


Abstract
A major metabolic pathway of haloperidol is glucuronidation catalyzed by UDP-glucuronosyltransferase (UGT). In this study, we found that two glucuronides were formed by the incubation of haloperidol with human liver microsomes (HLM) and presumed that the major and minor metabolites (>10-fold difference) were O- and N-glucuronide, respectively. The haloperidol N-glucuronidation was catalyzed solely by UGT1A4, whereas the haloperidol O-glucuronidation was catalyzed by UGT1A4, UGT1A9, and UGT2B7. The kinetics of the haloperidol O-glucuronidation in HLM was monophasic with Km and Vmax values of 85 μM and 3.2 nmol · min−1 · mg−1, respectively. From the kinetic parameters of the recombinant UGT1A4 (Km = 64 μM, Vmax = 0.6 nmol · min−1 · mg−1), UGT1A9 (Km = 174 μM, Vmax = 2.3 nmol · min−1 · mg−1), and UGT2B7 (Km = 45 μM, Vmax = 1.0 nmol · min−1 · mg−1), we could not estimate which isoform largely contributes to the reaction. Because the haloperidol O-glucuronidation in a panel of 17 HLM was significantly correlated (r = 0.732, p < 0.01) with zidovudine O-glucuronidation, a probe activity of UGT2B7, and the activity in the pooled HLM was prominently inhibited (58% of control) by gemfibrozil, an inhibitor of UGT2B7, we surmised that the reaction would mainly be catalyzed by UGT2B7. We could successfully estimate, using the concept of the relative activity factor, that the contributions of UGT1A4, UGT1A9, and UGT2B7 in HLM were approximately 10, 20, and 70%, respectively. The present study provides new insight into haloperidol glucuronidation, concerning the causes of interindividual differences in the efficacy and adverse reactions or drug-drug interactions.
Introduction
Haloperidol is a widely used antipsychotic drug for the treatment of schizophrenia and other psychiatric disorders. Haloperidol is extensively metabolized in the liver, and only approximately 1% of the administered dose is excreted unchanged in urine (Forsman and Ohman, 1976). In humans, several metabolic pathways have been reported, i.e., oxidative N-dealkylation (Gorrod and Fang, 1993), carbonyl reduction (Inaba and Kovacs, 1989), and glucuronidation (Oida et al., 1989) (Fig. 1). In plasma, the concentration of haloperidol glucuronide is highest among the metabolites, followed, in rank order, by unchanged haloperidol, reduced haloperidol, and reduced haloperidol glucuronide (Someya et al., 1992). Kudo and Ishizaki (1999) reported that glucuronidation accounts for 50 to 60% of biotransformation of haloperidol. Thus, glucuronidation would be a determinant of the steady-state plasma haloperidol concentration, which shows large interindividual differences (11-fold) (Hirokane et al., 1999). Oida et al. (1989) reported that the glucuronide detected in urine samples from patients receiving haloperidol was O-glucuronide, accounting for 18% of the dose, although O-glucuronide and N-glucuronide are surmised for haloperidol in view of the chemical structure. The UDP-glucuronosyltransferase (UGT) isoform(s) responsible for haloperidol glucuronidation remain to be clarified.

Proposed metabolic pathways of haloperidol in humans.
Some drug-drug interactions for haloperidol with carbamazepine, phenytoin, phenobarbital, fluoxetine, fluvoxamine, nefazodone, rifampicin, and quinidine have been reported (Linnoila et al., 1980; Jann et al., 1985; Goff et al., 1991). It has been recognized that the drug interactions would be mediated by CYP3A4, because these coadministered drugs have the potency to induce or inhibit CYP3A4 and the phase I metabolism of haloperidol is catalyzed by CYP3A4 (Pan and Belpaire, 1997; Yasui et al., 1999). It should be noted that carbamazepine, phenytoin, phenobarbital, and rifampicin have the potency to induce not only CYP3A4 but also UGT (Linnoila et al., 1980; Jann et al., 1985). Because the intrinsic clearances by glucuronidation appear to be greater than those by CYP3A4-mediated oxidations, there is a possibility that the drug interactions are through the induction of UGT. In the present study, we sought to identify the human UGT isoforms involved in haloperidol glucuronidation to obtain a clue for the underlying causes of the interindividual variability of pharmacokinetics or drug-drug interactions.
Materials and Methods
Materials.
Haloperidol, trifluoperazine dihydrochloride, propofol, zidovudine, niflumic acid, and gemfibrozil were purchased from Wako Pure Chemicals (Osaka, Japan). UDP-glucuronic acid (UDPGA) and alamethicin were from Sigma-Aldrich (St. Louis, MO). Zidovudine O-glucuronide and hecogenin were obtained from Toronto Research Chemicals (Toronto, ON, Canada) and Tokyo Kasei (Tokyo, Japan), respectively. Microsomes from 17 individual human livers (HG03, HG30, HG32, HG43, HG64, HG70, HG95, HH1, HH6, HH9, HH13, HH18, HH31, HH35, HH40, HH47, and HH89), pooled human liver microsomes (HLM) (n = 50), and recombinant human UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17 expressed in baculovirus-infected insect cells (Supersomes) were obtained from BD Gentest (Woburn, MA). Pooled rat liver microsomes (Sprague-Dawley rats, male, 7 weeks old, n = 5) were prepared previously (Shiratani et al., 2008). All other chemicals and solvents were of analytical grade or the highest grade commercially available.
