


Abstract
Substrates of aldehyde oxidase (AO), for which human clinical pharmacokinetics are reported, were selected and evaluated in pooled mixed-gender cryopreserved human hepatocytes in an effort to quantitatively characterize AO activity. Estimated hepatic clearance (Clh) for BIBX1382, carbazeran, O6-benzylguanine, zaleplon, and XK-469 using cryopreserved hepatocytes was 18, 17, 12, <4.3, and <4.3 ml · min−1 · kg−1, respectively. The observed metabolic clearance in cryopreserved hepatocytes was confirmed to be a result of AO-mediated metabolism via two approaches. Metabolite identification after incubations in the presence of H218O confirmed that the predominant oxidative metabolite was generated by AO, as expected isotope patterns in mass spectra were observed after analysis by high-resolution mass spectrometry. Second, clearance values were efficiently attenuated upon coincubation with hydralazine, an inhibitor of AO. The low exposure after oral doses of BIBX1382 and carbazeran (∼5% F) would have been fairly well predicted using simple hepatic extraction (fh) values derived from cryopreserved hepatocytes. In addition, the estimated hepatic clearance value for O6-benzylguanine was within ∼80% of the observed total clearance in humans after intravenous administration (15 ml · min−1 · kg−1), indicating a reasonable level of quantitative activity from this in vitro system. However, a 3.5-fold underprediction of total clearance was observed for zaleplon, despite the 5-oxo metabolite being clearly observed. These data taken together suggest that the use of cryopreserved hepatocytes may be a practical approach for assessing AO-mediated metabolism in discovery and potentially useful for predicting hepatic clearance of AO substrates.
Introduction
Aldehyde oxidase (AO) belongs to a family of enzymes referred to as molybdenum cofactor-containing enzymes that also includes xanthine oxidase. This drug-metabolizing enzyme is active as a homodimer composed of two identical ∼150-kDa subunits and demonstrates broad substrate selectivity, oxidizing a variety of aldehydes and heterocyclic-containing drug molecules (Beedham, 1987; Kitamura et al., 2006; Garattini et al., 2008). Of the known substrates of AO, it is heterocyclic ring-containing molecules that are of highest interest, because medicinal chemists commonly introduce these particular ring systems into chemical scaffolds in an effort to optimize toward improved solubility and lower lipophilicity, which, in general, also yields reduced metabolism by cytochrome P450 enzymes. This chemistry strategy, while generally successful, may ultimately lead to alternative metabolic clearance mechanisms such as those catalyzed by AO.
Aldehyde oxidase has numerous features that make studying this enzyme challenging in a preclinical setting. First, AO is present in the cytosolic fraction, and thus standard metabolic stability studies using liver microsomes do not capture AO-mediated metabolism. If one is not cautious, then this metabolic pathway could be, and in fact has been, completely overlooked (vida infra). Additional intricacies of this enzyme include apparent donor-to-donor variability in activity in humans (Al-Salmy, 2001; Sahi et al., 2008) and across preclinical species and strains, where rats generally have low activity and dogs are completely devoid of activity (Beedham et al., 1987; Garattini et al., 2008). Interest in this drug-metabolizing enzyme by the pharmaceutical industry has increased recently (Pryde et al., 2010; Garattini and Terao, 2011), with the recognition that extensive AO metabolism has led to severe clinical implications, either because of toxicological outcomes (Diamond et al., 2010) or higher-than-predicted clearance in humans (Kaye et al., 1984; Rosen et al., 1999; Dittrich et al., 2002; Akabane et al., 2011; Zhang et al., 2011), yielding unacceptable pharmacokinetic properties. In most of these cases, either metabolic pathways were only evaluated in liver microsomes or predictions of human clearance were extrapolated from preclinical species that possessed significantly lower AO activity compared with humans.
Scaling in vitro metabolism data to predict metabolic clearance is a critical component for half-life estimation and, ultimately, efficacious dose projections for new chemical entities moving toward clinical development. Scaling of in vitro metabolism data is a common approach for drug molecules that are primarily metabolized and cleared by cytochrome P450-mediated metabolic pathways (Houston, 1994; Obach et al., 1997), with numerous examples of successful predictions (Obach, 2000; Hutzler et al., 2010). However, collectively, there is inadequate information about the ability to scale in vitro clearance for compounds that are substrates of AO using hepatocytes, and assessment of AO metabolic activity is not included in characterization sheets when hepatocytes are received from vendors. To our knowledge, only one study has evaluated the ability to scale in vitro AO metabolism data using cytosol and S-9 fraction (Zientek et al., 2010), and underpredictions of free intrinsic clearance were noted. In addition, questions persist about the reported instability of the AO enzyme (Duley et al., 1985; Al-Salmy, 2001), which calls into question whether AO activity in current in vitro systems often used for scaling (e.g., cryopreserved hepatocytes) is representative of the in vivo situation. It also seems plausible that different proprietary hepatocyte isolation procedures by vendors may compromise AO activity. In an effort to address this current knowledge gap, the objective of this work was to evaluate and compare AO activity in cryopreserved hepatocytes from two different vendors using substrates that are reported to be primarily cleared by AO-mediated metabolism (Fig. 1), and for which human clinical pharmacokinetics have been reported. In vitro-in vivo correlations for AO activity following standard scaling approaches from cryopreserved hepatocytes are reported herein.

Structures of aldehyde oxidase substrates. Arrows indicate the major site of metabolism.
Materials and Methods
Chemicals.
Potassium phosphate buffer, O6-benzylguanine, XK-469, zaleplon, hydralazine, midazolam, propranolol, NADPH, and 18O-water (97% purity) were purchased from Sigma-Aldrich (St. Louis, MO). BIBX1382, BIBU1476, and carbazeran were acquired from the internal compound library at Boehringer Ingleheim USA (Ridgefield, CT). Pooled human intestinal cytosol was purchased from Celsis/In Vitro Technologies (Baltimore, MD), and cryopreserved human hepatocytes along with hepatocyte medium and reagents were purchased from both Celsis/In Vitro Technologies and Invitrogen (Carlsbad, CA). All other reagents and chemicals were of the highest purity available.
