) was purchased from Setareh Biotech (Eugene, OR). Gly-Pro-AMC was obtained from Bachem (Bubendorf, Switzerland). Human liver samples from 23 donors were obtained from the Human and Animal Bridging Research Organization (Chiba, Japan) (Supplemental Table 1). Primers were commercially synthesized at Life Technologies (Carlsbad, CA). All other chemicals were of the highest grade available.
Animals.
Male C57BL/6NCrSlc mice aged 8 weeks were purchased from Japan SLC (Shizuoka, Japan). All animals received food and water ad libitum, and mouse handling and experimental procedures were conducted in accordance with our animal care protocol approved by Kitasato University.
Pharmacokinetics Study of Vildagliptin In Vivo.
Vildagliptin (10 mg/kg, per os) was administered to each mouse. Plasma was collected at 0, 0.5, 1, 2, 4, and 8 hours after the administration. Plasma was mixed with twofold volume of acetonitrile and centrifuged at 15,000g for 5 minutes to remove protein. The plasma concentrations of vildagliptin and its main metabolite M20.7 were analyzed by liquid chromatography/tandem mass spectrometry (LC-MS/MS), as described below. The area under the plasma concentration-time curve from 0 hours to 8 hours (AUC0–8 hours) was calculated using the trapezoidal integration without extrapolation to infinity.
Establishment of STZ-Induced Diabetic Mice.
Mice were injected i.p. once with vehicle (saline) or STZ (250 mg/kg body weight). Seven days after the STZ injection, blood was collected from the submandibular vein into heparinized tubes and was immediately centrifuged at 3000g for 10 minutes at 4°C to obtain plasma. Plasma glucose concentrations were determined using the glucose CII-test Wako kit (Wako). The mice with plasma glucose levels exceeding 400 mg/dL were selected as STZ-induced diabetic mice.
Preparation of Tissue Samples.
Tissues of control mice and STZ-induced diabetic mice were collected 48 hours after administration of vildagliptin in the pharmacokinetics study, and then S9 fractions were prepared as pooled samples of each tissue in control mice (n = 4) and STZ-induced diabetic mice (n = 3). Mice were anesthetized by diethyl ether inhalation, and the livers were perfused with ice-cold 1.15% KCl. The liver, kidney, and small intestine were rinsed in cold 1.15% KCl and stored at −80°C. Tissue S9 fractions were prepared using the following procedure. Each tissue was homogenized in 3 volumes of a phosphate buffer (1.15% KCl, 0.1 M KH2PO4-K2HPO4 buffer, pH 7.4). The total cell homogenate was centrifuged at 600g for 10 minutes at 4°C. The pellet, which contained nuclear fraction, was discarded. The supernatant, which contained mostly cell protein, was further centrifuged at 9000g for 20 minutes at 4°C, and the supernatant was used as the S9 fraction. Protein concentrations were measured by the Bradford (1976) method using bovine serum albumin as a standard.
DPP-4 Activity Assays.
Several studies demonstrated that the DPP-4 activity measured using Gly-Pro-AMC as a substrate strongly correlates with the protein level of DPP-4 (Busso et al., 2005; Wagner et al., 2016). Therefore, we performed DPP-4 activity assays using the synthetic substrate Gly-Pro-AMC. DPP-4 activity was determined by cleavage rate of AMC from Gly-Pro-AMC, as described previously (Asakura et al., 2015), with slight modifications. Briefly, these assays were performed in 96-well flat-bottom plates in a total assay volume of 100 µL. The 2-µL plasma or diluted tissue samples (0.1 mg/mL) were incubated for 15 minutes at room temperature in assay buffer (50 mM glycine, pH 8.7, 1 mM EDTA). Fifteen minutes after the incubation, Gly-Pro-AMC was added to yield a final concentration of Gly-Pro-AMC of 50 µM, and the plates were incubated for 10 minutes at 25°C. Fluorescence was measured using a SpectraMax M5 96-well plate spectrophotometer (excitation, 360 nm; emission, 460 nm; Molecular Devices, Sunnyvale, CA). The DPP-4 activities in plasma and tissue samples were expressed as the amount of cleaved AMC per minute per mL (nmol/min/mL) and mg protein (nmol/min/mg protein), respectively.
Western Blot Analysis.