Haloperidol Glucuronidation.
Haloperidol glucuronide formation was determined according to the method of Narayanan et al. (2004) with slight modifications. In brief, a typical incubation mixture (200 μl of total volume) containing 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 25 μg/ml alamethicin, 0.5 mg/ml HLM, recombinant human UGTs, or rat liver microsomes (RLM) and 100 μM haloperidol was kept on ice for 10 min. The mixture was preincubated at 37°C for 3 min, and reaction was initiated by the addition of UDPGA (final concentration at 5 mM). After incubation at 37°C for 60 min, the reaction was terminated by the addition of 80 μl of ice-cold acetonitrile. After removal of the protein by centrifugation at 10,000g for 5 min, a 20- or 90-μl portion of the sample was subjected to LC-MS/MS or HPLC analysis.
Identification of Haloperidol Glucuronide by LC-MS/MS Analysis.
To identify haloperidol glucuronide, LC-MS/MS analysis was performed. This system was also used for the quantification of haloperidol N-glucuronide. The LC equipment consisted of an HP1100 system including a binary pump, an automatic sampler, and a column oven (Agilent Technologies, Santa Clara, CA), which was equipped with a Zorbax SB-C18 column (2.1 × 50 mm, 3.5 μm; Agilent Technologies). The column temperature was 25°C. The mobile phase was 0.1% formic acid (A) and methanol including 0.1% formic acid (B). The conditions for elution were as follows: 20% B (0–2.0 min), 20 to 90% B (2.01–3 min), 90% B (3.01–10 min), and 20% B (10.01–18 min). The flow rate was 0.2 ml/min. The LC system was connected to a PE Sciex API 2000 tandem mass spectrometer (Applied Biosystems, Foster City, CA) operated in the positive electrospray ionization mode. The turbo gas was maintained at 550°C. Nitrogen was used as the nebulizing, turbo, and curtain gas at 70, 40, and 50 psi, respectively. Parent and/or fragment ions were filtered in the first quadrupole and dissociated in the collision cell using nitrogen as the collision gas. The collision energy was 51 V for haloperidol. Two m/z ion transitions were monitored in the multiple reaction monitoring (MRM) mode: m/z 376.1 and 123.2 for haloperidol and m/z 552.1 and 123.2 for haloperidol glucuronide. The analytical data were processed using Analyst software (version 1.5; Applied Biosystems) in the API 2000 LC-MS/MS systems.
Quantification of Haloperidol Glucuronide by HPLC Analysis.
To quantify haloperidol O-glucuronide, HPLC analysis was performed according to the method of Narayanan et al. (2004) with slight modifications. The HPLC equipment consisted of an L-2130 pump (Hitachi, Tokyo, Japan), an L-2200 autosampler (Hitachi), an L-2400 UV detector (Hitachi), a D-2000 HPLC System Manager program (Hitachi), and an L-2300 column oven, which was equipped with a Mightysil RP-18GP column (4.6 × 150 mm, 5 μm; Kanto Chemical, Tokyo, Japan). The column temperature was 35°C. The mobile phase was 45% acetonitrile-20 mM ammonium dihydrogen phosphate, and the flow rate was 0.8 ml/min. The eluate was monitored at 270 nm.
For the quantification of haloperidol O-glucuronide, the eluate of the HPLC system from the incubation mixture with HLM, including haloperidol O-glucuronide, was collected with reference to the retention time. A part of the eluate was incubated with 1000 U/ml β-glucuronidase at 37°C for 24 h. The hydrolyzed haloperidol O-glucuronide was quantified as haloperidol by HPLC. Once we determined the peak area per known content of haloperidol O-glucuronide, the ratio was applied to the calculation of haloperidol O-glucuronide formed in the incubation mixtures. The quantified haloperidol O-glucuronide was also used as a reference for the quantification of haloperidol N-glucuronide in the LC-MS/MS analysis, on the assumption that the peak height ratios of the haloperidol O- and N-glucuronides would be equal.
Kinetic Analysis of Haloperidol O-Glucuronidation.
The haloperidol O-glucuronosyltransferase activities at substrate concentrations from 10 to 400 μM were determined as described above. Kinetic parameters were estimated from the fitted curves using a KaleidaGraph computer program (Synergy Software, Reading, PA) designed for nonlinear regression analysis. The following equations were applied for Michaelis-Menten kinetics (eq. 1) or substrate inhibition kinetics (eq. 2):
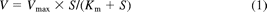
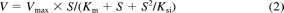 where V is the velocity of the reaction, S is the substrate concentration, Km is the Michaelis-Menten constant, Vmax is the maximum velocity, and Ksi is the substrate inhibition constant. Data are expressed as the mean ± S.D. of three independent determinations.
where V is the velocity of the reaction, S is the substrate concentration, Km is the Michaelis-Menten constant, Vmax is the maximum velocity, and Ksi is the substrate inhibition constant. Data are expressed as the mean ± S.D. of three independent determinations.
Other Glucuronidation Assays.