Hepatocyte Incubations.
Pooled mixed-gender cryopreserved human hepatocytes were obtained from Celsis/In Vitro Technologies (lot number OOO; 19 donors) and Life Technologies, Inc. (lot number HuE104; 10 donors) and stored in liquid nitrogen until use. Immediately before experiments, sufficient aliquots of hepatocytes were thawed rapidly (∼2 min) in a shaking water bath at 37°C. The contents of each vial were diluted 1:50 in prewarmed (37°C) cryopreserved hepatocyte recovery medium (lot HuE104; Life Technologies, Inc.) or Williams' Medium E (WME; lot OOO, pH 7.4) and gently mixed before centrifugation at 100g for 6 min at room temperature. After centrifugation, the supernatant was discarded, the hepatocyte pellet was resuspended in WME to provide 1 × 10e6 cells/ml by repeated gentle inversion in a capped tube, and the cell number and viability were determined using a hemocytometer after staining with trypan blue. Viabilities for each hepatocyte experiment were at least 80%. The cell suspension was diluted in WME to give two times the incubation concentration and prewarmed at 37°C for 15 min. Solutions of substrates (10% acetonitrile in H2O) were diluted in prewarmed WME (giving an incubation concentration of organic solvent ≤0.1%). Incubations (performed in quadruplicate) were initiated by the addition of substrate solution (250 μl) to the hepatocyte suspension (250 μl; 250,000 viable cells) in a 48-well tissue culture-treated polystyrene incubation plate (1-μM final substrate concentration), followed by gentle swirling in an atmosphere of 5% CO2 and 95% relative humidity (37°C). Incubations in the presence of the AO inhibitor hydralazine (50 μM) were conducted the same as described above, with hydralazine stock solutions prepared in water (10 mM). Reactions were terminated at 0, 5, 15, 30, 60, and 120 min by aliquoting 50 μl of incubate into 150 μl of ice-cold acetonitrile (0.1% acetic acid) containing an internal standard (0.25 μM tolbutamide). Quench plates were then centrifuged at 3000g (4°C) for 5 min, and supernatants were transferred to clean 96-well plates for LC/MS/MS bioanalysis.
Metabolite Profiling in Hepatocytes.
The metabolite profile of each AO substrate was also evaluated after incubation at 10 μM in cryopreserved human hepatocytes (lot OOO). Preparation of hepatocytes was similar to procedures described for in vitro clearance experiments, except that 2.0 × 10e6 cells/ml was the final concentration for metabolite generation. For incubations in the presence of 18O-water, substrate solutions were prepared in the 18O-water (250 μl), and 250 μl of cell suspension was added to initiate incubation (50% v/v, final). After a 120-min incubation in 24-well culture plates, reactions were terminated by the addition of 2 volumes of ice-cold acetonitrile, and the resulting mixture underwent centrifugation at 3000g (4°C) for 10 min. The supernatants were then transferred to clean glass test tubes and subsequently dried under a gentle stream of nitrogen (N2) gas. Dried samples were then reconstituted in 200 μl of mobile phase [85:15 (v/v) water/acetonitrile (0.1% formic acid)] and centrifuged again at 13,000g for 10 min before bioanalysis by high-resolution mass spectrometry.
Intestinal Cytosol Incubations.
Metabolic stability of carbazeran, BIBX1382, zaleplon, XK-469, and O6-benzylguanine was evaluated in human mixed-gender (six donors) intestinal cytosol in 96-well, deep-well assay plates. Incubations were conducted in 100 mM potassium phosphate buffer, pH 7.4, and 1 mg/ml intestinal cytosol with a final incubation volume of 0.4 ml. Reactions were initiated by addition of test compounds (1 μM; final organic, <0.1%). Aliquots (50 μl) were removed from the cytosol assay at 0, 5, 15, 30, 60, and 120 min, and added to 150 μl of ice-cold acetonitrile containing tolbutamide as an internal standard. Quenched samples were then centrifuged at 3000g (4°C) for 10 min to precipitate proteins, and the supernatant was transferred to clean 96-well plates for LC/MS/MS bioanalysis.
Liquid Chromatography-Mass Spectrometry Analysis.
Quantitation of all analytes was performed using an AB Sciex (Foster City, CA) API-5000 triple quadrupole mass spectrometer equipped with an electrospray ionization interface in positive ion mode and connected in-line to an Acquity UPLC system (Waters, Milford, MA). Separation of analytes was performed using a Waters Acquity BEH high-pressure C18 1.7 μm (2.1 × 50 mm) column held at 50°C. Mobile phase was flowed at 0.5 ml/min with a rapid gradient starting at 95% A (0.1% formic acid in water) and 5% B (0.1% formic acid in acetonitrile), held for 0.5 min, ramped linearly to 5:95 A/B, and held for 1.3 min, followed by returning to initial conditions (3 min total run time). The multiple reaction monitoring transitions for each analyte were as follows: carbazeran (m/z 360.9 > 272.1), BIBX1382 (m/z 388.0 > 98.0), zaleplon (m/z 306.2 > 236.2), XK-469 (m/z 345.0 > 273.0), O6-benzylguanine (m/z 242.0 > 199.1), and tolbutamide (m/z 271.3 > 91.1). Standard curves for each analyte were prepared with a concentration range of 0.003 to 2.0 μM, with a limit of quantitation of 0.008 μM. All data were analyzed using AB Sciex Analyst 1.4.2 software.
