


Abstract
Inhibition of the bile salt export pump (BSEP) has been recognized as a key factor in the development of drug-induced cholestasis (DIC). The risk of DIC in humans has been previously assessed using in vitro BSEP inhibition data (IC50) and unbound systemic drug exposure under assumption of the “free drug hypothesis.” This concept, however, is unlikely valid, as unbound intrahepatic drug concentrations are affected by active transport and metabolism. To investigate this hypothesis, we experimentally determined the in vitro liver-to-blood partition coefficients (Kpuu) for 18 drug compounds using the hepatic extended clearance model (ECM). In vitro–in vivo translatability of Kpuu values was verified for a subset of compounds in rat. Consequently, unbound intrahepatic concentrations were calculated from clinical exposure (systemic and hepatic inlet) and measured Kpuu data. Using these values, corresponding safety margins against BSEP IC50 values were determined and compared with the clinical incidence of DIC. Depending on the ECM class of a drug, in vitro Kpuu values deviated up to 14-fold from unity, and unbound intrahepatic concentrations were affected accordingly. The use of in vitro Kpuu-based safety margins allowed separation of clinical cholestasis frequency into three classes (no cholestasis, cholestasis in ≤2%, and cholestasis in >2% of subjects) for 17 out of 18 compounds. This assessment was significantly superior compared with using unbound extracellular concentrations as a surrogate for intrahepatic concentrations. Furthermore, the assessment of Kpuu according to ECM provides useful guidance for the quantitative evaluation of genetic and physiologic risk factors for the development of cholestasis.
Introduction
The liver is the major organ involved in the elimination of potentially harmful endogenous and xenobiotic substances, including pharmaceutical drugs, and is itself predisposed to toxicity resulting from high exposure to drugs and their metabolites. Drug-induced liver injury (DILI) is a leading cause of acute liver failure, termination of compounds in drug development, and drug withdrawal from the market (Lee, 2003; Food and Drug Administration, 2009). The severity of DILI ranges from asymptomatic elevations of liver enzymes to acute liver failure and manifests with hepatocellular, cholestatic, or mixed (hepatocellular/cholestatic) patterns.
Although drug-induced cholestasis (DIC) usually represents a less severe form of DILI, it is nevertheless reported to account for up to 26% of all hepatic adverse reactions (Bjornsson and Olsson, 2005; Hussaini and Farrington, 2007). Cholestasis is characterized by reduced bile flow, potentially resulting in accumulation of cytotoxic bile salts within hepatocytes, leading to liver damage (Stieger, 2010). The bile salt export pump (BSEP), a member of the ATP-binding cassette superfamily encoded by the ABCB11 gene, is expressed at the canalicular membrane of hepatocytes and plays a fundamental role in bile homeostasis by secreting bile acids from the hepatocyte into bile ducts. Impairment of BSEP function due to inhibition by drugs or genetic defects was previously identified as a key factor in the development of DIC, hereditary cholestatic syndromes, or intrahepatic cholestasis of pregnancy (Stieger et al., 2000; Fattinger et al., 2001; Funk et al., 2001; Pauli-Magnus et al., 2010; Dietrich and Geier, 2014). Several attempts have been made to predict DIC or DILI in humans from in vitro BSEP inhibition data due to limited translatability of hepatic adverse events from preclinical models (Olson et al., 2000). Recently, Dawson et al. (2012) and Morgan et al. (2013) demonstrated that potent BSEP in vitro inhibition and high systemic drug exposure correlate with the occurrence of DIC. However, the assessments did not allow clear separation of cholestatic/mixed from non-cholestatic drugs, and thus reliable prediction of DIC remains challenging.
Clinical drug toxicity, drug-drug interactions (DDIs), and pharmacological drug-target interactions are commonly anticipated by relating the in vitro target potency (IC50, Ki, or EC50 value) to the unbound (i.e., free) systemic drug concentration due to limited availability of tissue concentration data in humans (Muller and Milton, 2012; Zamek-Gliszczynski et al., 2013). This assessment is based on the “free drug hypothesis,” which assumes complete distribution equilibrium of the unbound drug between blood and tissue at steady state. This assumption, however, is unlikely to apply to organs such as the liver, where the distribution equilibrium is affected by active cellular transport and metabolic processes (Chu et al., 2013). Therefore, recent publications proposed estimating the unbound intracellular liver concentration using the liver-to-blood partition coefficient for unbound drug at steady state (Kpuu) in vitro (Yabe et al., 2011; Mateus et al., 2013; Pfeifer et al., 2013; Shitara et al., 2013; Nicolai et al., 2015). Following the concept of the hepatic extended clearance model (ECM), the in vitro (hepatocyte-to-medium) Kpuu can be derived from in vitro measurements of individual hepatic elimination process clearances (sinusoidal influx and efflux, metabolism, and biliary secretion), which govern hepatic elimination (Fig. 1) (Shitara et al., 2013; Camenisch et al., 2015; Camenisch, 2016). Measurements of individual hepatic process clearances additionally allow assignment of compounds into four distinct ECM categories to anticipate class-dependent effects on Kpuu and, as a consequence, on the unbound intrahepatic concentration (Fig. 2) (Camenisch et al., 2015; Camenisch, 2016).