Trifluoperazine N-glucuronide formation was determined according to the method of Fujiwara et al. (2007) with slight modifications. In brief, a typical incubation mixture (100 μl of total volume) contained 50 mM Tris-HCl buffer (pH 7.4), 10 mM MgCl2, 2.5 mM UDPGA, 25 μg/ml alamethicin, 0.25 mg/ml HLM or recombinant human UGT1A4, and 50 μM trifluoperazine (∼Km value of recombinant UGT1A4). Propofol O-glucuronide formation was determined according to the method of Fujiwara et al. (2007) with slight modifications. In brief, a typical incubation mixture (200 μl of total volume) contained 50 mM Tris-HCl buffer (pH 7.4), 10 mM MgCl2, 3 mM UDPGA, 25 μg/ml alamethicin, 0.5 mg/ml HLM or recombinant human UGT1A9, and 50 μM propofol (∼Km value of recombinant UGT1A9). Zidovudine O-glucuronide formation was determined according to a method of Court et al. (2003) with slight modifications. A typical incubation mixture (200 μl total volume) contained 50 mM Tris-HCl buffer (pH 7.4), 5 mM MgCl2, 5 mM UDPGA, 25 μg/ml alamethicin, 0.3 mg/ml HLM or recombinant human UGT2B7, and 100 μM zidovudine (8-fold lower than the Km value of recombinant UGT2B7 to minimize the contribution of UGT2B4). Quantification of zidovudine O-glucuronide was performed by comparing the HPLC peak height to that of the authentic standard. For quantification of the other glucuronides, the eluate from the HPLC column containing each glucuronide was collected, and a part of the eluate was hydrolyzed with NaOH at 75°C for 30 min (Hawes, 1998). The hydrolyzed glucuronides were quantified, using HPLC, by comparison of the peak heights with those of the external standard curve of the substrates.
Inhibition Analyses of Haloperidol O-Glucuronidation by Recombinant UGTs and HLM.
Hecogenin and niflumic acid, a selective inhibitor of UGT1A4 (Uchaipichat et al., 2006) and UGT1A9 (Miners et al., 2011), respectively, and gemfibrozil, a substrate for UGT2B7 (Mano et al., 2007), were used as inhibitors. Hecogenin was dissolved in dimethyl sulfoxide. Niflumic acid and gemfibrozil were dissolved in methanol. These compounds (10 μM) were added to the incubation mixtures described above to investigate their inhibitory effects on the haloperidol O-glucuronosyltransferase activities by recombinant UGT1A4, UGT1A9, and UGT2B7 (25 μM substrate concentration) or pooled HLM (25, 100, or 200 μM substrate concentration). The final concentration of the organic solvents in the incubation mixture was 2% (v/v). Control incubations contained the same concentration of organic solvent.
Contribution of UGT1A4, UGT1A9, and UGT2B7 to Haloperidol O-Glucuronidation in HLM.
The percentage contributions of UGT1A4, UGT1A9, and UGT2B7 to the haloperidol O-glucuronosyltransferase activity in human liver microsomes were estimated by application of the RAF approach proposed by Crespi (1995). This approach assumes that any effects on the rate of metabolism are independent of the substrate, i.e., the rank order of rates of metabolism is the same for a particular recombinant UGT isoform and the same UGT isoform present in human liver microsomes and any factor that affects the rate of metabolism for one substrate does so equally for other substrates. In the present study, we determined the RAF for UGT1A4 (i.e., RAFUGT1A4) as the ratio of the activity of trifluoperazine N-glucuronidation in human liver microsomes to that with recombinant human UGT1A4 (at 50 μM substrate concentration). Propofol O-glucuronidation (at 50 μM substrate concentration) was used for the calculation of the RAF for UGT1A9 (RAFUGT1A9). Zidovudine O-glucuronidation (at 100 μM substrate concentration) was used for the calculation of the RAF for UGT2B7 (RAFUGT2B7). Using the RAF values, the haloperidol O-glucuronidation by UGT1A4, UGT1A9, and UGT2B7 in human liver microsomes (VUGT1A4, VUGT1A9, and VUGT2B7, respectively) is expressed as follows:
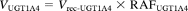
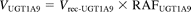
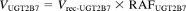 where Vrec-UGT1A4, Vrec-UGT1A9, and Vrec-UGT2B7 are haloperidol O-glucuronosyltransferase activities by recombinant UGT1A4, UGT1A9, and UGT2B7, respectively. The contributions of UGT1A4, UGT1A9, and UGT2B7 to the observed activity by human liver microsomes (VHLM) is estimated as follows:
where Vrec-UGT1A4, Vrec-UGT1A9, and Vrec-UGT2B7 are haloperidol O-glucuronosyltransferase activities by recombinant UGT1A4, UGT1A9, and UGT2B7, respectively. The contributions of UGT1A4, UGT1A9, and UGT2B7 to the observed activity by human liver microsomes (VHLM) is estimated as follows:
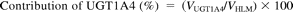
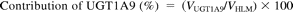

Statistical Analysis.
Correlation analysis was determined by Pearson's product moment method. p < 0.05 was considered statistically significant.
Results
Glucuronidation of Haloperidol by HLM, Recombinant UGTs, and RLM.