Liver, kidney, small intestine S9 fractions, and plasma (100 µg protein) of control mice and STZ-induced diabetic mice were subjected to NuPAGE 4–12% Bis-Tris gel (Life Technologies, Carlsbad, CA) and transferred to a polyvinylidene difluoride membrane Immobilon-P (Millipore, Bedford, MA) following the manufacurer’s protocol. The membrane was blocked for 1 hour with 50 mg/mL skimmed milk in phosphate-buffered saline (PBS) and then incubated with anti-human DPP-4 antibody (AF1180) diluted with PBS (1:1000) as a primary antibody overnight. The membrane was washed with PBS three times and incubated with horseradish peroxidase–labeled secondary antibody diluted with PBS (1:10,000) for 1 hour. The bands were detected using Chemi-Lumi One L Western-blotting detection reagents (Nacalai Tesque, Kyoto, Japan).
Hydrolysis Assays of the Cyano Group of Vildagliptin.
M20.7 formation was determined according to the method of Asakura et al. (2015) with slight modifications. Briefly, a typical incubation mixture (200 µL total volume) contained 50 mM Tris-HCl buffer (pH 7.4), 1 µM vildagliptin, and various enzyme sources. Because the therapeutic maximum plasma concentration of vildagliptin after the therapeutic dose of 100 mg/d is approximately 1 µM (He, 2012), 1 µM vildagliptin was used as a substrate. The final concentration of mouse liver, kidney, and small intestine S9 fraction was 2.0 mg/mL, whereas the final concentration of human liver microsomes (HLMs) was 0.3 mg/mL. The reaction was initiated by adding vildagliptin after a 5-minute preincubation at 37°C, and the reaction mixture was incubated for 2 hours at 37°C. The reaction was terminated by an addition of 200 µL cold acetonitrile. After removal of protein by centrifugation at 15,000g for 5 minutes, a 10-µL portion of the sample was subjected to LC-MS/MS assay to measure the concentration of M20.7. The nonreaction control samples were kept on ice for the full period before centrifugation. The concentration of M20.7 obtained with the nonreaction control was subtracted from that obtained with each enzyme source. The vildagliptin cyano group-hydrolyzing activity was expressed as an M20.7 formation rate (pmol/h/mg protein).
LC-MS/MS Conditions.
We used a Waters Xevo TQD mass spectrometer (Waters, Milford, MA) to measure the concentrations of vildagliptin and M20.7. A Waters ACQUITY UPLC system (Waters) was connected to the Waters Xevo TQD mass spectrometer operated in the positive electrospray ionization mode. Vildagliptin and M20.7 were separated on a Polaris 5 µm C18-A 50 × 2.0-mm column (25°C) (Agilent Technologies, Amstelveen, The Netherlands) with a MetaGuard 2.0 mm Polaris 5 µm C18-A guard column (Agilent Technologies). The mobile phase of A/B (1:3, v/v) was used, in which A was methanol/10 mM ammonium acetate, pH 8.0 (5:95, v/v), and B was acetonitrile/methanol (10:90, v/v). The flow rate was adjusted to 0.2 mL/min, and an eluent between 0 and 5 minutes was introduced into the mass spectrometer. Vildagliptin and M20.7 were analyzed during the multiple reaction monitoring mode of the mass spectrometer using m/z 304.1 > 154.2 and m/z 323.1 > 173.3, respectively, as the transition. The ionization conditions were as follows: capillary voltage, 3.4 kV; cone voltage, 42 V; collision energy, 18 V; source temperature, 150°C; desolvation temperature, 200°C; collision gas, argon. Data acquisition, instrument control, and data handling were performed with MassLynx Software (version 4.1; Waters).
Real-Time Reverse Transcription-Polymerase Chain Reaction.
Total RNA was extracted from the mouse liver, kidney, and small intestine using TRIzol reagent (Life Technologies). Complementary DNA was synthesized from total RNA using ReverTra Ace qPCR RT Master Mix (Toyobo, Tokyo, Japan), according to the manufacturer's protocol. Real-time reverse transcription-polymerase chain reaction (RT-PCR) was performed with THUNDERBIRD SYBR qPCR Mix (Toyobo), and the reactions were run in a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA). After an initial denaturation at 95°C for 30 seconds, amplification was performed by denaturation at 95°C for 5 seconds and annealing and extension at 60°C for 30 seconds for 45 cycles. The primer sequences are summarized in Supplemental Table 2. Expression was normalized with the expression of cyclophilin.
Statistical Analysis.
Data were presented as means ± S.D. and were assessed for statistical significance using the unpaired t test. The correlation analysis was performed using the Spearman rank method. A value of P < 0.05 was considered statistically significant.
Results
Pharmacokinetics Study of Vildagliptin In Vivo.