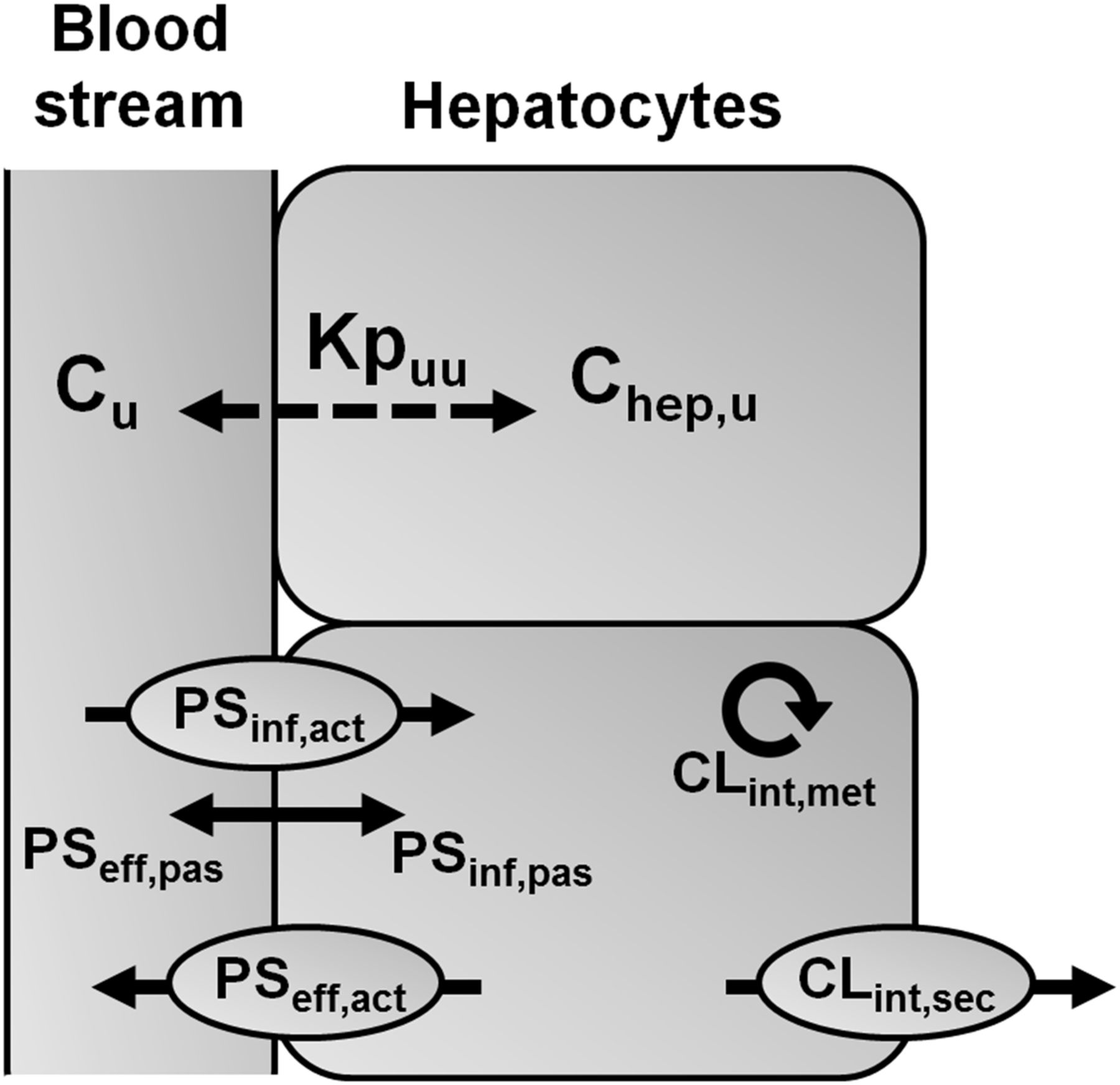
ECM and Kpuu. Schematic diagram of hepatic clearance processes determining the liver-to-blood partition coefficient for unbound drug at steady state (Kpuu). The unbound intrahepatic concentration (Chep,u) is determined by the unbound extracellular concentration (Cu) and the interplay between all hepatic process clearances, where Chep,u = Cu × Kpuu. Unbound drug in the blood stream is taken up into hepatocytes (PSinf) by transporters (PSinf,act) and/or by passive diffusion (PSinf,pas). Elimination of drug from the hepatocyte (CLint) occurs via hepatic metabolism (CLint,met), by active secretion into bile (CLint,sec), and by sinusoidal efflux (PSeff) via active transport (PSeff,act) and/or passive diffusion (PSeff,pas).

Drug classification according to ECM and expected impact on Kpuu and Chep,u. ECM class 1/2 was assigned if PSeff < CLint; otherwise, class 3/4 was assigned. Class 1/3 was assigned if PSeff = PSinf; otherwise, class 2/4 was assigned. In the present study, PSeff was assumed to occur only via passive diffusion and to be equal to PSinf,pas (PSeff = PSinf,pas). Adapted from Camenisch et al. (2015).
The aim of the present study was to identify the reference drug concentration (unbound systemic, unbound hepatic inlet, or unbound intrahepatic concentration) that provides the best anticipation of DIC risk due to BSEP inhibition. Upon assessing in vitro–in vivo correlation (IVIVC) for Kpuu in rat using literature data, we experimentally determined human hepatic in vitro Kpuu values using the ECM concept for 18 drug compounds with diverse physicochemical and pharmacokinetic properties. Unbound intrahepatic concentrations in humans were estimated by applying ECM-based Kpuu to either unbound systemic or hepatic inlet concentrations. The resulting safety margins between in vitro BSEP IC50 and the various reference concentrations were compared with the clinical incidence of DIC. In addition, the potential impact of genetic and physiologic risk factors on the induction of cholestasis is discussed using bosentan as tool compound.
Materials and Methods
Materials.
Radiolabeled test compounds ([3H] or [14C]) were obtained from PerkinElmer (Boston, MA), American Radiolabeled Chemicals (St. Louis, MO), and Moravek Biochemicals (Brea, CA). Radiochemical purity of all compounds was ≥95% as determined in-house by high-performance liquid chromatography analysis. All other chemicals were purchased from commercial sources and were of analytical grade.
Determination of Hepatic In Vitro Kpuu.
Previous work performed by our group has shown that in vivo hepatic organ clearances (CLh) were correctly predicted by feeding up-scaled in vitro hepatic process clearances into the ECM (eq. 1) and by applying the “well stirred” liver model (eq. 2) (Camenisch and Umehara, 2012; Umehara and Camenisch, 2012; Kunze et al., 2015):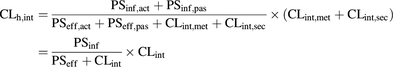 (1)
(1) (2)where CLh,int is the intrinsic hepatic clearance, PSinf is the sum of active (PSinf,act) and passive uptake membrane permeability (PSinf,pas), PSeff is the sum of active (PSeff,act) and passive sinusoidal efflux membrane permeability (PSeff,pas), CLint is the sum of intrinsic metabolic (CLint,met) and biliary clearances (CLint,sec), Qh is the hepatic blood flow, and fub is the unbound fraction in blood.
(2)where CLh,int is the intrinsic hepatic clearance, PSinf is the sum of active (PSinf,act) and passive uptake membrane permeability (PSinf,pas), PSeff is the sum of active (PSeff,act) and passive sinusoidal efflux membrane permeability (PSeff,pas), CLint is the sum of intrinsic metabolic (CLint,met) and biliary clearances (CLint,sec), Qh is the hepatic blood flow, and fub is the unbound fraction in blood.
This in vitro–in vivo extrapolation approach for human and rat hepatic clearance provided a good prediction accuracy for a diverse data set of 13 compounds with ∼80% within 2-fold error (Camenisch and Umehara, 2012; Umehara and Camenisch, 2012). According to the concept of the ECM, the intrinsic clearance is driven by the intracellular concentration (Shitara et al., 2013), and eq. 1 can be rearranged as follows:
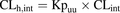 (3)
(3)Hepatic Process Clearances.
Hepatic process clearances for 18 test compounds were experimentally determined as previously described in full detail elsewhere (Camenisch and Umehara, 2012; Umehara and Camenisch, 2012; Kunze et al., 2014, 2015). All presented in vitro data for atorvastatin, cerivastatin, cyclosporine A, fluvastatin, ketoconazole, lovastatin acid, pitavastatin, pravastatin, rosuvastatin, simvastatin acid, and verapamil were taken from Camenisch and Umehara (2012) and Kunze et al. (2015).
In brief, PSinf,act and PSinf,pas were determined in pooled suspended human hepatocytes using the oil-spin method. PSinf,act and PSinf,pas represent single time-point measurements within the linear time and concentration range, and PSinf,pas was determined in the presence of uptake transporter inhibitors or at high substrate concentrations where active transport processes are known to be saturated. Measured (apparent) uptake clearances were corrected for nonspecific binding to the assay device using radioactivity recoveries and for saturable binding to cell surfaces using data from control incubations at 4°C (Kunze et al., 2014). For the highly lipophilic compounds ketoconazole and atazanvir (logD7.4 > 4), PSinf was determined from the slope of initial uptake velocity (3 time points between 0.5 and 3 minutes), taking into account initial cellular binding and nonspecific binding to the assay device.