When the incubation mixture containing HLM, UDPGA, and haloperidol (at concentrations of 100 μM) was analyzed by LC-MS/MS, the monitoring of Q1 MS at m/z 552.1 corresponding to haloperidol glucuronide showed two peaks, peak A and peak B, at the retention times at 8.4 and 9.1 min, respectively (Fig. 2A). The monitoring of Q1 MS at m/z 376.1 corresponding to haloperidol showed a peak, peak C, at the retention time at 8.6 min. Peaks A and B were not observed when haloperidol and HLM were incubated without UDPGA (data not shown). The product ion spectra of peaks A and B showed an ion m/z 376.1 corresponding to haloperidol, suggesting the loss of glucuronic acid (176 atomic mass units). Other product ions such as m/z 358.4, 165.0, and 123.2 were the same as those observed in peak C. These results suggest that both peaks A and B represent glucuronides (metabolite A and metabolite B). For the subsequent study, MRM at m/z 552.1 and 123.2 was used to detect haloperidol glucuronide (Fig. 2, B–D). In the HLM (Fig. 2B), the velocity of metabolite A formation (1.64 nmol · min−1 · mg−1) was dominant over that of the metabolite B formation (0.10 nmol · min−1 · mg−1). Because it has been reported that the haloperidol O-glucuronide was a major metabolite in urine samples from patients receiving haloperidol (Oida et al., 1989; Luo et al., 1995), we speculated that metabolite A might be the haloperidol O-glucuronide.

LC-MS/MS analysis of haloperidol glucuronidation by HLM (A and B), recombinant UGTs (C), and RLM (D) and HPLC analysis to quantify haloperidol O-glucuronide (E). Q1 MS at m/z 552.1 corresponding to haloperidol glucuronide showed peaks A and B at 8.4 and 9.1 min, respectively. Q1 MS at m/z 376.1 corresponding to haloperidol showed peak C at 8.6 min. Product ion spectra of the peaks A, B, and C are shown in a box. MRM at m/z 552.1 and 123.2 was used to detect haloperidol glucuronides formed by HLM (B), recombinant UGT1A4, UGT1A9, and UGT2B7 (C), and RLM (D). In the HPLC analysis, the metabolite A was well separated from haloperidol, although the peak of metabolite B was faint (E). amu, atomic mass units.
The haloperidol glucuronidation by human recombinant UGTs was also determined at a concentration of 100 μM haloperidol. As shown in Fig. 2, C and UGT1A4 produced two metabolites A and B, whereas UGT1A9 and UGT2B7 produced only metabolite A. The other isoforms used in this study did not show any metabolites (Fig. 3). UGT1A4 is an isoform that prefers catalyzing the N-glucuronidation of tertiary amines (Green and Tephly 1996; King et al., 2000). Therefore, it was surmised that metabolite B might be the haloperidol N-glucuronide. In rats, the ortholog of human UGT1A4 is not functional (King et al., 2000). When we incubated haloperidol with RLM, only metabolite A was produced (Fig. 2D). Although we did not further confirm the structure of the metabolites A and B, we analyzed metabolite A and B formation as haloperidol O-glucuronidation and N-glucuronidation, respectively. Because the retention times of haloperidol O-glucuronide and haloperidol were close in the LC-MS/MS system that was optimized for the detection of haloperidol N-glucuronide, we established an HPLC condition to easily detect and quantify the haloperidol O-glucuronide (Fig. 2E). Under this condition, haloperidol O-glucuronide, haloperidol, and haloperidol N-glucuronide were eluted at 6.5, 13.9, and 16.0 min, respectively. As summarized in Fig. 3, for haloperidol O-glucuronidation, UGT1A9 showed the highest activity (1.63 nmol · min−1 · mg−1), followed by UGT2B7 (1.29 nmol · min−1 · mg−1) and UGT1A4 (0.44 nmol · min−1 · mg−1). Haloperidol N-glucuronide formation by UGT1A4 was 0.21 nmol · min−1 · mg−1. In the subsequent experiment, we pursued the study, focusing on haloperidol O-glucuronidation as the major metabolic pathway of haloperidol.