Our in vitro study previously demonstrated that DPP-4 greatly contributed to the cyano group hydrolysis of vildagliptin in mouse, rat, and human livers and that vildagliptin cyano group-hydrolyzing activity of DPP-4 in liver samples was strongly inhibited by sitagliptin, which is another selective DPP-4 inhibitor (Asakura et al., 2015). Therefore, to investigate whether DPP-4 is involved in the cyano group hydrolysis of vildagliptin in vivo, we conducted pharmacokinetics studies using sitagliptin as a competitive inhibitor of the DPP-4–mediated cyano group hydrolysis of vildagliptin. Although the AUC0–8 hours value of vildagliptin in mice coadministered with vildagliptin (10 mg/kg) and sitagliptin (1000 mg/kg) was comparable with that in mice administered vildagliptin alone (Fig. 2A), the AUC0–8 hours value of M20.7 in mice coadministered with vildagliptin and sitagliptin was significantly lower than that in mice administered vildagliptin alone (P < 0.01; Fig. 2B; Table 2). These results indicate that DPP-4 could greatly contribute to the cyano group hydrolysis of vildagliptin in vivo.

Pharmacokinetics study of vildagliptin and M20.7. Pharmacokinetics of vildagliptin (A) and its main metabolite M20.7 (B) in control mice administered with vildagliptin (10 mg/kg, per os) (n = 4), mice coadministered with vildagliptin (10 mg/kg, per os) and sitagliptin (1000 mg/kg) (n = 3), and STZ-induced diabetic mice administered with vildagliptin (10 mg/kg, per os) (n = 3) are shown. Results are shown as means ± S.D. of the plasma concentrations of vildagliptin and M20.7.
AUC0–8 hours in three mouse groups following oral administration of vildagliptin (10 mg/kg)
We next conducted pharmacokinetics studies using STZ-induced diabetic mice to investigate the effect of increasing DPP-4 activity level in blood on the blood concentrations of vildagliptin and M20.7. In this model, body weight was decreased and plasma glucose level was significantly increased 7 days after the STZ injection (P < 0.01; Supplemental Fig. 1, A and B). These data indicated that STZ-induced diabetic mice were established as reported (Das et al., 2014). Additionally, plasma DPP-4 level was significantly increased 7 days after the STZ injection (P < 0.01; Supplemental Fig. 1C), which is consistent with previous reports (Das et al., 2014). Because plasma DPP-4 expression level was increased 1.9-fold in STZ-induced diabetic mice (Supplemental Fig. 1C), the AUC0–8 hours value of M20.7 in those mice could be higher than that in control mice. However, the AUC0–8 hours value of M20.7 in STZ-induced diabetic mice was 2.6-fold lower than that in control mice (P < 0.01; Fig. 2B; Table 2). These findings suggest that DPP-4 expression in blood does not have an effect on the pharmacokinetics of vildagliptin in vivo.
DPP-4 Expression Levels and Vildagliptin Cyano Group-Hydrolyzing Activities in Liver, Kidney, Small Intestine Tissues, and Plasma of Control Mice and STZ-Induced Diabetic Mice.
Our in vivo pharmacokinetics study using STZ-induced diabetic mice indicated that the DPP-4 expression level in a tissue that is important in the vildagliptin cyano group-hydrolyzing reaction should have been decreased in STZ-induced diabetic mice. DPP-4 is highly expressed in liver, kidney, and small intestine in mice (Asakura et al., 2015). To evaluate the difference of DPP-4 expression levels between liver, kidney, small intestine, and plasma of control mice and those of STZ-induced diabetic mice, we performed DPP-4 activity assays using the synthetic DPP-4 substrate Gly-Pro-AMC. The DPP-4 activity in liver, kidney, small intestine, and plasma of control mice was 2.8 ± 0.1, 2.4 ± 0.1, 2.3 ± 0.1, and 0.074 ± 0.002 nmol/min/mg protein, respectively, and that of STZ-induced diabetic mice was 1.8 ± 0.1, 2.9 ± 0.1, 3.1 ± 0.1, and 0.14 ± 0.01 nmol/min/mg protein, respectively (Fig. 3A). Not only DPP-4 activities in plasma, but also DPP-4 activities in kidney and small intestine of STZ-induced diabetic mice were statistically higher than those of control mice (P < 0.01). In contrast, the DPP-4 activity in the liver of STZ-induced diabetic mice was significantly lower than that of control mice (P < 0.01). Additionally, the DPP-4 protein expression level in the liver of STZ-induced diabetic mice was also lower than that of control mice (Fig. 3B). The band reacting with the anti-human DPP-4 antibody was observed in the liver, kidney, and small intestine S9 fractions of both control mice and STZ-induced diabetic mice. However, the band in plasma was not observed, suggesting that DPP-4 expression level in blood is significantly lower compared with that in those tissues in line with the DPP-4 activity measured using Gly-Pro-AMC (Fig. 3, A and B). We further performed hydrolysis assays of the cyano group of vildagliptin using liver, kidney, small intestine S9 fractions, and plasma. M20.7 formation rate in liver, kidney, and small intestine of control mice was 0.31 ± 0.01, 0.23 ± 0.01, and 0.24 ± 0.01 pmol/h/mg S9 protein, respectively, and that of STZ-induced diabetic mice was 0.22 ± 0.02, 0.30 ± 0.01, and 0.40 ± 0.02 pmol/h/mg S9 protein, respectively (Fig. 3C). In line with the DPP-4 activities, M20.7 formation rates in kidney and small intestine of STZ-induced diabetic mice were statistically higher than those of control mice (P < 0.01). Meanwhile, M20.7 formation rate in the liver of STZ-induced diabetic mice was statistically lower than that of control mice (P < 0.01). M20.7 was not formed by incubating vildagliptin with mouse plasma. Our data indicate that DPP-4 in blood does not play an important role in vildagliptin metabolism.