Metabolite profiling of in vitro samples after incubation with cryopreserved human hepatocytes was performed using a high-resolution LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA) connected to a Thermo Accela UPLC pump and CTC PAL auto-sampler (LEAP Technologies, Carrboro, NC). The Orbitrap was equipped with an electrospray ionization source in positive polarity and was calibrated using ProteoMass LTQ/FT-Hybrid Calibration Mix (Supelco, Bellefonte, PA) for mass accuracies <5 ppm in external calibration mode. The source voltage was set at 5 kV, tube lens voltage was set at 90 V, and capillary voltage was set at 5 V, with the capillary temperature set at 325°C. Sheath gas flow was at 70 U, aux gas was at 15 U, and sweep gas flow was at 5. Analytes were loaded (20 μl injection) onto a Supelco Discovery C18 column (5 μm, 2.1 × 150 mm; Supelco) and separated using a gradient elution profile consisting of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). The gradient was initiated at a 95% A/5% B ratio at a flow rate of 0.4 ml/min, held for 5 min, ramped linearly from 5% B to 48% B over 10 min, then to 95% B over the next minute, and held for 2 min. The gradient was then returned to initial conditions, and the column was allowed to re-equilibrate for 3 min (total run time, 22 min). The high-performance liquid chromatography eluent was introduced first into a photodiode array detector set at 254 nm and subsequently via electrospray ionization directly into the Orbitrap. Mass spectrometry analyses were carried out using full-scan MS with a mass range of 100 to 1000 Da, and MS/MS spectra were collected in a data-dependent fashion with relative collision energies of 35% and collision-induced dissociation. Resolution was set at 30,000 for Fourier Transform MS and 15,000 for Fourier transformation MS/MS. All mass spectral data were analyzed using Xcalibur version 2.1 software (Thermo Fisher Scientific), and chemical structures and calculated exact mass estimates for fragments were generated using ChemDraw Ultra version 12 software (CambridgeSoft Corporation, Cambridge, MA).
Hepatic Clearance Estimates.
In vitro intrinsic clearance (Clint) was estimated from cryopreserved human hepatocytes using substrate depletion methodologies and eq. 1:
 where t1/2 is the observed in vitro substrate depletion half-life (in minutes) and 25.7 g liver weight/kg b.wt. and 120 × 10e6 hepatocytes per gram liver were used as scaling factors. Hepatic clearance (Clh) was predicted using Clint values and the well stirred model according to eq. 2:
where t1/2 is the observed in vitro substrate depletion half-life (in minutes) and 25.7 g liver weight/kg b.wt. and 120 × 10e6 hepatocytes per gram liver were used as scaling factors. Hepatic clearance (Clh) was predicted using Clint values and the well stirred model according to eq. 2:
 where Qh is human hepatic blood flow (20.7 ml · min−1 · kg−1), fu is fraction unbound in plasma, and Clint is in vitro intrinsic clearance derived from in vitro experiments in hepatocytes. Hepatic extraction ratio (Eh) is a measure of the liver's ability to extract drug before reaching the systemic circulation and can be estimated using eq. 3:
where Qh is human hepatic blood flow (20.7 ml · min−1 · kg−1), fu is fraction unbound in plasma, and Clint is in vitro intrinsic clearance derived from in vitro experiments in hepatocytes. Hepatic extraction ratio (Eh) is a measure of the liver's ability to extract drug before reaching the systemic circulation and can be estimated using eq. 3:
 where Clh is hepatic clearance estimated from in vitro metabolic stability data from cryopreserved hepatocytes and Qh is hepatic liver blood flow (20.7 ml · min−1 · kg−1 in human). Oral bioavailability may be estimated using eq. 4: F = Fabs × Fg × Fh, where Fabs is the fraction of the dose that is absorbed from the intestinal lumen to the enterocytes, Fg is the fraction of the dose that escapes intestinal first-pass metabolism, and Fh is the fraction of the dose that passes through the liver without being metabolized, also estimated by 1 − Eh. Assuming complete absorption (Fabs = 1), and limited intestinal metabolism (Fg = 1), oral bioavailability would be comparable to Fh (F ≈ Fh). Thus, oral bioavailability was estimated using the following relationship (eq. 5): F = 1 − Eh.
where Clh is hepatic clearance estimated from in vitro metabolic stability data from cryopreserved hepatocytes and Qh is hepatic liver blood flow (20.7 ml · min−1 · kg−1 in human). Oral bioavailability may be estimated using eq. 4: F = Fabs × Fg × Fh, where Fabs is the fraction of the dose that is absorbed from the intestinal lumen to the enterocytes, Fg is the fraction of the dose that escapes intestinal first-pass metabolism, and Fh is the fraction of the dose that passes through the liver without being metabolized, also estimated by 1 − Eh. Assuming complete absorption (Fabs = 1), and limited intestinal metabolism (Fg = 1), oral bioavailability would be comparable to Fh (F ≈ Fh). Thus, oral bioavailability was estimated using the following relationship (eq. 5): F = 1 − Eh.
Results
Hepatocyte Intrinsic Clearance.
BIBX1382, carbazeran, O6-benzylguanine, zaleplon, and XK-469 were incubated in pooled mixed-gender cryopreserved human hepatocytes from two different vendors to estimate hepatic clearance and quantitatively evaluate AO activity by comparing with known human pharmacokinetic data (Table 1). First, there seemed to be negligible differences in activity in the two lots of hepatocytes tested, because predicted hepatic clearance values (in milliliters per minute per kilogram) for each of the substrates tested were within 87% of each other. In addition, data among the four replicates displayed high consistency with low S.D.s (Table 1). BIBX1382 and carbazeran both displayed high predicted hepatic clearances of 17 to 18 ml · min−1 · kg−1, which equates to 84 to 88% of human liver blood flow (20.7 ml · min−1 · kg−1). This is consistent with what has been observed clinically in humans after intravenous and/or oral dosing (e.g., high clearance and low oral bioavailability). Hepatic clearance of O6-benzylguanine ranged from 11.2 to 12.8 ml · min−1 · kg−1 in cryopreserved hepatocytes, which is 72 to 82% of the observed total clearance reported in humans (15.6 ml · min−1 · kg−1). Zaleplon was a substrate for which low rates of metabolism were observed in intrinsic clearance studies (<4.3 ml · min−1 · kg−1), despite the fact that it is a moderate to high clearance drug in humans with a reported total clearance of 15.7 ml · min−1 · kg−1 and oral bioavailability of 30% (Table 1). In our intrinsic clearance studies in cryopreserved hepatocytes, maximum incubation times were 120 min, and thus an upper limit of 790 min was selected as the threshold for how far an estimated half-life could be extrapolated beyond the length of the incubation, assuming 10% bioanalytical variability. This half-life equates to a predicted hepatic clearance of 4.3 ml · min−1 · kg−1, and thus if the half-life was >790 min, then the predicted hepatic clearance is reported as <4.3 ml · min−1 · kg−1. The observed in vitro-in vivo disconnect for zaleplon remains unresolved. XK-469 is a low-clearance drug in humans, and this is reflected in our studies, with predicted hepatic clearance values <4.3 ml · min−1 · kg−1 (Table 1). Data confirming that the observed intrinsic clearance in cryopreserved hepatocytes is mediated by AO are reported below.