PSeff was assumed to occur only via passive diffusion and to be equal to PSinf,pas (PSeff,act = 0, PSeff,pas = PSinf,pas).
Apparent metabolic clearance (CLint,met,app) was determined using human liver microsomes. Incubations with all test compounds were performed in the presence of nicotinamide adenine dinucleotide phosphate (NADPH). The known uridine diphosphate (UDP) glucuronosyltransferase (substrates cerivastatin, fluvastatin, ibuprofen, pitavastatin, and simvastatin acid were additionally incubated in the presence of UDP, and CLint,met represents the sum of NADPH and UDP incubations. CLint,met,app values were corrected for the unbound fraction in liver microsomes (fumic) as follows: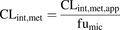 (4)Values for fumic and corresponding literature references are provided in Supplemental Table 1.
(4)Values for fumic and corresponding literature references are provided in Supplemental Table 1.
Apparent biliary clearance (CLint,sec,app) was determined in sandwich-cultured human hepatocytes (B-CLEAR method; Qualyst, Inc., Durham, NC) and was corrected for the unbound fraction in hepatocytes (fuhep) as given in eq. 5: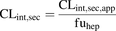 (5)Values of fuhep were derived from logD7.4 as follows (Yabe et al., 2011):
(5)Values of fuhep were derived from logD7.4 as follows (Yabe et al., 2011): (6)Values for logD7.4 and corresponding literature references are provided in Supplemental Table 1.
(6)Values for logD7.4 and corresponding literature references are provided in Supplemental Table 1.
The in vitro clearances were up-scaled to human organ level (ml/min/kg) using the following scaling factors: 99 (106 cells/g liver) for suspended hepatocytes, 53 (mg protein/g liver) for human liver microsomes, 116 (mg protein/g liver) for sandwich-cultured hepatocytes, and 25.7 (g liver/kg body weight) for liver weight.
Calculation of Unbound Drug Concentrations in the Systemic Circulation, at the Hepatic Inlet, and in the Hepatocyte.
Unbound drug concentrations in blood and plasma are equal (Kwon, 2001) and herein are referred to as unbound systemic drug concentrations (Csys,u). Csys,u was calculated from the total maximum available drug plasma concentration (Cmax) at steady state upon oral administration of the maximum recommended dose in healthy human subjects and the fraction unbound in plasma (fup):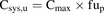 (7)According to eq. 8, unbound drug concentrations at the hepatic inlet (Cinlet,u) were calculated as the sum of drug in the systemic circulation reaching the liver via the hepatic artery (i.e., Csys,u) and drug that is delivered by the portal vein upon intestinal absorption (Giacomini et al., 2010):
(7)According to eq. 8, unbound drug concentrations at the hepatic inlet (Cinlet,u) were calculated as the sum of drug in the systemic circulation reaching the liver via the hepatic artery (i.e., Csys,u) and drug that is delivered by the portal vein upon intestinal absorption (Giacomini et al., 2010):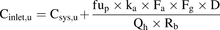 (8)where ka is the absorption rate constant, Fa is the fraction absorbed, Fg is the fraction escaping gut metabolism, D is the maximum recommended single oral dose, Qh is the hepatic blood flow (1.45 l/min), and Rb is the blood-to-plasma partition coefficient.
(8)where ka is the absorption rate constant, Fa is the fraction absorbed, Fg is the fraction escaping gut metabolism, D is the maximum recommended single oral dose, Qh is the hepatic blood flow (1.45 l/min), and Rb is the blood-to-plasma partition coefficient.
Additionally, maximum unbound hepatic inlet concentrations (Cinlet,max,u), representing a “worst-case,” were calculated according to eq. 8 assuming complete and fast drug absorption (i.e., Fa × Fg = 1 and ka = 0.1 min−1) (Ito et al., 1998).
Unbound intracellular drug concentrations in the hepatocyte [hereafter referred to as unbound intrahepatic concentration (Chep,u)] were calculated on the basis of Csys,u (eq. 9), Cinlet,u (eq. 10), or Cinlet,max,u (eq. 11):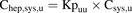 (9)
(9)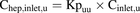 (10)
(10)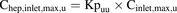 (11)All clinical exposure data, pharmacokinetic parameters, and calculations thereof are provided in the Supplemental Material together with the corresponding literature references (Supplemental Tables 1–3).
(11)All clinical exposure data, pharmacokinetic parameters, and calculations thereof are provided in the Supplemental Material together with the corresponding literature references (Supplemental Tables 1–3).
Human BSEP IC50 Values.
In vitro IC50 data for BSEP inhibition were collected from the literature. In the case of multiple reported values, the lowest one was used for risk assessments (Table 1), whereas Supplemental Table 4 shows the range of reported data. All data were determined under comparable conditions using membrane vesicles expressing human BSEP with [3H]taurocholate as probe substrate.
In vitro process clearances, in vitro Kpuu, and human drug exposure
ECM classes were derived as described in Fig. 2. Unbound extracellular (systemic and hepatic inlet) and intrahepatic concentrations were calculated as described in Materials and Methods, and all required pharmacokinetic parameters and corresponding literature references are summarized in Supplemental Tables 1–3. Literature references for BSEP IC50 values are provided in Supplemental Table 4; presented data represent the lowest available IC50 value.
Cholestasis Classification.
Cholestasis annotation was carried out exclusively on the basis of clinical studies, drug labels, and a comprehensive literature search where cholestasis or mixed liver injury was observed under controlled conditions (i.e., declaration of comedication, underlying disease state, known dosing regimen). Drugs were categorized as “cholestatic” based on reports of one of the following adverse events: cholestasis, cholestatic liver injury, cholestatic jaundice, cholestatic hepatitis, mixed liver injury, or biochemical evidence of cholestasis or mixed liver injury in the form of elevated serum alkaline phosphatase (AP) [AP ≥ 2× upper limit of normal (ULN) and ratio between alanine aminotransferase ULN and AP ULN <5] (Council for International Organizations of Medical Sciences, 1999). Based on the reported cholestasis incidence, drugs were assigned to the subclasses “common” (>2% of subjects) or “rare” (≤2% of subjects). In the absence of the previously defined cholestasis events, drugs were categorized as “no cholestasis.” The threshold of 2% was selected based on literature information from clinical reports or from the drug label, where rare adverse events were commonly defined as <2%. Detailed cholestasis annotations and literature references are summarized in Supplemental Table 4.
Data Analysis.
The DIC risk was assessed based on safety margins, calculated as the ratio between BSEP IC50 and the unbound intrahepatic drug concentration. Following the static R-value approach for reversible enzyme or transporter inhibition and using Cmax,u as the basis for unbound intrahepatic concentrations, the assessment represents a “worst-case” scenario for DIC (Rowland and Matin, 1973). Receiver operating characteristic (ROC) curve analysis (OriginPro 2016; OriginLab Corporation, Northampton, MA) was used to determine the optimal cut-off values (safety margin thresholds) between the three different cholestasis classes and to evaluate the accuracy with which cholestasis classes can be separated. ROC analysis calculates the sensitivity (fraction of true positive (TP) classifications) and specificity (fraction of true negative (TN) classifications) for any possible cut-off value and plots sensitivity against 1 − specificity, representing the ROC curve. The optimal cut-off values correspond to the maximum rate of correct positive and negative classifications and were determined by minimizing the distance (d) between point [0,1] in the ROC space (where sensitivity and specificity are maximum) and any point on the ROC curve (Kumar and Indrayan, 2011): (12)The separation of cholestasis classes by the estimated safety margin thresholds was compared using the area under the ROC curves (ROC AUCs), where 1.0 represents a perfect discrimination between two classes, and 0.5 represents the poorest discrimination.