DPP-4 activities and vildagliptin cyano group-hydrolyzing activities in liver, kidney, small intestine, and plasma in control mice and STZ-induced diabetic mice. Tissues were collected 48 hours after administration of vildagliptin in the pharmacokinetics study, and then pooled samples of each tissue in control mice (n = 4) and STZ-induced diabetic mice (n = 3) were prepared. (A) DPP-4 activities in S9 fractions and plasma were measured using Gly-Pro-AMC as a substrate. Data represent the means ± S.D. of three independent experiments. **P < 0.01. (B) The DPP-4 protein expression levels were determined by Western blot analysis. Liver, kidney, small intestine S9 fractions, and plasma (100 µg protein) of control mice and STZ-induced diabetic mice were subjected to NuPAGE 4–12% Bis-Tris gel and probed with the anti–DPP-4 antibody. (C) The vildagliptin cyano group-hydrolyzing activities in S9 fractions were measured. The substrate concentration was 1 µM. Data represent the means ± S.D. of three independent experiments. **P < 0.01. ND, not detectable. (D) The estimated DPP-4 activities (nmol/min/tissue or plasma) in whole liver, kidney, small intestine tissues, and plasma are shown. Data represent the means ± S.D. **P < 0.01. (E) The estimated M20.7 formation rates (pmol/h/tissue) in whole liver, kidney, and small intestine tissues are shown. Data represent the means ± S.D. **P < 0.01. ND, not detectable.
To estimate the DPP-4 activities and M20.7 formation rates in each tissue, we calculated the DPP-4 activity per tissue (nmol/min/tissue) and M20.7 formation rate per tissue (pmol/h/tissue) using organ weights and S9 protein yields. In our present study, the average organ weights of mouse liver, kidneys, and small intestine were 1.07, 0.315, and 0.869 g, respectively. S9 protein yields of mouse liver, kidneys, and small intestine were 121, 90.0, and 49.0 mg S9 protein/g tissue, respectively. These parameters were consistent with the values in previous reports (Davies and Morris, 1993; Abdullah et al., 2016). We also calculated the DPP-4 activity in whole plasma (nmol/min/plasma) using a reported plasma volume in mouse, which is 1.0 mL (Davies and Morris, 1993). The estimated DPP-4 activity in whole liver, kidney, small intestine tissues, and plasma of control mice was 367 ± 1.0, 67.4 ± 1.3, 97.5 ± 1.4, and 4.5 ± 0.15 nmol/min/tissue or plasma, respectively (Fig. 3D), indicating that the liver has the highest DPP-4 activity among these tissues and plasma. The estimated M20.7 formation rate in whole liver, kidney, and small intestine tissues was 40.4 ± 1.2, 6.5 ± 0.4, and 10.2 ± 0.1 pmol/h/tissue, respectively (Fig. 3E), indicating that the liver has the highest vildagliptin cyano group-hydrolyzing activity among these tissues. These findings indicate that the liver is the main organ responsible for vildagliptin cyano group hydrolysis in vivo.
Correlation Analyses between Hepatic DPP-4 Activities and Vildagliptin Cyano Group-Hydrolyzing Activities in Mice.
To further investigate whether the hepatic DPP-4 greatly contributes to the formation of M20.7 in vivo, we performed a correlation analysis between DPP-4 activities and M20.7 formation rates in the individual mouse liver S9 fractions. The DPP-4 activities measured using Gly-Pro-AMC positively correlated with M20.7 formation rates (Rs = 0.886, P < 0.05; Fig. 4A). We also performed a correlation analysis between hepatic DPP-4 activities and AUC0–8 hours values of M20.7 in the individual mice. The hepatic DPP-4 activities significantly positively correlated with the AUC0–8 hours values of M20.7 (Rs = 0.943, P < 0.05; Fig. 4B). These findings indicate that the decrease in AUC0–8 hours value of M20.7 in STZ-induced diabetic mice was attributed to the decreased hepatic DPP-4 activity (Fig. 2B; Table 2). These results also indicate that hepatic DPP-4 is primarily responsible for vildagliptin cyano group hydrolysis in the whole body.