Summary of predicted hepatic clearance of aldehyde oxidase substrates in cryopreserved human hepatocytes compared with reported human pharmacokinetic properties
Hepatic clearance estimates (ml · min−1 · kg−1) were calculated as described under Materials and Methods. Data are presented as mean (S.D.) from n = 4 replicates.
Intestinal Cytosol.
Incubations of each AO substrate with human intestinal cytosol resulted in no measurable metabolism as determined by substrate depletion methodologies (data not shown). After oral dosing, assuming that fraction absorbed (fa) is equal to 1 (for top limit estimate), and limited intestinal metabolism (fg = 1), the predicted oral bioavailability using hepatic clearance (Clh) estimates and eq. 5 for BIBX1382, carbazeran, and zaleplon (dosed orally in the clinic) is ∼13, 15, and >79%, respectively (Table 1).
Metabolite Profiling.
The LC-mass spectrometry data demonstrating the metabolite profiles of AO substrates after incubation with cryopreserved human hepatocytes are shown in Fig. 2. For each substrate, the AO-generated metabolite was most predominant. The metabolites generated by aldehyde oxidase for the tested AO substrates in these studies have previously been characterized and reported, with the exception of BIBX1382. Thus, this is the first reported characterization of the AO-mediated metabolism of BIBX1382, where in vitro-generated metabolite has been demonstrated as being produced by aldehyde oxidase, matching that of the synthetically prepared metabolite (BIBU1476). Because authentic standard metabolites of the other tested AO substrates were not acquired, a thorough characterization was performed, including incubation in the presence of ∼50% H218O (see Fig. 4) in an effort to confirm oxidation by molybdenum hydroxylases and not cytochrome P450. In theory, the maximal percentage incorporation of 18O would be 48.5% (incubations conducted in 50:50 mixtures of H216O and H218O, and H218O of 97% purity). In addition, MS/MS fragmentation using high-resolution mass spectrometry was acquired for each metabolite of interest (supplemental data).
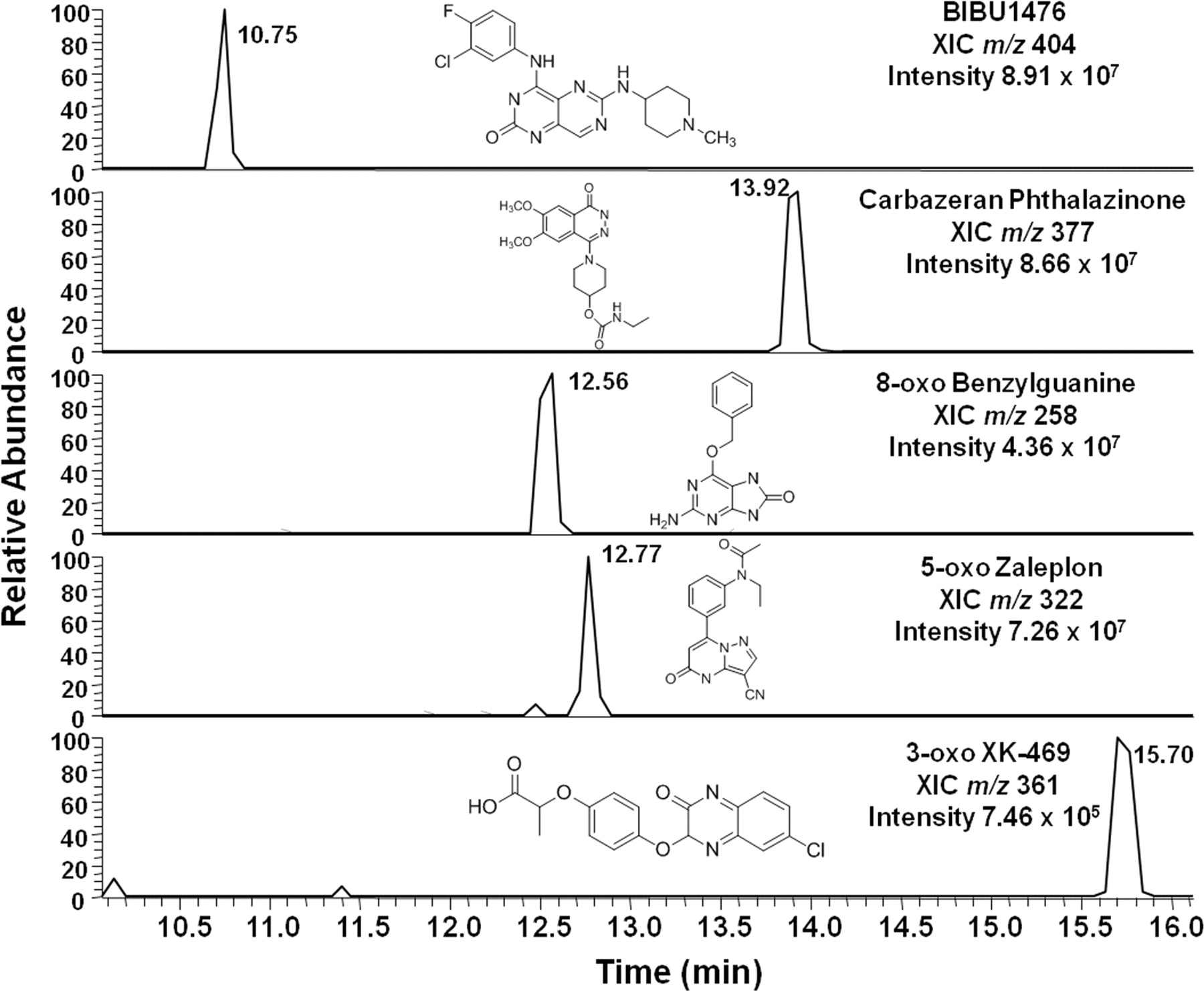
Representative LC-mass spectrometry XICs of aldehyde oxidase-mediated metabolites after incubation of 10 μM substrate in cryopreserved human hepatocytes (lot OOO).