(12)The separation of cholestasis classes by the estimated safety margin thresholds was compared using the area under the ROC curves (ROC AUCs), where 1.0 represents a perfect discrimination between two classes, and 0.5 represents the poorest discrimination.
Quantification of Physiologic and Genetic Factors.
A theoretical assessment for the impact of disease state and polymorphic pathways on DIC was conducted using bosentan as an example drug compound. The impact of disease state on Csys,u concentrations was estimated based on literature observations that pulmonary arterial hypertension (PAH) patients show 2-fold increased systemic bosentan concentrations compared with healthy volunteers (Dingemanse and van Giersbergen, 2004). The resulting hepatic inlet and intrahepatic concentrations were calculated according to eqs. 8 and 10 using a Csys,u value of 0.1327 µM.
The effect of nonsynonymous polymorphisms of hepatic metabolic enzymes and transporters on the in vitro Kpuu of bosentan was calculated according to eq. 13 using measured in vitro process clearances (Table 1), their fractional in vivo contribution (fn), and reported fold changes in activity compared with the reference genotype (α): 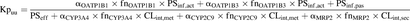 (13) The α values used were determined from the following data. Increased activity was reported for the organic anion transporting polypeptide (OATP) variants OATP1B1*1b and OATP1B3*2 (αOATP1B1 = αOATP1B3 = 2) (Rowland and Matin, 1973; Letschert et al., 2004). The cytochrome P450 variants CYP3A4*20 and CYP2C9*3 are associated with loss of function (αCYP3A4 = αCYP2C9 = 0) (Lee et al., 2002; Werk and Cascorbi, 2014), similar to the multidrug resistance protein (MRP) 2 variant MRP2*16 (c.2302C>T) (αMRP2 = 0) (Hulot et al., 2005; Pratt et al., 2015). According to the literature, OATP1B1 and OATP1B3 equally contribute to the total active hepatic uptake of bosentan (fnOATP1B1 = fnOATP1B3 = 0.5) (Treiber et al., 2007). Hepatic metabolism is mediated by CYP3A4 (fnCYP3A4 = 0.6) and CYP2C9 (fnCYP2C9 = 0.4) (Dingemanse and van Giersbergen, 2004), and biliary secretion is mediated by MRP2 (fnMRP2 = 1.0) (Fahrmayr et al., 2013).
(13) The α values used were determined from the following data. Increased activity was reported for the organic anion transporting polypeptide (OATP) variants OATP1B1*1b and OATP1B3*2 (αOATP1B1 = αOATP1B3 = 2) (Rowland and Matin, 1973; Letschert et al., 2004). The cytochrome P450 variants CYP3A4*20 and CYP2C9*3 are associated with loss of function (αCYP3A4 = αCYP2C9 = 0) (Lee et al., 2002; Werk and Cascorbi, 2014), similar to the multidrug resistance protein (MRP) 2 variant MRP2*16 (c.2302C>T) (αMRP2 = 0) (Hulot et al., 2005; Pratt et al., 2015). According to the literature, OATP1B1 and OATP1B3 equally contribute to the total active hepatic uptake of bosentan (fnOATP1B1 = fnOATP1B3 = 0.5) (Treiber et al., 2007). Hepatic metabolism is mediated by CYP3A4 (fnCYP3A4 = 0.6) and CYP2C9 (fnCYP2C9 = 0.4) (Dingemanse and van Giersbergen, 2004), and biliary secretion is mediated by MRP2 (fnMRP2 = 1.0) (Fahrmayr et al., 2013).
To estimate the potential impact of BSEP polymorphisms on the DIC safety margin, reduced BSEP activity was assumed to mimic an increased inhibition potential, where 100% activity corresponds to the BSEP IC50 value of bosentan (22 µM). According to the literature, BSEP G855R (c.2563G>A) has <20% transport activity compared with nonpolymorphic BSEP (Lang et al., 2007), which was simulated using a 5-fold reduced BSEP IC50 value (4.4 µM).
Results
Kpuu and ECM Class Assignment.
In vitro measured hepatic process clearances for the 18 investigated drug compounds are summarized in Table 1 together with the resulting in vitro Kpuu values, which range from 0.07 to 2.41. Figure 3 displays the corresponding fold change in intrahepatic unbound drug concentrations (Chep,u) calculated from extracellular concentrations and in vitro Kpuu, where atazanavir and pravastatin represent the extremes with ∼14-fold decreased and ∼2.4-fold increased Chep,u, respectively.
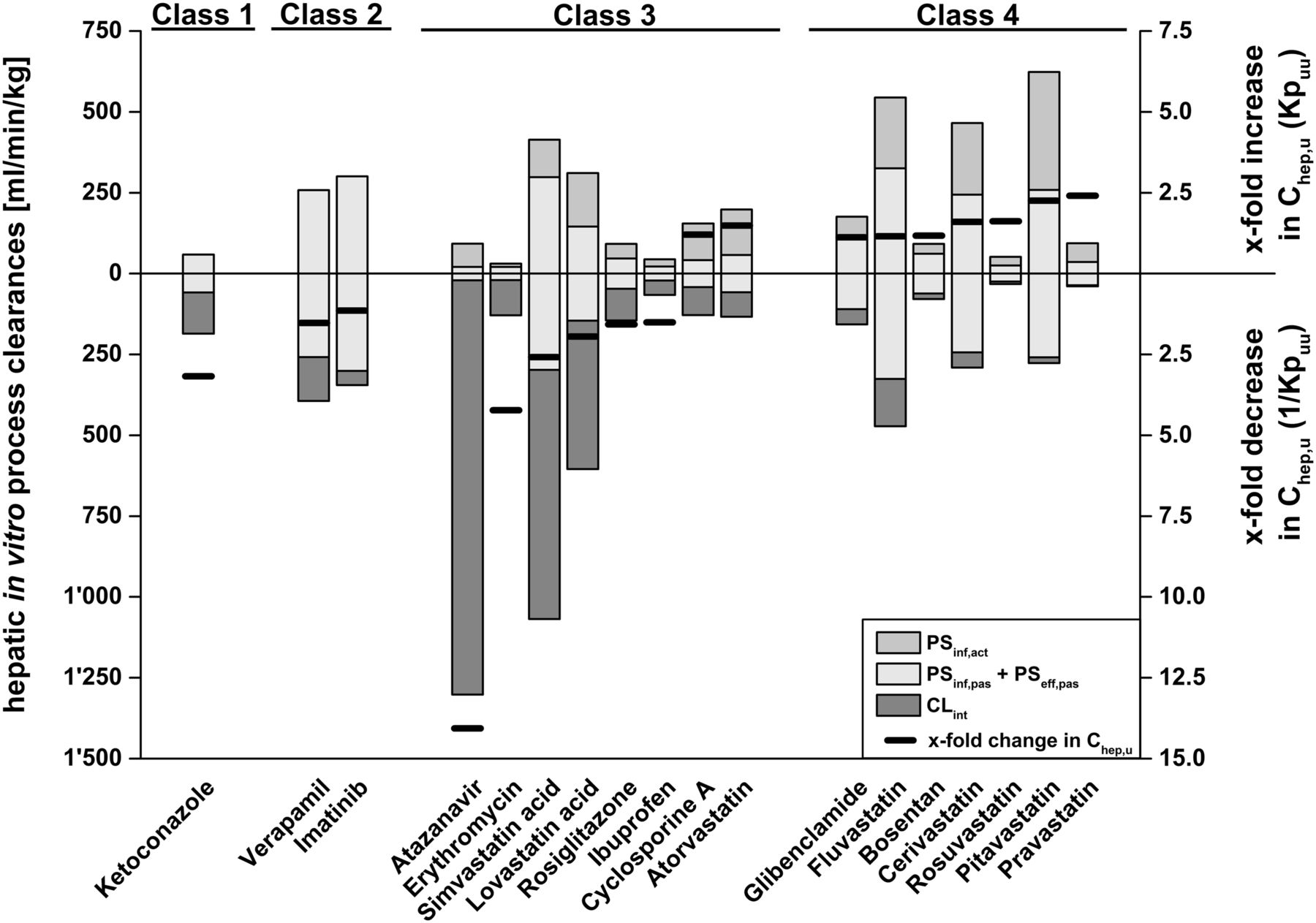
ECM class-dependent impact of experimentally determined hepatic process clearances on Kpuu and Chep,u. The black lines represent the x-fold change of intrahepatic concentrations (Chep,u) depending on Kpuu. x-fold increase and decrease in Chep,u correspond to Kpuu and 1/Kpuu, respectively. Gray, white, and dark-gray bars represent the underlying process clearances (active uptake, sum of passive uptake and efflux, and intrinsic clearance, respectively), which affect Kpuu, as described in Fig. 2. Hepatic process clearances refer to the left y-axis, whereas changes in Chep,u refer to the right y-axis.