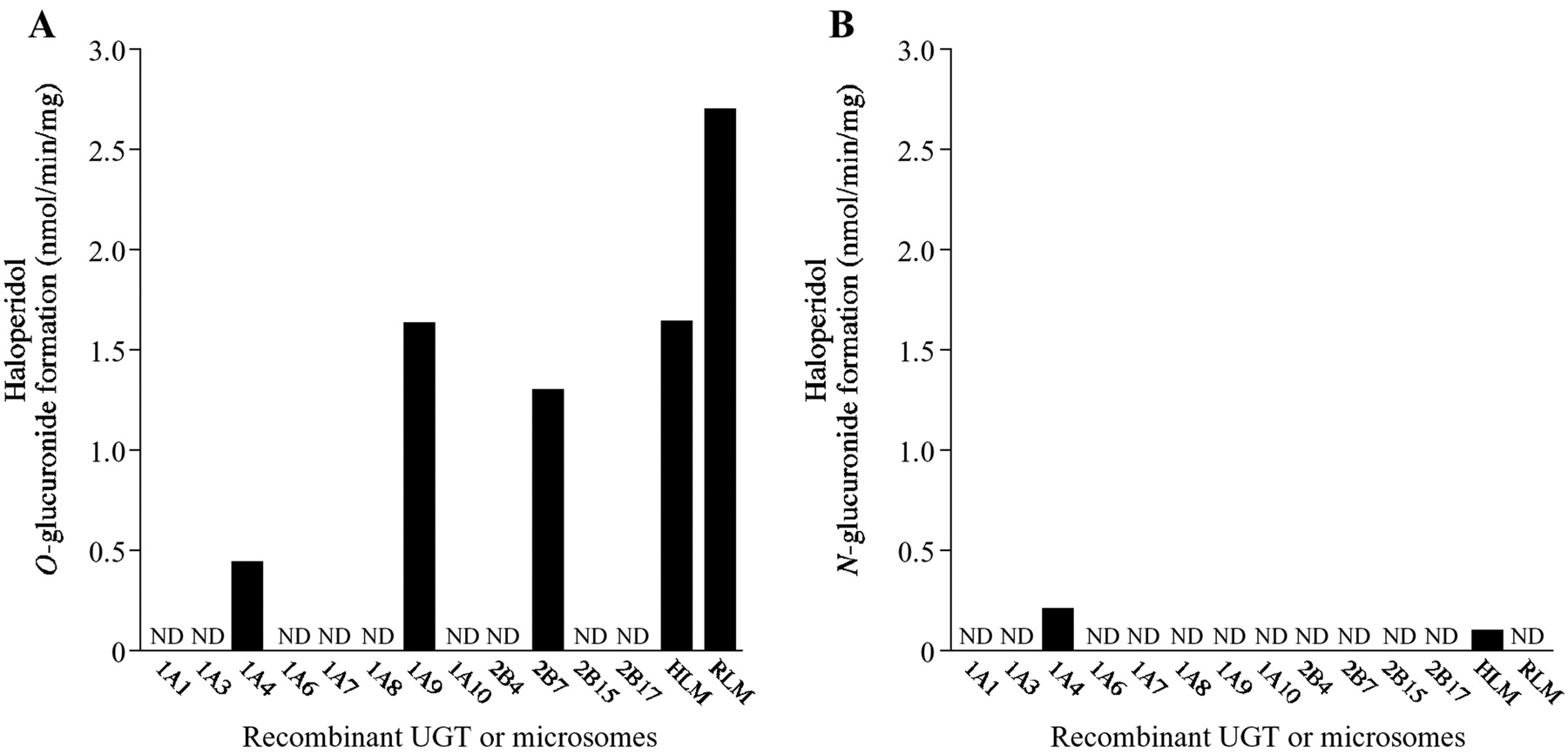
Haloperidol O-glucuronide (A) and N-glucuronide (B) formation by recombinant human UGTs expressed in baculovirus-infected insect cells, pooled HLM, and RLM. Haloperidol (100 μM) was incubated with each enzyme (0.5 mg/ml) for 60 min. Each column represents the mean of duplicate determinations.
Kinetic Analyses of Haloperidol O-Glucuronidation by HLM and Recombinant UGTs.
Kinetic analyses of haloperidol O-glucuronidation in pooled HLM were performed (Fig. 4A). The kinetics was best described by the Michaelis-Menten equation, which was supported by a monophasic Eadie-Hofstee plot. The apparent Km, Vmax, and intrinsic clearance (CLint) values were 85 ± 10 μM, 3.2 ± 0.1 nmol · min−1 · mg−1, and 38 ± 6 μl · min−1 · mg−1, respectively. Next, kinetic analyses by recombinant UGT1A4, UGT1A9, and UGT2B7 were performed (Fig. 4B). The kinetics by UGT1A4 was best described by the Michaelis-Menten equation with Km, Vmax, and CLint values of 64 ± 9 μM, 0.6 ± 0.0 nmol · min−1 · mg−1, and 9 ± 2 μl · min−1 · mg−1, respectively. The kinetics by UGT1A9 and UGT2B7 were best described by the substrate inhibition kinetics. The Km, Vmax, CLint, and Ksi values for UGT1A9 were 174 ± 59 μM, 2.3 ± 0.3 nmol · min−1 · mg−1, 16 ± 7 μl · min−1 · mg−1, and 344 ± 98 μM, respectively. The Km, Vmax, CLint, and Ksi values for UGT2B7 were 45 ± 9 μM, 1.0 ± 0.1 nmol · min−1 · mg−1, 23 ± 6 μl · min−1 · mg−1, and 590 ± 150 μM, respectively.

Kinetic analyses of haloperidol O-glucuronidation in pooled HLM (A) and recombinant UGTs (B). The Eadie-Hofstee plot is shown as an inset. Microsomes (0.5 mg/ml) were incubated with 10 to 400 μM haloperidol and 5 mM UDPGA at 37°C for 60 min. Data are mean ± S.D. of three independent determinations.
Correlation Analysis of Haloperidol O-Glucuronidation and Glucuronidation of Typical Substrates for UGT1A4, UGT1A9, and UGT2B7 in HLM.