BIBX1382.
Figure 3A shows the LC-MS extracted ion chromatogram (XIC) of the hepatocyte-generated metabolite of BIBX1382 (m/z 404), with the retention time (10.7 min) matching that of the synthetically prepared authentic standard metabolite, BIBU1476. The protonated molecular ion of BIBX1382 was observed at [M+H]+ at m/z 388.14 with a retention time of 12.5 min. Fragmentation of BIBX1382 produced three predominant fragment ions at m/z 357.1028, m/z 345.1027, and m/z 291.0559, which all correspond to daughter ions after fragmentation of the methyl-piperidine moiety (Fig. 3B). For the predominant oxidized metabolite of BIBX1382, which had a retention time of 10.7 min and produced a protonated molecular ion at [M+H]+ at m/z 404.14 (BIBU1476), each of the aforementioned fragment ions shifted 16 Da to m/z 373.0976, m/z 361.0978, and m/z 307.0508, respectively (Fig. 3B). In addition, the fragmentation pattern of the metabolically generated oxidative metabolite of BIBX1382 was consistent with the synthetically prepared standard BIBU1476 (data not shown). Incorporation of 18O into the product metabolite was observed after hepatocyte incubations, as evidenced by the presence of the molecular ion at m/z 406.1432 (68% relative abundance), which is two mass units higher than the metabolite molecular ion at m/z 404.1398 (Fig. 4A). Incorporation of 18O was approximately 33% (48.5% × 0.68). The resolution of the Orbitrap was increased to 100,000 to derive molecular formulas, distinguish the mass difference between the 37Cl isotope and the 18O isotope, and ensure accuracy in the incorporation calculation (Supplemental Fig. 1).

A, XIC showing the metabolite of BIBX1382 generated after incubation in cryopreserved human hepatocytes matching that of the authentic standard (BIBU1476). B, spectra of product ions obtained by mass fragmentation of BIBX1382 at m/z 388 (top) and the aldehyde oxidase-mediated metabolite BIBU1476 at m/z 404 (bottom). RT, retention time; NL, normalized level; ESI, electrospray ionization.

High-resolution mass spectra of aldehyde oxidase metabolites generated in cryopreserved human hepatocytes after incubation in 48.5% H218O with BIBX1382 (A), carbazeran (B), O6-benzylguanine (C), zaleplon (D), XK-469 (E), and midazolam (negative control; F) demonstrating a characteristic isotope pattern after incorporation of oxygen from water into the product metabolites. The insets demonstrate spectra from incubations lacking 18O-water. ESI, electrospray ionization. F:FTMS, Fourier Transform mass spectrometry; NL, normalized level.
Carbazeran.
The protonated molecular ion of carbazeran was observed at [M+H]+ at m/z 361 with a retention time of 10.9 min. Fragmentation of carbazeran produced a predominant fragment ion at m/z 272.1395, corresponding to loss of the ethylcarbamate moiety (−89 Da) (Supplemental Fig. 2). The known predominant oxidized metabolite of carbazeran (phthalazinone) had a retention time of 13.9 min and produced a protonated molecular ion at [M+H]+ at m/z 377. Fragmentation of this oxidative metabolite yielded a predominant fragment ion at m/z 288.1347, 16 Da higher than the key fragment observed in the parent spectrum. Incorporation of 18O into the phthalazinone metabolite of carbazeran after incubation in hepatocytes was apparent (∼47.5% incorporation), with a characteristic isotope pattern showing molecular ions at m/z 377.1817 (+16 Da) and 379.1858 (+18 Da) at approximately equivalent abundances (Fig. 4B).
O6-Benzylguanine.
The protonated molecular ion of O6-benzylguanine was observed at [M+H]+ at m/z 242 with a retention time of 10.5 min. Fragmentation of O6-benzylguanine generated a product ion at m/z 225.0772, corresponding to loss of the amino moiety (−17 Da) (Supplemental Fig. 3). A second diagnostic fragment ion was observed at m/z 91.0539, corresponding to the remaining methyl-benzene moiety. The oxidized metabolite of O6-benzylguanine (8-oxo benzylguanine) had a retention time of 12.5 min, produced a protonated molecular ion at [M+H]+ at m/z 258, and upon fragmentation yielded a fragment ion m/z 241.0722 that was 16 Da higher than what was observed in the parent spectrum. Remaining intact was the fragment ion observed at m/z 91.0539. Incorporation of 18O into the 8-oxo metabolite of O6-benzylguanine after incubation in hepatocytes was observed (∼42% incorporation), with a characteristic isotope pattern showing molecular ions at m/z 258.0987 (+16 Da) and m/z 260.1028 (+18 Da) (Fig. 4C).
Zaleplon.