Compound classification according to ECM is based on the extent of individual hepatic in vitro clearance processes (Fig. 2). As hepatic clearance processes significantly impact intrahepatic concentrations, as illustrated in Figs. 2 and 3, ECM class-dependent effects on Kpuu values can be expected. For the ECM class 1 compound ketoconazole, a Kpuu value <1 was obtained as a result of a predominant intrinsic clearance contribution (i.e., PSeff + CLint > PSinf). On the other hand, for ECM class 2 compounds imatinib and verapamil, Kpuu was mainly determined by passive uptake and efflux processes and, consequently, approached values of 1 (i.e., PSeff + CLint ≈ PSinf). For the ECM class 3 compounds with predominant intrinsic clearance (atazanavir, erythromycin, ibuprofen, lovastatin acid, rosiglitazone, and simvastatin acid), Kpuu values <1 were obtained (i.e., PSeff + CLint > PSinf). The ECM class 3 compounds with substantial active hepatic uptake — namely, atorvastatin and cyclosporine A — exhibit Kpuu values >1 (i.e., PSeff + CLint < PSinf). Similarly, the ECM class 4 compounds bosentan, cerivastatin, fluvastatin, glibenclamide, pitavastatin, pravastatin, and rosuvastatin reveal Kpuu values >1 due to predominant hepatic uptake (i.e., PSeff + CLint < PSinf).
In Vitro–In Vivo Correlation of Kpuu in Rat.
Estimation of Chep,u using ECM-based in vitro Kpuu data implies that the in vitro Kpuu directly translates to in vivo Kpuu. We made this assumption based on previous successful applications of the ECM approach for hepatic clearance and DDI predictions (Camenisch and Umehara, 2012; Umehara and Camenisch, 2012; Kunze et al., 2015). To further validate the ECM concept, we performed an IVIVC for Kpuu in rat using published in-house data (Umehara and Camenisch, 2012) (Fig. 4). In addition, IVIVC of reported in vitro Kpuu values from initial rate hepatic uptake clearances in suspended hepatocytes (Yabe et al., 2011; Shitara et al., 2013) and from sandwich-cultured hepatocytes (Pfeifer et al., 2013) is shown in Fig. 4. Observed (in vivo) and predicted (in vitro) Kpuu data following the ECM concept were in good agreement, with all five compounds deviating by less than 2.5-fold. Also, Kpuu from rat sandwich-cultured hepatocytes demonstrated close IVIVC for three compounds. In contrast, in vitro Kpuu from suspended hepatocytes generally provided overestimations of in vivo Kpuu, most likely due to the absence of intrinsic clearance processes (metabolism and biliary secretion).

In vitro–in vivo correlation of hepatic Kpuu in rat. Rat in vitro Kpuu values were calculated according to ECM (eqs. 1 and 3) using published in-house in vitro hepatic process clearance data or were taken from the literature. Rat in vivo Kpuu values were derived from reported liver partition and drug-binding data. Detailed calculations of Kpuu and literature references are summarized in Supplemental Table 5. Black diamonds refer to ECM-based Kpuu (in-house data); white squares, triangles, and circles represent in vitro Kpuu obtained by Pfeifer et al. (2013), Yabe et al. (2011), and Shitara et al. (2013), respectively. The solid line is the line of unity and dotted lines are 3-fold deviations. Numbers represent the investigated drugs: 1, atorvastatin; 2, cyclosporine A; 3, furamidine; 4, ketoconazole; 5, pravastatin; 6, ritonavir; 7, rosuvastatin; 8, verapamil.
For atorvastatin, cyclosporine A, and verapamil, different ECM classes were assigned for rat and human due to different contributions of the individual clearance processes. The impact of in vitro and in vivo Kpuu values in rats, however, was in line with the ECM theory described earlier (ECM class 1: Kpuu < 1; ECM class 4: Kpuu > 1).
Correlation between BSEP Inhibition, Drug Concentrations, and Drug-Induced Cholestasis.
The assignments of the 18 test drug compounds into the three cholestasis frequency classes (“no,” “rare,” “common”) are shown in Table 2. Risk assessments with regard to DIC were conducted using safety margins calculated as the ratio of BSEP IC50 and either unbound extracellular drug concentrations (Csys,u or Cinlet,u) or unbound intrahepatic concentrations upon application of Kpuu (Chep,sys,u or Chep,inlet,u) (Fig. 5). The risk assessment with unbound systemic concentrations showed a separation between drugs in cholestasis classes “common” and “no cholestasis,” whereas drugs in the class “rare” markedly overlapped with drugs in the other classes (Fig. 5A). The separation of drugs in cholestasis class “rare” from “common” or “no cholestasis” was not improved by using the substantially higher unbound drug concentrations at the hepatic inlet (Fig. 5B). This incomplete separation of the cholestasis classes based on extracellular concentrations is reflected by ROC AUC values of 0.83–0.94. In contrast, the use of unbound intrahepatic drug concentrations following Kpuu correction markedly enhanced the separation between the three cholestasis classes. The risk assessment based on Chep,sys,u provided a good separation between the three classes, with only one drug (rosiglitazone, class “no”) being clearly mispredicted (Fig. 5C). The risk assessment was further improved by using Kpuu-corrected unbound hepatic inlet concentrations. Using this reference concentration, an almost complete separation of all drugs into classes “no,” “rare,” and “common” was achieved (Fig. 5D), as supported by ROC AUC values ≥0.97. Using Chep,inlet,u as a reference, safety margin thresholds between classes “common”/”rare” and “rare”/”no” of 26 and 529, respectively, were obtained (Fig. 5D). These thresholds are clearly lower than those obtained from risk assessments using unbound extracellular concentrations (Fig. 5, A and B; “common”/”rare”: 100 and 59, “rare”/”no”: 3821 and 728 for Csys,u and Cinlet,u, respectively). Using Chep,inlet,u, the safety margin of simvastatin was reduced to 584-fold compared with 69,667-fold based on Chep,sys,u, representing the largest safety margin change within our test set.