Haloperidol O-glucuronidation in a panel of 17 HLM was determined at 25, 100, and 200 μM substrate concentrations. The activities at 25 μM haloperidol varied from 0.16 to 0.63 nmol · min−1 · mg−1, showing 3.9-fold variability. The activities at 100 μM haloperidol varied from 0.67 to 2.04 nmol · min−1 · mg−1, showing 3.0-fold variability. The activities at 200 μM haloperidol varied from 0.90 to 2.77 nmol · min−1 · mg−1, showing 3.1-fold variability. Thus, the interindividual variability of the haloperidol O-glucuronidation was not so large. We measured trifluoperazine, propofol, and zidovudine glucuronidation in the panel of HLM as specific activities for UGT1A4, UGT1A9, and UGT2B7. Although the propofol O-glucuronidation is catalyzed by UGT1A8 and UGT1A9, UGT1A8 is absent in human livers. Zidovudine O-glucuronidation, which is known as a probe activity for UGT2B7, is also catalyzed by UGT2B4 and UGT2B17 (Court et al., 2003). We used a substrate concentration lower than the Km value of UGT2B7 to minimize the contribution of UGT2B4 and UGT2B17. Trifluoperazine N-glucuronidation varied from 0.26 to 0.75 nmol · min−1 · mg−1, showing 2.9-fold variability. Propofol O-glucuronidation varied from 2.07 to 7.64 nmol · min−1 · mg−1, showing 3.7-fold variability. Zidovudine O-glucuronidation varied from 0.14 to 0.57 nmol · min−1 · mg−1, showing 4.1-fold variability. Using the data, we performed a correlation analysis (Table 1). Haloperidol O-glucuronidation at a 25 μM substrate concentration was significantly correlated with zidovudine O-glucuronidation (r = 0.732, p < 0.001) but not with the glucuronidation of the other two compounds, indicating that UGT2B7 would mainly contribute to haloperidol O-glucuronidation. When the haloperidol concentrations were increased to 100 or 200 μM, a significant correlation with trifluoperazine N-glucuronidation and propofol O-glucuronidation in addition to the zidovudine O-glucuronidation was observed, indicating that UGT1A4 and UGT1A9, in addition to UGT2B7, would contribute to haloperidol O-glucuronidation at a higher substrate concentration.
Correlation coefficients between haloperidol O-glucuronidation and typical activities in a panel of 17 HLM
Inhibition Analyses.
To investigate which UGT isoform mainly contributes to haloperidol O-glucuronidation in HLM, we performed inhibition studies. First, we validated the inhibition selectivity of hecogenin, niflumic acid, and gemfibrozil using recombinant enzymes (Fig. 5A). Hecogenin (at 10 μM) strongly inhibited the haloperidol O-glucuronidation by UGT1A4 to 4% of control, but its effects on UGT1A9 and UGT2B7 activities were minor (77 and 82% of control, respectively). Niflumic acid (at 10 μM) almost completely inhibited the UGT1A9 activity (1% of control), whereas it hardly affected the UGT1A4 and UGT2B7 activities (99% of control). Gemfibrozil (at 10 μM) prominently inhibited the UGT2B7 activity (13% of control), whereas it hardly inhibited the UGT1A9 activity (79% of control) and increased the UGT1A4 activity (120% of control). Under the condition of inhibitors, we performed an inhibition study for haloperidol O-glucuronidation in HLM (Fig. 5B). At 25 μM haloperidol, the inhibitory effects of hecogenin and niflumic acid were minor (92 and 83% of control, respectively), but gemfibrozil substantially inhibited the activity (58% of control). The results suggest that the contribution of UGT2B7 may be predominant for haloperidol O-glucuronidation, supporting the results of the correlation analysis. When the haloperidol concentrations were 100 and 200 μM, although the inhibitory effects of hecogenin and niflumic acid were almost the same as that at 25 μM haloperidol, the inhibitory effects of gemfibrozil were diminished (68 and 71% of control, respectively). It was implied that the contribution of UGT2B7 might decrease with increased haloperidol concentration.

Inhibition studies of haloperidol O-glucuronidation by recombinant UGTs (A) and HLM (B). The effect of hecogenin (10 μM), niflumic acid (10 μM), and gemfibrozil (10 μM) on haloperidol O-glucuronidation by recombinant UGT1A4, UGT1A9, and UGT2B7 was evaluated at 25 μM substrate concentration. Using these inhibitors, we investigated the inhibitory effects on the haloperidol O-glucuronidation in HLM. Haloperidol (25, 100, and 200 μM) was incubated with pooled HLM in the absence or presence of hecogenin (10 μM), niflumic acid (10 μM), and gemfibrozil (10 μM). Each column represents the mean of duplicate determinations.
Estimation of the Percentage Contributions of UGT1A4, UGT1A9, and UGT2B7 to Haloperidol O-Glucuronidation in HLM Using RAF.