The protonated molecular ion of zaleplon was observed at [M+H]+ at m/z 306 with a retention time of 14.5 min. Fragmentation of zaleplon produced three major product ions at m/z 264.1248, m/z 288.1248, and m/z 236.0934, all corresponding to ions generated by fragmentation of the ethylacetamide moiety (Supplemental Fig. 4). The oxidized metabolite of zaleplon (5-oxo zaleplon) had a retention time of 12.7 min and produced a protonated molecular ion at [M+H]+ at m/z 322. Upon fragmentation of this metabolite, the three aforementioned fragment ions in the parent spectrum shifted 16 Da to m/z 280.1196, m/z 304.1197, and m/z 252.0884, respectively. Incorporation of 18O into the 5-oxo metabolite of zaleplon after incubation in hepatocytes was observed (∼40% incorporation), with a characteristic isotope pattern showing molecular ions at m/z 322.1300 (+16 Da) and m/z 324.1343 (+18 Da) (Fig. 4D).
XK-469.
The protonated molecular ion of XK-469 was observed at [M+H]+ at m/z 345 with a retention time of 17.4 min. Fragmentation of XK-469 produced a predominant fragment ion at m/z 299.0585, corresponding to loss of the acid moiety (−46 Da) (Supplemental Fig. 5). The oxidized metabolite of XK-469 (3-oxo XK-469) had a retention time of 15.7 min and produced a protonated molecular ion at [M+H]+ at m/z 361. Upon fragmentation of this metabolite, a predominant fragment ion was observed at m/z 315.0533, or 16 Da higher than what was observed in the parent spectrum. Incorporation of 18O into the 3-oxo metabolite of XK-469 after incubation in hepatocytes was observed (∼50% incorporation), with a characteristic isotope pattern showing molecular ions at m/z 361.0590 (+16 Da) and m/z 363.0631 (+18 Da) (Fig. 4E). Figure 4F shows the MS/MS spectra of the 1-hydroxy metabolite of midazolam as a negative control. As expected, incorporation of 18O into the 1-hydroxy metabolite was not observed, consistent with a cytochrome P450-mediated oxidative mechanism where the source of oxygen in product comes from molecular oxygen.
Phenotyping Using Hydralazine.
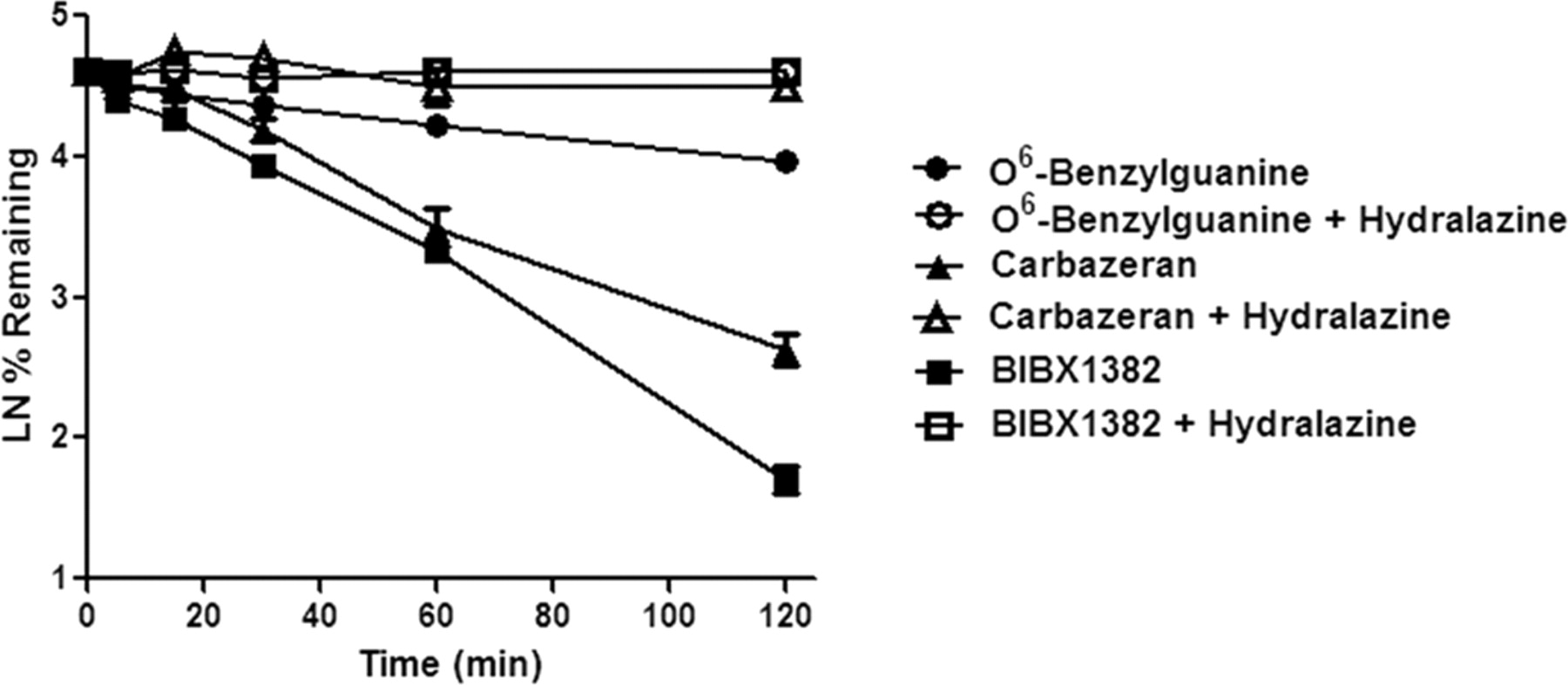
Hydralazine is a known inhibitor of AO and thus was used in these studies for phenotyping to confirm that the observed metabolic activity in cryopreserved hepatocytes was predominantly mediated by AO. Unfortunately, the metabolic turnover of both zaleplon and XK-469 was too low in our in vitro system to enable this conclusion when coincubated with hydralazine. However, upon coincubation of BIBX1382, carbazeran, and O6-benzylguanine with hydralazine (50 μM), metabolic clearance was substantially attenuated (Fig. 5). Predicted hepatic clearance (Clh) values for O6-benzylguanine, carbazeran, and BIBX1382 was reduced from 12.4, 17.2, and 18.2 ml · min−1 · kg−1 to <4.3, <4.3, and 7 ml · min−1 · kg−1, respectively, when coincubated with hydralazine. Meanwhile, when the cytochrome P450 substrate propranolol was incubated in the presence of hydralazine, negligible inhibition of metabolism was observed (data not shown).