Cholestasis classification
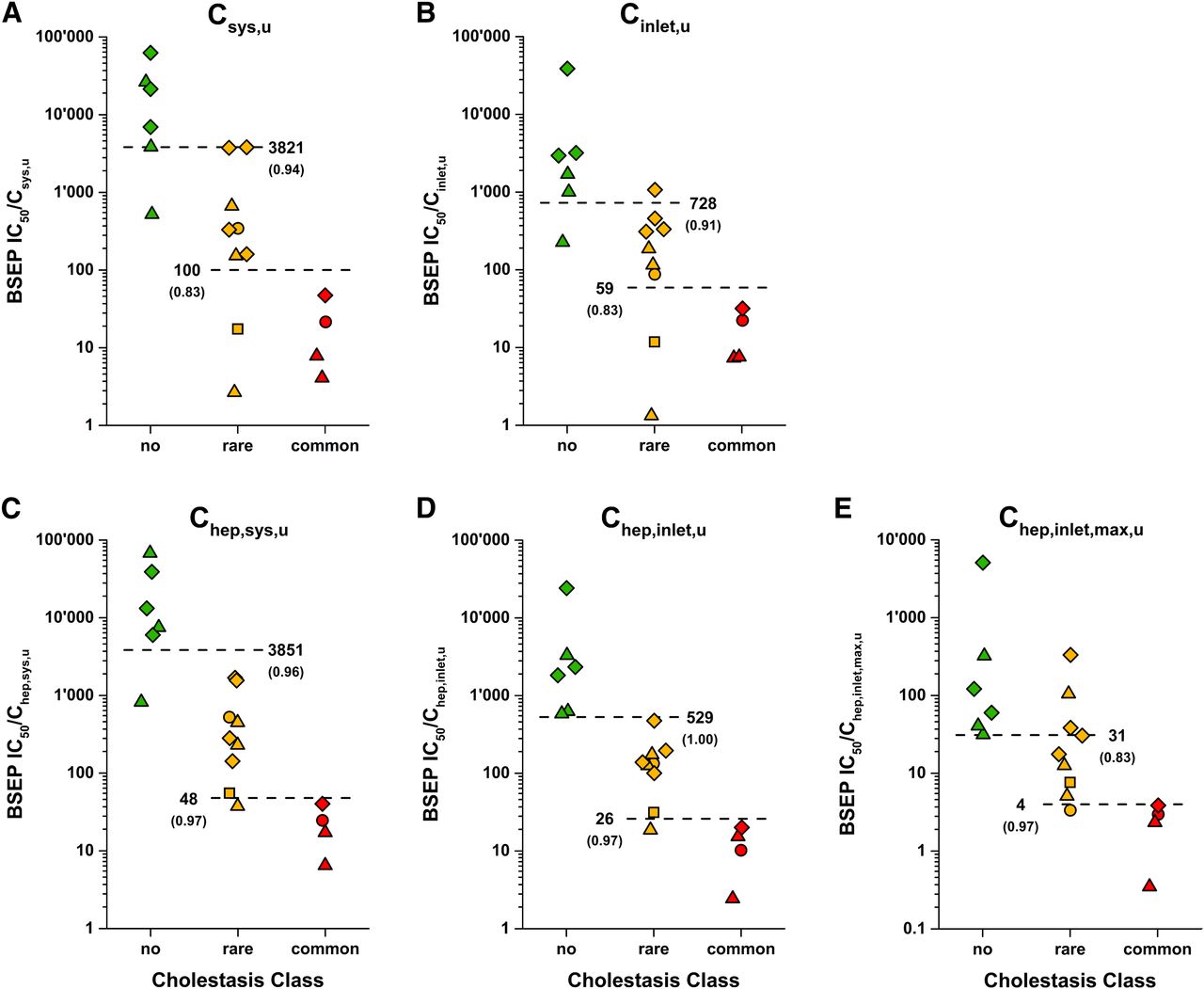
Correlation between BSEP inhibition, drug concentration, and drug-induced cholestasis. Safety margins for all 18 drug compounds representing the ratio of BSEP IC50 value and unbound systemic concentration (Csys,u) (A), unbound concentration at the hepatic inlet (Cinlet,u) (B), unbound intrahepatic concentration on basis of Csys,u (Chep,sys,u) (C), unbound intrahepatic concentration on basis of Cinlet,u (Chep,inlet,u) (D), and worst-case assessment of maximum unbound intrahepatic concentration (Chep,inlet,max,u) (E). Red, yellow, and green symbols represent the cholestasis classes “common,” “rare,” and “no cholestasis,” respectively. Squares, circles, triangles, and diamonds represent the ECM classes 1, 2, 3, and 4, respectively. Estimated safety margin thresholds between the cholestasis classes “common”/“rare” and “rare”/“no cholestasis” are shown next to dashed lines with ROC AUCs in brackets.
Additionally, maximum (worst-case) unbound hepatic inlet concentrations (Cinlet,max,u) were calculated to represent the situation during early drug development, where no clinical data are available. Assuming fast and complete intestinal absorption, substantially higher unbound intrahepatic concentrations were obtained compared to using measured clinical parameters. Whereas the resulting ratios still allowed separation of “common” from “rare” cholestasis events, the separation between “no” and “rare” was poorer than that obtained in risk assessments based on measured clinical parameters (Fig. 5D vs. Fig. 5E).
Quantification of Risk Factors for DIC.
The theoretical impact of disease state and polymorphic hepatic enzymes and transporters on bosentan-induced cholestasis was estimated based on clinical systemic exposure and in vitro enzyme and transporter polymorphism data (Fig. 6). The systemic exposure of bosentan is reported to be ∼2-fold higher in PAH patients compared with healthy subjects (Dingemanse and van Giersbergen, 2004). Based on this, we calculated the corresponding Chep,inlet,u in PAH patients (0.1901 µM), which is 69% higher than in healthy subjects (Fig. 6, healthy vs. PAH patients). Next, we calculated Kpuu, Chep,inlet,u, and corresponding safety margins for bosentan using altered transporter and enzyme activities due to genetic polymorphisms according to eq. 13. Our assessment indicates that the increased transport activity of OATP1B1*1b or OATP1B3*2 variants would increase Kpuu and Chep,inlet,u by 17% (Fig. 6, “PAH patients” vs. “OATP1B1*1b” or “OATP1B3*2”). Loss-of-function mutations in CYP3A4, CYP2C9, and MRP2 would result in increases in both Kpuu and Chep,inlet,u by 14, 9, and 1%, respectively (Fig. 6, “PAH patients” vs. “CYP3A4*20”, “CYP2C9*3”, or “MRP2*16”). The assumption of concurrence of all these genetic variants leads to predicted increases of Kpuu and Chep,inlet,u by 70% and a reduction of the safety margin from 116 to 68 (Fig. 6, “PAH patients” vs. “CYP3A4*20, CYP2C9*3, MRP2*16, OATP1B1*1b, OATP1B3*2”). Additionally, we evaluated the potential impact of BSEP polymorphisms on the DIC risk assessment. Presence of the low-activity variant BSEP G885R was simulated using a 5-fold reduced BSEP IC50 value (4.4 µM). This estimation leads to a 23-fold safety margin (Fig. 6, “PAH patients” vs. “BSEP G885R”). The theoretical combination of the BSEP G885R variant with genetic variants that affect the Kpuu of bosentan would further increase the DIC risk, as shown in Fig. 6 (“PAH patients” vs. “BSEP G885R, CYP3A4*20, CYP2C9*3, MRP2*16, OATP1B1*1b, OATP1B3*2”).