Correlation between hepatic DPP-4 activities and vildagliptin cyano group-hydrolyzing activities in individual mice. The relationships between hepatic DPP-4 activities and hepatic M20.7 formation rates (A) and the AUC0–8 hours values of M20.7 (B) in individual mice are shown. The correlation analysis was performed using the Spearman rank method. Data represent the means of triplicate determinations.
mRNA Expression Levels of Dpp-4 and Other Genes in Liver, Kidney, and Small Intestine of Control and STZ-induced Diabetic Mice.
In the STZ-induced diabetic mice, the DPP-4 protein expression levels in the liver were lower than those in control mice, whereas the expression in kidney and small intestine was higher in the diabetic mice (Fig. 3, A and B). The real-time RT-PCR analysis showed that the Dpp-4 mRNA expression level in the liver of STZ-induced diabetic mice was statistically lower than that of control mice (P < 0.01; Fig. 5A). MicroRNA-29, adipocyte content rate in tissues, or hepatocyte nuclear factor (HNF)1α can be involved in the decreased DPP-4 expression in the liver of STZ-induced diabetic mice (Erickson et al., 1999; Kanasaki et al., 2014; Zilleßen et al., 2016). The forkhead box (FOX) transcription factor FOXA2 positively regulates the expression of microRNA-29 (Kurtz et al., 2014). Additionally, the gene expression of not only DPP-4, but also collagen, type I, α1 is negatively regulated by microRNA-29 (Roderburg et al., 2011). Whereas Foxa2 mRNA was not detectable in the tissues (Fig. 5B), the real-time RT-PCR analysis showed that the collagen, type I, α1 mRNA expression level in the liver of STZ-induced diabetic mice was not decreased as observed in the Dpp-4 mRNA expression level in the liver (Fig. 5C). Although fatty acid–binding protein (FABP) 4 and adiponectin have been reported as markers of adipose tissue (Hu et al., 1996; Westerbacka et al., 2007), the mRNA expression levels of Fabp4 and adiponectin did not correlate with the Dpp-4 mRNA expression level in the liver of control and diabetic mice (Fig. 5, D and E). These data suggested that microRNA-29 and adipocyte content rate in tissues were not associated with the DPP-4 expression levels in the livers. Interestingly, although the Hnf-1α mRNA expression levels were increased in the liver of STZ-induced diabetic mice (Fig. 5F), mRNA expression of Fabp1, which is a target of HNF-1α (Akiyama et al., 2000), in the liver of STZ-induced diabetic mice was lower more than 50-fold compared with that of control mice (Fig. 5G). Therefore, the lowered HNF-1α activity might have caused the decrease of Dpp-4 mRNA expression level in the liver of STZ-induced diabetic mice.

The mRNA expression levels of Dpp-4 (A), Foxa2 (B), collagen, type I, α1 (C), Fabp4 (D), adiponectin (E), Hnf-1α (F), and Fabp1 (G) in liver, kidney, and small intestine of control mice and STZ-induced diabetic mice. Expression was normalized with the expression of cyclophilin, and the expression level in each tissue of control mice was defined as 1. Data represent the means ± S.D. of three independent experiments. *P < 0.05; **P < 0.01. ND, not detectable.
Interindividual Variability of Hepatic DPP-4 Activities and Vildagliptin Cyano Group-Hydrolyzing Activities in Humans.