Metabolic stability of AO substrates O6-benzylguanine (circles), carbazeran (triangles), and BIBX1382 (squares) in human cryopreserved hepatocytes (lot HuE104) in the absence (filled symbols) and in the presence (open symbols) of the AO inhibitor hydralazine (50 μM). Predicted hepatic clearance (Clh) values for O6-benzylguanine, carbazeran, and BIBX1382 was attenuated from 12.4, 17.2, and 18.2 to <4.3, <4.3, and 7 ml · min−1 · kg−1, respectively, when coincubated with hydralazine.
Discussion
AO is a cytosolic drug-metabolizing enzyme that recently has been highlighted as having relevance to drug discovery and development (Pryde et al., 2010; Garattini and Terao, 2011). The ideal in vitro system for comprehensively evaluating drug metabolism is, theoretically, hepatocytes, which contain the full complement of drug-metabolizing enzymes and are readily available from numerous vendors. However, although AO activity has been demonstrated in cryopreserved human hepatocytes (Sahi et al., 2008), an appropriate characterization of quantitative AO activity has not been reported. Thus, a number of probe substrates of AO were selected for investigational studies to evaluate this activity.
BIBX1382 was a drug candidate evaluated in the clinic as an inhibitor of epidermal growth factor receptor for the treatment of cancer (Solca et al., 2004). In human liver microsomes, it was observed that BIBX1382 was relatively stable and metabolized by CYP2D6 (Dittrich et al., 2002). In addition, in preclinical pharmacokinetic evaluations in rats and mice, oral bioavailability ranged between 50 and 100%. After oral dosing to human patients, plasma levels were far below that expected to be efficacious (∼5% average absolute oral bioavailability), and a previously uncharacterized metabolite was observed in the urine of one patient that was also circulating in human plasma at high concentrations (Dittrich et al., 2002). Retrospective experiments by Dittrich et al. (2002) suggested that BIBX1382 was metabolized by hepatic AO, although details were not reported. In these studies, BIBX1382 was confirmed to be stable in human liver microsomes but highly unstable in human liver cytosol, with the AO inhibitor raloxifene completely eliminating metabolism (Supplemental Fig. 6). In addition, we demonstrate that estimated hepatic clearance of BIBX1382 in cryopreserved hepatocytes is high (17.6–18.2 ml · min−1 · kg−1), a clearance that approaches human liver blood flow. The predominant metabolite after incubation of BIBX1382 in cryopreserved human hepatocytes had a retention time (Fig. 3) and MS/MS fragmentation pattern matching that of the authentic standard metabolite, BIBU1476, with oxidation occurring on the pyrimido-pyrimidine core. Incorporation of 18O-oxygen was observed after hepatocyte incubations (Fig. 4A), supporting an AO-mediated mechanism. Total clearance of BIBX1382 after intravenous dosing in humans is reported to be high (25–55 ml · min−1 · kg−1), which exceeds liver blood flow and suggests that the potential exists for either extrahepatic clearance or some other phenomena such as partitioning into blood cells. However, if the predicted hepatic clearance obtained from cryopreserved hepatocytes (Clh = 17.6–18.2 ml · min−1 · kg−1) was used to estimate oral bioavailability using eqs. 3 and 5 (assuming fabs and fg = 1), then low oral exposure (12–15% F; Table 1) would have been predicted after first-pass extraction (actual range was 1.8–12.3%). Meanwhile, no measurable metabolism was observed in human intestinal cytosol, suggesting that AO activity in the gut would play a minor role in impacting oral absorption.
The inotropic agent carbazeran was previously reported to be almost completely cleared presystemically in human via 4-hydroxylation to the phthalazinone metabolite by AO (Kaye et al., 1984), whereas bioavailability in dog was ∼68%, with the predominant metabolic pathway being O-demethylation (Kaye et al., 1985). Carbazeran demonstrated a similar profile to BIBX1382, with high clearance observed in cryopreserved hepatocytes (∼17.5 ml · min−1 · kg−1). The predominant oxidative metabolite observed after metabolite profiling eluted at 13.9 min and had a protonated molecular ion at [M+H]+ at m/z 377 (Fig. 2). In addition, incubations in the presence of 18O-water lead to incorporation of 18O as evidenced by a characteristic mass spectral isotope pattern with approximately equivalent abundance of ions at m/z 377.1817 and m/z 379.1858 (Fig. 4B). Similar to BIBX1382, if calculated hepatic clearance from cryopreserved hepatocytes is used to estimate oral bioavailability, then low oral exposure (∼15% F) would be predicted (Table 1). This is a slight underprediction from what was observed in humans (<5% F), which suggests that there may be other mechanisms contributing to low oral exposure of carbazeran (e.g., absorption), or that AO activity in cryopreserved hepatocytes may be modestly impacted during the process of homogenization of liver, harvesting of cells, and cryopreservation. This is currently being investigated in our laboratory and will be reported in due course. One parameter that has not been included in this analysis is the effect of plasma protein binding on predicted hepatic clearance. In previous studies by Zientek et al. (2010), in which scaling of in vitro clearance from human liver cytosol and S-9 fraction was evaluated, clearance values were corrected for free fraction. However, as a general rule, the hepatic clearance for high-extraction-ratio drugs should be primarily driven by liver blood flow and not by fraction unbound to plasma proteins (e.g., Clh = Qh). The efficient hepatic extraction of BIBX1382 and carbazeran after oral dose (e.g., ≤5% F) demonstrates this concept.