Theoretical impact of polymorphic hepatic enzymes and transporters on DIC safety margins for bosentan. Chep,inlet,u of bosentan (125 mg twice daily) in PAH patients in various enzyme/transporter polymorphism scenarios was calculated from extracellular Cinlet,u and in vitro Kpuu using in vitro activity data (eq. 13). Contributions of transporters and cytochrome P450 enzymes to the hepatic bosentan clearance as well as functional impact of polymorphisms are described in Materials and Methods. The dashed line represents the safety margin threshold between the cholestasis classes “common”/“rare” (26-fold).
Discussion
In the present work, we compare the use of unbound extracellular (systemic and hepatic inlet) or Kpuu–based intrahepatic concentrations in risk assessments of cholestasis upon BSEP inhibition.
Our work follows up on recent studies, which investigated the impact of systemic drug exposure on the clinical manifestation of DIC or DILI. Using safety margin ratios of BSEP IC50 to systemic drug concentration, our analyses revealed a generally increased risk of DIC among drugs with lower safety margins (Fig. 5A). However, no reliable separation of cholestatic and non-cholestatic drugs was obtained from this assessment. The outcome was not improved when hepatic inlet concentrations were used for safety margin calculations (Fig. 5B), even though this concentration is considered most relevant with regard to inhibition of hepatic enzymes or transporters (Zamek-Gliszczynski et al., 2013). These observations based on extracellular concentrations are in line with previous studies using similar approaches (Dawson et al., 2012; Morgan et al., 2013; Shah et al., 2015). However, several differences from previous work are important to highlight. First, following the “free drug hypothesis,” we used only unbound drug concentrations rather than total drug concentrations. Indeed, we obtained poorer predictions of DIC using total drug concentrations in plasma or blood (data not shown). Second, we evaluated the association of BSEP inhibition with DIC only and not with other types of liver toxicity. Hepatocellular DILI cases, which are included in other studies, are rather caused by direct toxicity or immune-mediated reactions (Chen et al., 2015) and are likely not explained by BSEP inhibition. Third, we classified the investigated drugs according to the DIC incidence instead of a commonly used DILI severity grading (Chen et al., 2011; Pedersen et al., 2013; Aleo et al., 2014). Fourth, the present assessment represents a worst-case scenario using the lowest reported in vitro BSEP IC50 (variability in IC50 values was within a 3-fold range for the majority of dugs with the exception of cyclosporine A, erythromycin, glibenclamide, and rosiglitazone) and the maximum reported systemic drug concentration at steady state after administration of the highest recommended oral dose. At least for certain drugs (e.g., Csys,u of atorvastatin and rosiglitazone), our parameters significantly deviate from previous studies (Dawson et al., 2012; Morgan et al., 2013; Shah et al., 2015).
However, relating extracellular drug concentrations to inhibition of an intracellular liver target (BSEP) is not expected to provide meaningful risk estimations if intracellular drug concentrations are affected by active transport or metabolic processes (Dawson et al., 2012; Chu et al., 2013; Camenisch et al., 2015; Camenisch, 2016). Indeed, applying the in vitro Kpuu values to obtain unbound intrahepatic drug concentrations markedly improved the separation of cholestasis classes. This became particularly evident for the correlation with unbound intrahepatic concentrations based on hepatic inlet concentrations, where almost complete separation between the different cholestasis classes was obtained (Fig. 5D). The use of hepatic inlet concentrations is most important for drugs with high hepatic first-pass elimination and significantly reduced systemic concentrations, as observed for simvastatin (115-fold difference between Csys,u and Cinlet,u). We therefore conclude that the unbound intrahepatic drug concentration based on hepatic inlet concentrations clearly represents the most reliable reference concentration for prediction of the DIC risk using BSEP inhibition assays.
BSEP in vitro inhibition data are commonly generated in early drug development. However, at this stage, clinical drug exposure data are rarely available, and DIC risk assessments based on the presented approach are not possible. We therefore performed an alternative risk assessment assuming that the clinical systemic exposure can be accurately predicted and assuming complete and rapid absorption to obtain worst-case hepatic inlet concentrations (Fig. 5E) (Ito et al., 1998; Giacomini et al., 2010). While the cholestasis classes “common” and “rare” were reasonably well separated by this approach, the separation between classes “no cholestasis” and “rare” failed compared with risk approaches using measured clinical input parameters (Fig. 5D vs. Fig. 5E). Therefore, for DIC risk assessments during preclinical development, we suggest applying unbound intrahepatic drug concentrations derived from in vitro Kpuu and predicted systemic exposure (e.g., using physiologically based pharmacokinetic modeling).
Kpuu and corresponding unbound intrahepatic drug concentrations are affected by the individual contributions of active hepatic transport and metabolic processes and correlate well with the four ECM classes (Figs. 2 and 3). For drugs in ECM classes 1 and 3, where hepatic uptake is the rate-limiting elimination step, Kpuu is likely to be below 1 (Chep,u < Cu). On the other hand, class 3 drugs can also approach Kpuu above 1 if active hepatic uptake is extensive (Chep,u > Cu). As illustrated in Fig. 7, unbound extracellular concentrations of ketoconazole and simvastatin acid markedly overestimated the DIC risk. Use of Kpuu-corrected unbound intrahepatic concentrations shifted ketoconazole (Kpuu = 0.32) and simvastatin acid (Kpuu = 0.39) into their appropriate risk zones (“rare” and “no” cholestasis, respectively). For class 4 drugs, passive uptake/efflux permeability exceeds intrinsic hepatic clearance. In combination with active hepatic uptake processes, drugs accumulate within the hepatocytes, resulting in Kpuu values greater 1 (Figs. 2 and 3). For these drugs, the DIC risk is underestimated by unbound extracellular concentrations, as highlighted for pitavastatin in Fig. 7. After correction with Kpuu, pitavastatin was assigned to the correct risk zone (“rare”). Only for class 2 drugs, such as imatinib, do unbound extracellular concentrations represent an appropriate surrogate for the unbound intrahepatic concentration (Kpuu ≈ 1.0, Chep,u ≈ Cu) (Fig. 7).