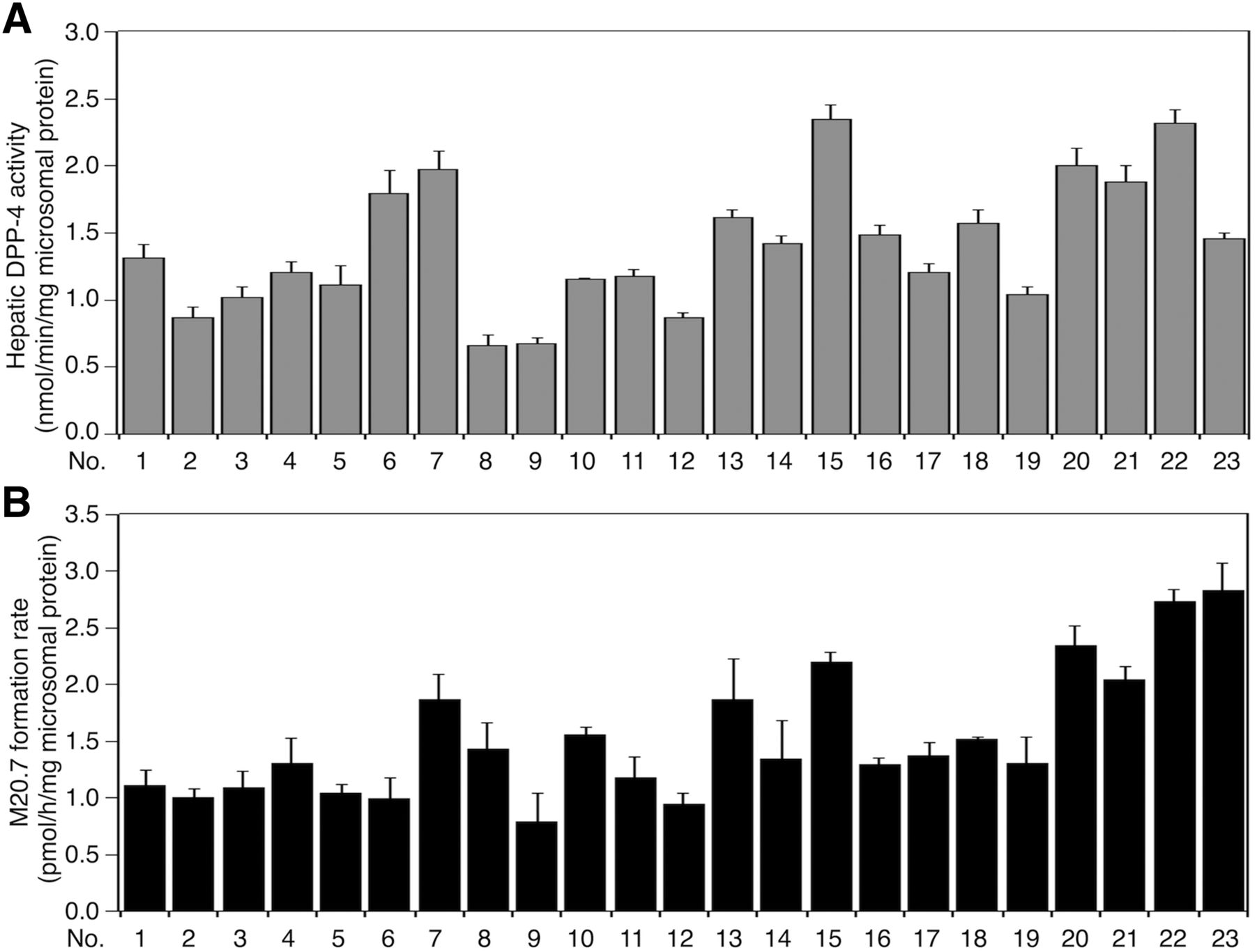
Our in vivo study indicated that hepatic DPP-4 could be primarily responsible for vildagliptin cyano group hydrolysis. To investigate the interindividual variability of DPP-4 activities and vildagliptin cyano group-hydrolyzing activities in humans, we measured DPP-4 activities using Gly-Pro-AMC as a substrate and M20.7 formation rates in 23 individual HLMs. The DPP-4 activities varied from 0.66 to 2.32 nmol/min/mg microsomal protein, showing a 3.5-fold variation (Fig. 6A). The M20.7 formation rates varied from 0.79 to 2.83 pmol/h/mg microsomal protein, showing a 3.6-fold variation (Fig. 6B). The hepatic DPP-4 activity positively correlated with M20.7 formation rate (Rs = 0.678, P < 0.01; Fig. 7A). These results indicate that there is a more than 3.5-fold interindividual variability of hepatic DPP-4 activities in humans.

Interindividual variability of hepatic DPP-4 activities and vildagliptin cyano group-hydrolyzing activities. (A) The hepatic DPP-4 activities in a panel of 23 individual HLMs were measured using a Gly-Pro-AMC as a substrate. Data represent the means ± S.D. of three independent experiments. (B) The vildagliptin cyano group-hydrolyzing activities in a panel of 23 individual HLMs were measured. The substrate concentration was 1 µM. Data represent the means ± S.D. of three independent experiments.

Correlation analyses between hepatic vildagliptin cyano group-hydrolyzing activities and other drug-metabolizing enzyme activities in individual HLMs and characteristics of 23 donors. The relationships between hepatic vildagliptin cyano group-hydrolyzing activities and DPP-4 (A), arylacetamide deacetylase (B), CES1 (C), and CES2 (D) activities of individual HLMs from 23 donors are shown. The relationships between hepatic vildagliptin cyano group-hydrolyzing activities and age (E) and BMI (F) of 23 donors are also shown. The correlation analysis was performed using the Spearman rank method. Data represent the means of triplicate determinations.