We sought to estimate the percentage contributions of UGT1A4, UGT1A9, and UGT2B7 to the haloperidol O-glucuronidation in HLM using RAF (Fig. 6; Table 2). For this analysis, we randomly selected 6 of 17 HLM. Trifluoperazine N-glucuronidation in the 6 HLM ranged from 0.258 to 0.729 nmol · min−1 · mg−1, and that in recombinant UGT1A4 was 0.702 nmol · min−1 · mg−1. Thus, RAFUGT1A4 was estimated to range from 0.66 to 1.85. Propofol O-glucuronidation in the 6 HLM ranged from 2.07 to 5.89 nmol · min−1 · mg−1, and that in recombinant UGT1A9 was 12.3 nmol · min−1 · mg−1. Thus, RAFUGT1A9 was estimated to range from 0.17 to 0.48. Zidovudine O-glucuronidation in the 6 HLM ranged from 0.145 to 0.290 nmol · min−1 · mg−1, and that in recombinant UGT2B7 was 0.226 nmol · min−1 · mg−1. Thus, RAFUGT2B7 was estimated to range from 0.64 to 1.29. With these RAF values, the percentage contributions of UGT1A4, UGT1A9, and UGT2B7 to haloperidol O-glucuronidation in human liver microsomes were estimated as described under Materials and Methods. At a 25 μM haloperidol concentration, the contributions of UGT1A4, UGT1A9, and UGT2B7 were calculated to be 13.7 ± 6.1, 20.9 ± 11.3, and 83.2 ± 7.2%, respectively (mean ± S.D. of 6 HLM). At a 100 μM haloperidol concentration, the contributions of UGT1A4, UGT1A9, and UGT2B7 were calculated to be 18.9 ± 6.6, 27.6 ± 10.6, and 66.5 ± 4.2%, respectively. At a 200 μM haloperidol concentration, the contributions of UGT1A4, UGT1A9, and UGT2B7 were calculated to be 19.2 ± 6.6, 23.4 ± 10.1, and 42.6 ± 5.3%, respectively. These results suggest that UGT2B7 is the major enzyme responsible for haloperidol O-glucuronidation in human liver. Interestingly, we noticed that the interindividual differences in activities as well as percentage contributions of each UGT isoform were not large.

The percentage contributions of UGT1A4, UGT1A9, and UGT2B7 to haloperidol O-glucuronidation in six HLM at substrate concentrations of 25 μM (A), 100 μM (B), and 200 μM (C). The prediction method was based on RAF, as described under Materials and Methods. Each column represents the mean of duplicate determinations.
Contribution of UGT1A4, UGT1A9, and UGT2B7 to haloperidol O-glucuronidation in HLM
Discussion
Glucuronidation accounts for 50 to 60% of the metabolic pathways of haloperidol in humans. The potency of glucuronidation would be a factor controlling the plasma concentration of haloperidol and thereby the clinical efficacy and adverse effects. The purpose of this study was to identify the human UGT isoforms responsible for haloperidol glucuronidation to understand the causes of interindividual variability in the plasma concentration and drug interactions.
We found that two glucuronides, which could be O-glucuronide and N-glucuronide, were formed by the incubation of haloperidol with HLM and UDPGA. To determine which peak is for O-glucuronide or N-glucuronide, we used a feature of UGT1A4; that is, human UGT1A4 prefers tertiary amines as substrates (Green and Tephly 1996; King et al., 2000) and rat UGT1A4 is not functional (Mackenzie et al., 2005). The result that metabolite B was formed by UGT1A4, but not by UGT1A9, UGT2B7, or RLM, led us to conclude that metabolite B would be the N-glucuronide. As a supportive finding, we demonstrated in a previous study that trifluoperazine N-glucuronidation, a specific activity of human UGT1A4, was not detected by RLM (Shiratani et al., 2008). In addition, the finding that metabolite A formation was much higher than metabolite B formation also supported the assumption that metabolite A would be the O-glucuronide, based on previous studies reporting that the haloperidol O-glucuronide is a major metabolite (Oida et al., 1989; Luo et al., 1995). It was surprising that the haloperidol N-glucuronide was detected in this in vitro study, because the metabolite was not detected in urine samples from patients (Oida et al., 1989; Luo et al., 1995). Oida et al. (1989) reported that the haloperidol O-glucuronide is stable against enzymic degradation, probably because the bond is surrounded by three rings to sterically hinder the approach of β-glucuronidase. It seems that the stability of quaternary N-glucuronides under acidic conditions and susceptibility to β-glucuronidase vary among compounds (Hawes, 1998). A likely explanation for the discrepancy between in vivo and in vitro is that the formation of haloperidol N-glucuronide might be too low to be detected in urine or plasma or that the N-glucuronide might be unstable in urine.
We found that UGT1A4, UGT1A9, and UGT2B7, which are all expressed in human liver, have the catalytic activity for haloperidol O-glucuronidation (Fig. 3) with their CLint values of 9, 16, and 23 μl · min−1 · mg−1, respectively. However, because the absolute expression level of each UGT isoform in the recombinant system and HLM is not elucidated, we could never determine which UGT isoform is the major contributor from the values of intrinsic clearance of recombinant UGTs. Therefore, to evaluate the UGT isoform that is mainly responsible for the haloperidol O-glucuronidation, correlation analysis and an inhibition study using HLM were performed. In the correlation analysis, the haloperidol O-glucuronidation in HLM was significantly correlated with the zidovudine O-glucuronidation, which is a probe reaction of UGT2B7 (Table 1), suggesting the major contribution of UGT2B7. When a relatively higher concentration of haloperidol was used, the activity was also correlated with trifluoperazine N-glucuronidation and propofol O-glucuronidation, which are specifically catalyzed by UGT1A4 and UGT1A9, respectively, in addition to zidovudine O-glucuronidation. The results suggest the additional contribution of UGT1A4 and UGT1A9. However, when the three probe reactions were compared, a significant correlation was observed between the trifluoperazine N-glucuronidation and propofol O-glucuronidation (r = 0.518; p < 0.05) or zidovudine O-glucuronidation (r = 0.708; p < 0.01), as such a correlation beyond the UGT isoforms has been reported previously (Ramírez et al., 2008). Thus, a correlation analysis alone does not provide compelling evidence. However, the result of the inhibition study supplemented the results of the correlation study to suggest the major contribution of UGT2B7 and minor contribution of UGT1A4 and UGT1A9 to the haloperidol O-glucuronidation.