ECM class-dependent effect of Kpuu on the DIC risk assessment. BSEP IC50 values were plotted against unbound hepatic inlet concentrations before (Cinlet,u) and after correction with in vitro Kpuu to unbound intrahepatic concentrations (Chep,inlet,u). Ketoconazole (class 1), imatinib (class 2), simvastatin acid (class 3), and pitavastatin are shown as yellow squares, red circles, green triangles, and yellow diamonds, respectively. Red, yellow, and green symbols represent the cholestasis classes “common”, “rare,” and “no cholestasis,” respectively. Dashed lines represent the safety margin thresholds between cholestasis classes “common”/“rare” (26-fold) and “rare”/“no” (529-fold), respectively.
For the in vitro Kpuu assessment, we generally assumed absence of active sinusoidal efflux. Kpuu values of substrates for basolateral MRP3 and MRP4, which have been shown to be upregulated in human and rat under cholestatic conditions (Soroka et al., 2001; Gradhand et al., 2008), might therefore be overpredicted. However, except for rosuvastatin, the relevance of MRP3 and MRP4 in transport of pharmaceutical drugs is unknown and requires further research. In addition, we assumed that the ECM-based in vitro Kpuu directly translates to in vivo Kpuu. The assumption was based on the good IVIVC for Kpuu in rat (Fig. 4) and on previous hepatic clearance predictions using the ECM, where no systematic underprediction of in vivo hepatic clearance was observed. Similarly, in vitro Kpuu using sandwich-cultured hepatocytes recently provided a close IVIVC in rats for rosuvastatin, ritonavir, and furamidine (Pfeifer et al., 2013), whereas Kpuu from suspended hepatocytes generally overpredicted Kpuu in vivo, likely due to the absence of intrinsic clearance processes. Partial loss of activity in certain in vitro systems has previously been described (Lundquist et al., 2014); however, the need of scaling factors for pharmacokinetic modeling is controversially discussed and likely compound-dependent, requiring clinical data (Jones et al., 2012; Morse et al., 2015; Yoshikado et al., 2016). Especially in an early drug development stage, the direct use of in vitro Kpuu is therefore expected to enable estimations of unbound intrahepatic drug concentrations.
Our risk assessment using unbound hepatic inlet concentrations predicted a DIC risk in more than 2% of subjects for safety margins below ∼25 (Fig. 5D). Since unbound intrahepatic concentrations are 25-fold below the IC50 of BSEP, no relevant inhibition of BSEP would be expected. However, the occurrence of DIC in a low percentage of patients leads us to the hypothesis that these subjects react more sensitively to BSEP inhibition than the rest of the population. This variability might be explained by various physiologic (e.g., age, gender, underlying diseases), exogenous (e.g., comedication, nutrition), and genetic factors (polymorphisms) that potentially modify intrahepatic drug concentrations or BSEP activity, resulting in increased cholestasis risk. Using bosentan as an example, we evaluated the theoretical impact of disease state and polymorphic hepatic enzymes and transporters on DIC risk (Fig. 6). Physiologic factors, such as increased age and gender, are not associated with altered risk of bosentan-induced cholestasis (Markova et al., 2013). Similarly, increased systemic bosentan exposure in PAH patients could not be linked to higher incidence of liver injury (Dingemanse and van Giersbergen, 2004), which is in line with our assessment (Fig. 6). The present analysis suggests that genetic variants of transporters and enzymes involved in bosentan elimination only marginally affect the DIC risk assessment, even upon the unlikely concurrence of all polymorphisms (Fig. 6). Indeed, bosentan-induced liver injury could not be associated with polymorphic variants of OATP1B1, OATP1B3, CYP2C9, or MRP2 (Markova et al., 2013, 2014; Roustit et al., 2014). In addition, we evaluated the impact of the BSEP variant G885R, which was recently associated with DIC, likely due to substantially reduced activity (<20%) (Lang et al., 2007). Based on these data, we estimated a significantly increased risk of bosentan-induced cholestasis in PAH patients carrying the G885R variant, as indicated by a 23-fold safety margin and the change in cholestasis class from “rare” to “common” (Fig. 6). In summary, the presented risk assessment for bosentan illustrates the utility of ECM-based Kpuu assessments to define the relevance of polymorphic enzymes and transporters in the hepatic elimination of a drug. Especially for drugs with high risk of DIC, such information could guide the selection of genotyping targets in clinics and ultimately allow personalized dosing regimens. However, further research on frequency, global distribution, and in vivo effects of polymorphisms will be required to estimate the incidence of DIC in heterogenic populations.
In conclusion, we demonstrated that the incidence of DIC upon BSEP inhibition correlates with Kpuu-based unbound intrahepatic drug concentrations. To the best of our knowledge, this approach represents the most reliable prediction of DIC available so far, which also allows to account for polymorphisms on hepatic enzymes and transporters associated with DIC risk. Our study represents a proof-of-concept for estimating the inhibition potential of an intracellular transporter and is therefore expected to likewise improve risk assessments for other intrahepatic targets involved in DDI, pharmacologic efficacy, and toxicology, such as hyperbilirubinemia upon MRP2 or UGT1A1 inhibition. The validation of ECM-based risk assessments for other target enzymes or transporters as well as the extension of the approach to other organs, such as the kidney, will require extensive future research.
Acknowledgments
The authors thank the many Novartis Drug Metabolism and Pharmacokinetics scientists of Basel, Switzerland, who supported this work. Special thank goes to Marc Witschi, Judith Streckfuss, and Hans-Joachim Handschin for technical assistance; to Kenichi Umehara and Ewa Gatlik for scientific advice; and to David Pearson for critical evaluation of this manuscript.
Authorship Contributions
Participated in research design: Riede, Poller, Camenisch.
Conducted experiments: Riede.
Performed data analysis: Riede, Poller, Camenisch.
Wrote or contributed to the writing of the manuscript: Riede, Poller, Huwyler, Camenisch.
Footnotes
- Received November 16, 2016.
- Accepted March 1, 2017.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AP
- alkaline phosphatase
- AUC
- area under the curve
- BSEP
- bile salt export pump
- Chep,inlet,u
- unbound intrahepatic concentration on the basis of Cinlet,u
- Chep,sys,u
- unbound intrahepatic concentration on the basis of Csys,u
- Chep,u
- unbound intrahepatic concentration
- Cinlet,max,u
- “worst-case assessment” of unbound concentration at the hepatic inlet
- Cinlet
- u, unbound concentration at the hepatic inlet
- CLint
- intrinsic clearance
- CLint,met
- intrinsic metabolic clearance
- CLint
- sec intrinsic biliary clearance
- Csys,u
- unbound systemic concentration
- Cu
- unbound extracellular concentration
- DDI
- drug-drug interaction
- DIC
- drug-induced cholestasis
- DILI
- drug-induced liver injury
- ECM
- extended clearance model
- fn
- fractional in vivo contribution
- IVIVC
- in vitro–in vivo correlation
- Kpuu
- liver-to-blood partition coefficient for unbound drug at steady state
- MRP
- multidrug resistance protein
- OATP
- organic anion transporting polypeptide
- PAH
- pulmonary arterial hypertension
- PSeff
- sinusoidal efflux clearance
- PSeff
- act active sinusoidal efflux clearance
- PSeff
- pas passive sinusoidal efflux clearance
- PSinf
- total sinusoidal uptake clearance
- PSinf,act
- active sinusoidal uptake clearance
- PSinf,pas
- passive sinusoidal uptake clearance
- ROC
- receiver operating characteristic curve
- ULN
- upper limit of normal
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}