O6-Benzylguanine was reported to be rapidly metabolized in human liver cytosol by AO to the O6-benzyl-8-oxoguanine metabolite (Roy et al., 1995). Subsequently, O6-benzylguanine was shown to be converted primarily by metabolism to the 8-oxo metabolite after intravenous administration to cancer patients (Dolan et al., 1998). Total clearance reported following intravenous dosing was 15.6 ml · min−1 · kg−1. Given that the 8-oxo metabolite was observed in plasma of patients at levels far exceeding the parent drug levels (2.2-fold higher Cmax and 12- to 29-fold higher area under the curve), and that urinary excretion of O6-benzylguanine only accounted for 3.2% of the administered dose in humans (Dolan et al., 1998), one can assume that the AO-mediated pathway contributes primarily to the observed clearance. When incubated in cryopreserved human hepatocytes, hepatic clearance values ranged from 11.2 to 12.8 ml · min−1 · kg−1, representing 72 to 82% of the observed total clearance in humans. On the basis of these findings, we conclude that approximately quantitative AO activity is apparent in cryopreserved hepatocytes and that scaling clearance to humans seems reasonable.
One of the more perplexing findings from these studies was that zaleplon, despite demonstrating moderate to high clearance in humans (15.7 ml · min−1 · kg−1) and 30% oral bioavailability (Rosen et al., 1999), had low intrinsic clearance in hepatocytes (Table 1). Because of the clearance observed with BIBX1382, carbazeran, and O6-benzylguanine, it is unlikely that the low intrinsic clearance of zaleplon is due to compromised AO activity in the hepatocytes. In addition, the AO-derived metabolite of zaleplon (5-oxo) was clearly observed and characterized in our analyses (Fig. 2), with incorporation of 18O (Fig. 4D), consistent with previous reports regarding AO-mediated metabolism of zaleplon (Lake et al., 2002). For an unknown reason, the AO-mediated metabolism is not reflected in intrinsic clearance. Saturation of the AO enzyme at only 1 μM is highly unlikely, because the reported Km for the 5-oxo pathway in human liver cytosol is 124 μM (Renwick et al., 2002). In addition, intestinal metabolism of zaleplon was not observed in our studies. In short, zaleplon was investigated and shown to have high permeability in Caco-2 cells (data not shown), suggesting that access to the hepatocyte was also not likely to be a limitation. Additional studies would have to be performed to understand the lack of correlation with the observed clearance of zaleplon in humans.
XK-469 is an antitumor agent that was reported to be metabolized by AO, with the 3-oxo metabolite being the predominant metabolite in both urine of dosed patients and in human hepatocytes (Anderson et al., 2005). However, upon dosing in humans, low clearance was observed (Alousi et al., 2007). Results from our studies using cryopreserved hepatocytes were consistent with these reports. We clearly observed an AO-mediated metabolite (Fig. 2), with incorporation of 18O (Fig. 4E), but in vitro clearance values were extremely low (<4.3 ml · min−1 · kg−1). It was somewhat expected that we would not be able to adequately predict the low observed clearance of XK-469, and this points to the fact that additional hepatocyte systems, such as plated hepatocytes, need to be investigated where longer incubations can be conducted to enable predicting clearance for slowly metabolized drugs, an issue that plagues low-turnover substrates in general.
Reaction phenotyping studies were also conducted in cryopreserved hepatocytes to confirm that the observed clearance values were driven primarily by AO. One practical approach would be to coincubate with a chemical inhibitor of the enzyme of interest. Several inhibitors of AO have been characterized and may be used in vitro, including raloxifene (Obach et al., 2004; Obach, 2004) and menadione (Obach et al., 2004; Sahi et al., 2008). Raloxifene is complicated in that it also is a mechanism-based inactivator of cytochrome P450 3A4 (Baer et al., 2007), and menadione nonselectively inhibits many cytochrome P450 enzymes (R. S. Obach, unpublished observations). Hydralazine, an antihypertensive agent, has been reported to be an efficient inhibitor of AO, but not of xanthine oxidase (Johnson et al., 1985) or the cytochrome P450 enzymes. Thus, this inhibitor was coincubated (50 μM) in the presence of AO substrate in hepatocytes. Unfortunately, intrinsic clearance values for zaleplon and XK-469 were already too low to decipher a difference when coincubated with hydralazine, but the observed clearance values for BIBX1382, carbazeran, and O6-benzylguanine were all substantially attenuated upon coincubation with hydralazine (Fig. 5), corroborating AO as the metabolic enzyme involved in the clearance of these substrates in cryopreserved hepatocytes.
In summary, the two lots of cryopreserved hepatocytes tested seem to maintain approximately quantitative AO activity, as evidenced by chemical inhibition (e.g., hydralazine), 18O-incoporation studies, and the ability to adequately scale hepatic clearance for known AO substrates. However, given the reports of the potential instability of AO (Duley et al., 1985; Al-Salmy, 2001), a factor that has not been completely resolved, one probably should not assume that all hepatocyte lots from vendors would possess similar quantitative AO activity. Thus, the recommendation would be to characterize each lot of hepatocytes acquired with control AO substrates before implementing them into a screening strategy. Data reported here should help provide a benchmark for expected clearance values when using cryopreserved hepatocytes. Future efforts from our lab in this area will include evaluation of additional hepatocyte systems such as plated hepatocytes for AO activity.
Authorship Contributions
Participated in research design: Hutzler, Yang, Albaugh, and Fisher.
Conducted experiments: Yang, Albaugh, Fullenwider, and Schmenk.
Performed data analysis: Yang, Albaugh, Fullenwider, and Schmenk.
Wrote or contributed to the writing of the manuscript: Hutzler, Yang, and Albaugh.
Acknowledgments
We thank J. Scott Daniels (Vanderbilt Center for Neuroscience Drug Discovery, Nashville, TN) for helpful comments on data presentation and Matthew Cerny for thoughtful review of our manuscript.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- AO
- aldehyde oxidase
- Clint
- intrinsic clearance
- Clh
- hepatic clearance
- Qh
- liver blood flow
- XIC
- extracted ion chromatogram
- WME
- Williams' Medium E
- LC
- liquid chromatography
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry.

- Received September 21, 2011.
- Accepted October 26, 2011.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}