We also performed correlation analyses between vildagliptin cyano group-hydrolyzing activities and other drug-metabolizing enzyme activities in these HLM samples. Arylacetamide deacetylase, carboxylesterase (CES)1, and CES2 activities in these HLM samples were previously measured using their selective substrates, phenacetin, fenofibrate, and irinotecan (Shimizu et al., 2014). The hepatic vildagliptin cyano group-hydrolyzing activity did not correlate to activities of arylacetamide deacetylase (Rs = −0.0168, P = 0.939), CES1 (Rs = −0.226, P = 0.299), and CES2 (Rs = −0.0929, P = 0.673) (Fig. 7, B–D). We also performed correlation analyses between vildagliptin cyano group-hydrolyzing activities and protein expression levels of CYP3A4 in these HLM samples. The hepatic vildagliptin cyano group-hydrolyzing activity did not correlate to the previously reported protein expression level of CYP3A4 (Takagi et al., 2008) (Rs = 0.433, P = 0.184; Supplemental Fig. 2). These data indicate that the correlation between the DPP-4 activities measured using Gly-Pro-AMC as a substrate and vildagliptin cyano group-hydrolyzing activities in HLMs was not attributed to deactivation or degradation of the HLM samples.
We further performed correlation analyses between hepatic vildagliptin cyano group-hydrolyzing activities and characteristics of 23 donors. The hepatic vildagliptin cyano group-hydrolyzing activity did not correlate to age (Rs = 0.122, P = 0.609) or BMI (Rs = 0.394, P = 0.0631) (Fig. 7, E and F). These findings indicate that age and BMI might not be predictive factors of hepatic vildagliptin cyano group-hydrolyzing activities.
Discussion
In six healthy subjects following the administration of a single 100 mg oral dose of vildagliptin, there were 2.4-fold and 1.7-fold interindividual variabilities in Cmax (approximately 450–1100 ng/mL) and AUC (approximately 2000–3300 ng × h/mL) of vildagliptin, respectively (He et al., 2007). Because the renal function of these six healthy subjects was normal, the variability of vildagliptin cyano group-hydrolyzing activity might have caused the interindividual variability in Cmax and AUC of vildagliptin. There are nonresponders to vildagliptin, who do not show any significant decrease of hemoglobin A1c (Nakamura and Terauchi, 2013; Matsui et al., 2015). Additionally, vildagliptin can cause rare but serious adverse reactions, such as liver injury (Deacon, 2011; Kurita et al., 2014), bullous pemphigoid (Béné et al., 2015; Mendonça et al., 2016), and interstitial pneumonia (Kuse et al., 2016), in patients. Recently, we demonstrated that not only parental vildagliptin but also M20.7 might be involved in the onset of vildagliptin-associated liver injury (Asakura et al., 2016). More accurate prediction of the variability of vildagliptin and M20.7 exposure can improve efficacy and safety of vildagliptin. However, to date, there was no in vivo study to investigate the predictive factors of the variability of vildagliptin and M20.7 exposure.
Previously, we demonstrated that DPP-4 mainly contributes to the hepatic metabolism of vildagliptin in vitro (Asakura et al., 2015). In the present study, pharmacokinetics studies, DPP-4 activity assays, and vildagliptin cyano group-hydrolyzing assays indicate that hepatic DPP-4 greatly contributes to the formation of M20.7 even in vivo (Figs. 2–4). Although there is an interindividual variability of blood DPP-4 expression levels in humans (Lamers et al., 2011; Sell et al., 2013), our results suggested that DPP-4 in blood does not play an important role in the clearance of vildagliptin in vivo (Figs. 2 and 3). Additionally, in a panel of 23 individual HLMs, we found that there is an approximately 3.5-fold variation in vildagliptin cyano group-hydrolyzing activities (Fig. 6). Therefore, predetermination of the interindividual variability of hepatic vildagliptin cyano group-hydrolyzing activity might be useful for the prediction of blood vildagliptin concentrations in vivo.