To extrapolate the data of recombinant enzymes to the activity in human liver, a concept of RAF was proposed for P450s (Crespi, 1995). We applied the RAF to quantitatively estimate the contributions of each UGT isoform to the haloperidol O-glucuronidation. For the calculation of the RAF, V, Vmax, or CLint values could be used. When we validated these RAFs using V, Vmax, or CLint to predict the contribution of cytochrome P450 isoforms in our previous study (Nakajima et al., 1999), we found that the RAF using Vmax or CLint resulted in the overestimation of the contribution of given enzyme, if the probe substrates used are not specific for the enzyme. The RAF using V at an appropriate substrate concentration could obtain a reasonable prediction. If the probe substrates used were specific, the results based on the RAFs using V, Vmax, or CLint were almost the same. In this study, we adopted the RAF using V, considering the relatively low specificity of UGT substrates. As the results, the contributions of UGT1A4, UGT1A9, and UGT2B7 to haloperidol O-glucuronidation with a substrate concentration of 25 μM were estimated to be 13.7, 20.9, and 83.2%, respectively, on average. With substrate concentrations of 100 and 200 μM, the contributions of UGT1A4, UGT1A9, and UGT2B7 were calculated to be 18.9 to 19.2%, 23.4 to 27.6%, and 42.6 to 66.5%, respectively, indicating the increasing contributions of UGT1A4 and UGT1A9.
If UGT2B7 and UG1A9, which showed substrate inhibition kinetics, were the main contributors to haloperidol O-glucuronidation in human liver, one would expect the kinetics in HLM to also show substrate inhibition kinetics. However, the kinetics of HLM followed the Michaelis-Menten equation. The inconsistency of the kinetic model between recombinant UGTs and HLM is no rare matter (Staines et al., 2004; Mano et al., 2006). In such cases, the substrate inhibition was observed in recombinant UGTs, but not in HLMs. Thus, it seems that the substrate inhibition kinetics is a unique feature of recombinant UGTs. Although the explanations for the differences in the kinetics of recombinant UGTs and HLMs are largely speculative, they may include 1) the difference in membrane compositions (lipid or fatty acids) of HLMs and recombinant UGTs, 2) the difference in the extent of post-translational modification (glycosylation or phosphorylation), and 3) the presence or absence of other UGTs (or enzymes). In addition, the difference in the kinetic models between the recombinant UGTs and HLM would explain why the sum of the percentage contributions of the three isoforms was less than 100% at higher substrate concentrations. However, we thought the estimation was successful on the whole because the results were consistent with the results of the correlation analysis and inhibition study.
We found no dramatic interindividual difference in the percentage contribution of the three UGT isoforms. In addition, the interindividual differences in the haloperidol glucuronidation were 3- to 4-fold, and those in trifluoperazine, propofol, and zidovudine glucuronidation representing UGT1A4, UGT1A9, and UGT2B7, respectively, were also 3- to 4-fold. It was demonstrated previously that the plasma haloperidol concentrations corrected by the daily dose and body weight showed 11-fold interindividual variability among 231 patients with schizophrenia (Hirokane et al., 1999). An in vitro study using nine HLM reported that the interindividual variability of the N-dealkylation of haloperidol catalyzed by CYP3A4 was 10-fold (Pan et al., 1997). Because the intrinsic clearance by glucuronidation is much greater than those by CYP3A4-mediated oxidations (50–60% versus 20–30% of the biotransformation) (Kudo and Ishizaki, 1999), the difference in the glucuronidation potency may also be one of the factors for the pharmacokinetics of haloperidol.
In conclusion, we thoroughly characterized the glucuronidation of haloperidol in HLM and found that the haloperidol O-glucuronidation, a major pathway, was mainly catalyzed by UGT2B7 and, to a minor extent, by UGT1A4 and UGT1A9, and the haloperidol N-glucuronidation, a minor pathway, was catalyzed solely by UGT1A4. The variability of the glucuronidation potency would be one of the factors for the interindividual difference in the plasma haloperidol concentration. Consideration of the inhibition or induction of these UGT enzymes might be informative to predict drug-drug interactions.
Authorship Contributions
Participated in research design: Kato, Nakajima, Oda, Fukami, and Yokoi.
Conducted experiments: Kato and Oda.
Contributed new reagents or analytic tools: Kato.
Performed data analysis: Kato, Nakajima, and Oda.
Wrote or contributed to the writing of the manuscript: Kato, Nakajima, and Yokoi.
Acknowledgments
We acknowledge Brent Bell for reviewing the manuscript.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
ABBREVIATIONS:
- UGT
- UDP-glucuronosyltransferase
- UDPGA
- UDP-glucuronic acid
- HLM
- human liver microsomes
- RLM
- rat liver microsomes
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- HPLC
- high-performance liquid chromatography
- RAF
- relative activity factor
- MS
- mass spectrometry
- MRM
- multiple reaction monitoring.
- Received August 4, 2011.
- Accepted October 25, 2011.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}