Plasma protein binding can be an important factor to predict the in vivo clearance of drugs. Although the affinity of vildagliptin for DPP-4 is high, binding of vildagliptin to plasma proteins is very low (9.3%) (He et al., 2009a). Therefore, it has been suggested that protein binding of vildagliptin is clinically negligible (He et al., 2009a; Golightly et al., 2012). Importantly, vildagliptin and M20.7 are ultimately excreted by the kidney (He et al., 2009a; He, 2012). In our pharmacokinetics study, the AUC0–8 hours values of M20.7 were significantly (62%) decreased in STZ-induced diabetic mice (Fig. 2B; Table 2). The AUC0–8 hours value of vildagliptin in STZ-induced diabetic mice was also decreased (by 24%) compared with that in control mice (Table 1). These data suggest that the renal excretion of parental vildagliptin in STZ-induced diabetic mice might have been slightly increased compared with that in control mice. However, the percent decrease in the AUC values of M20.7 (62%) is significantly higher than that of parental vildagliptin (24%) in STZ-induced diabetic mice. These suggest that the decrease of the AUC value of M20.7 was mainly attributed to the decreased DPP-4 expression in the liver. To support this hypothesis, it was observed that the AUC0–8 hours value of parental vildagliptin in mice coadministered with sitagliptin was increased by 20% compared with that in mice administered vildagliptin alone (Table 1). In humans, the contribution of metabolism to the clearance of vildagliptin is much higher compared with mice (Table 1). Therefore, a decreased or lower expression of DPP-4 in the liver might result in an increase of blood vildagliptin concentrations in humans. Differences in hepatic DPP-4 clearance can be expected to translate to vildagliptin and M20.7 exposure, which can be further used as predeterminating factor of efficacy and safety of vildagliptin.
We failed to establish the link between hepatic DPP-4 and parental vildagliptin pharmacokinetics. We demonstrated that hepatic DPP-4 is the main enzyme for the formation of M20.7 (Figs. 3 and 4). However, the change of hepatic DPP-4 resulted in no significant change of the parent drug concentration in mice (Fig. 2A; Table 1). Because the contribution of the renal excretion of parental vildagliptin to the clearance of vildagliptin in mice is much higher compared with humans (Table 1), mice might not be the useful model animals for the in vivo pharmacokinetics studies of parental vildagliptin. Although rats and dogs can be better model animals (Table 1), it was demonstrated that the contribution rate of DPP-4 to vildagliptin cyano group hydrolysis in rats was too high (approximately 90%) (Asakura et al., 2015). These suggest that dogs might be better model animals for the in vivo pharmacokinetics studies of parental vildagliptin. However, in the case of understanding in vivo pharmacokinetics of M20.7, mice might still be useful animal models.
It was previously reported that BMI could be a predictive factor of the DPP-4 expression level in blood (Lamers et al., 2011). However, our data showed that blood DPP-4 would not play a key role in the vildagliptin metabolism in humans (Fig. 3). Furthermore, the hepatic vildagliptin cyano group-hydrolyzing activity did not correlate to BMI (Fig. 7F). Therefore, BMI of individuals would not be a useful predictive factor of blood vildagliptin concentrations. We previously demonstrated that the polymorphism in the DPP-4 gene significantly reduced the enzyme activity of DPP-4 (Asakura et al., 2015). In addition, it has been reported that hepatic DPP-4 expression level was higher in patients with hepatitis C virus infection and nonalcoholic fatty liver disease (Itou et al., 2008; Miyazaki et al., 2012). Therefore, such genetic and clinical condition, instead of BMI, of patients needs to be determined prior to administration of vildagliptin to improve their quality of life.
In conclusion, DPP-4 greatly contributes to the formation of M20.7 in vivo. Not plasma, but hepatic DPP-4 controls the pharmacokinetics of vildagliptin. There is an at least 3.5-fold interindividual variability in hepatic vildagliptin cyano group-hydrolyzing activity in humans. Therefore, predetermination of hepatic vildagliptin cyano group-hydrolyzing activities as well as genetic polymorphisms of DPP-4 is required to predict blood vildagliptin concentrations in humans.
Authorship Contributions
Participated in research design: Asakura, Fukami, Nakajima, Fujii, Atsuda, Itoh, Fujiwara.
Conducted experiments: Asakura, Fujiwara.
Performed data analysis: Asakura, Fujiwara.
Wrote or contributed to the writing of the manuscript: Asakura, Fukami, Nakajima, Fujii, Atsuda, Itoh, Fujiwara.
Footnotes
- Received October 10, 2016.
- Accepted November 23, 2016.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AMC
- 7-amino-4-methylcoumarin
- AUC
- area under the plasma concentration-time curve
- BMI
- body mass index
- CES
- carboxylesterase
- CYP
- cytochrome P450
- DPP-4
- dipeptidyl peptidase-4
- FABP
- fatty acid–binding protein
- Gly-Pro-AMC
- H-glycyl-prolyl-7-amino-4-methylcoumarin
- HLM
- human liver microsome
- HNF
- hepatocyte nuclear factor
- LC-MS/MS
- liquid chromatography/tandem mass spectrometry
- PBS
- phosphate-buffered saline
- RT-PCR
- reverse transcription-polymerase chain reaction
- STZ
- streptozotocin
- T2DM
- type 2 diabetes mellitus
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}