


Abstract
Olmesartan medoxomil (OM) is a prodrug-type angiotensin II type 1 receptor antagonist. The OM-hydrolyzing enzyme responsible for prodrug bioactivation was purified from human plasma through successive column chromatography and was molecularly identified through N-terminal amino acid sequencing, which resulted in a sequence of 20 amino acids identical to that of human paraoxonase 1 (PON1). Two recombinant allozymes of human PON1 (PON1192QQ and PON1192RR) were constructed and were clearly demonstrated to hydrolyze OM; hydrolysis by the latter allozyme was slightly faster than that by the former. In addition, we evaluated the contribution of PON1 to OM bioactivation in human plasma. Enzyme kinetic studies demonstrated that OM was hydrolyzed more effectively by the recombinant PON1 proteins than by purified albumin. The OM-hydrolyzing activities of the recombinant PON1 proteins and diluted plasma were greatly reduced in the absence of calcium ions. Immunoprecipitation with anti-PON1 IgG completely abolished the OM-hydrolyzing activity in human plasma, whereas the activity was partially inhibited with anti-albumin IgG. The distribution pattern of the OM-hydrolyzing activity in human serum lipoprotein fractions and lipoprotein-deficient serum was examined and showed that most of the OM-hydrolyzing activity was located in the high-density lipoprotein fraction, with which PON1 is closely associated. In conclusion, we identified PON1 as the OM-bioactivating hydrolase in human plasma on a molecular basis and demonstrated that PON1, but not albumin, plays a major role in OM bioactivation in human plasma.
Introduction
Several prodrug strategies have been developed to enable drugs to exhibit optimal pharmacokinetics and pharmacological actions by overcoming a number of barriers to drug-like properties. In particular, an esterification strategy has been used historically to increase transcellular absorption of poorly permeable drugs administrated orally. Esterases, which are involved in the prodrug bioactivation process, are widely distributed in the blood, liver, intestine, and many other biological fluids and tissues (Testa and Mayer, 2003). In most cases, intestinal esterases serve as the major enzymes in bioactivation of prodrugs during the first pass through the gut after absorption. In some cases, however, prodrug molecules escape the activation process catalyzed by intestinal esterases, enter the blood circulation as the prodrug, and then are activated by serum (plasma) and liver esterases. Several esterases in human plasma have been investigated as key enzymes responsible for prodrug bioactivation (Ettmayer et al., 2004; Testa, 2004; Li et al., 2005; Satoh and Hosokawa, 2006), including paraoxonase/arylesterase 1 (PON1), cholinesterase, and albumin. For example, human PON1, which is localized predominantly in plasma and is associated with high-density lipoprotein (HDL), was reported to be a major bioactivating enzyme for the antibacterial agent prulifloxacin (Tougou et al., 1998).
Olmesartan medoxomil (OM) is a prodrug-type angiotensin receptor blocker that is prescribed throughout the world as monotherapy and in combination with a thiazide diuretic and/or a calcium channel blocker (Chrysant, 2008; Scott and McCormack, 2008; Rump and Sellin, 2010; Ram, 2011). As shown in Fig. 1, OM is one of the exemplary cases of bioavailability improvement through derivatization into the (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl ester (medoxomil ester) prodrug (Scott and McCormack, 2008). It was reported that multiple enzymes are capable of OM bioactivation in humans, including plasma albumin (Ma et al., 2005) and an intestinal and liver hydrolase carboxymethylenebutenolidase homolog (CMBL) (Ishizuka et al., 2010).
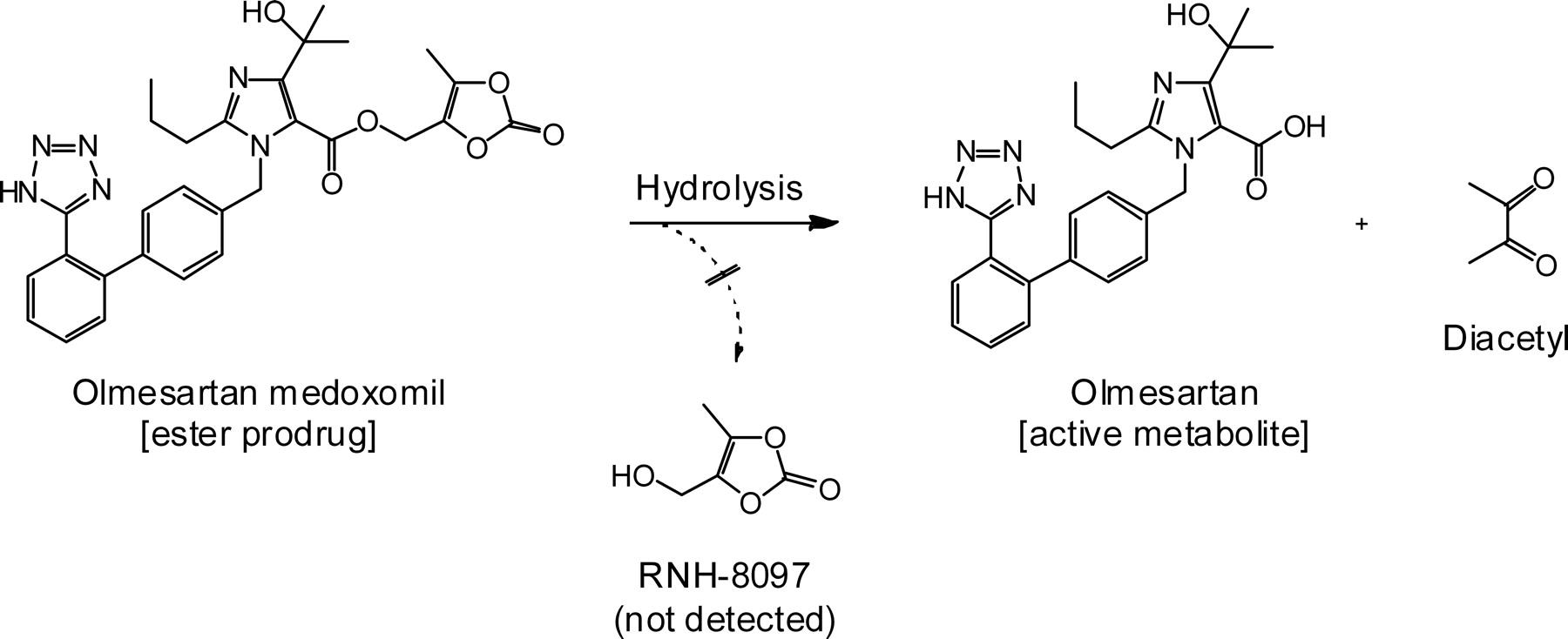
Bioactivation of olmesartan medoxomil. Hydrolysis liberates its active metabolite, olmesartan, and generates a diketone. Another possible product, RNH-8097, was not detected in the in vitro reaction mixture.
The identification and characterization of enzymes responsible for prodrug bioactivation are important because their properties become key determinants for the pharmacokinetics of the pharmacologically active metabolites and thus the pharmacodynamics of the drug entities, such as the onset of drug action and the therapeutic efficacy. In this study, we purified the OM-bioactivating hydrolase from human plasma, molecularly identified it as PON1, and directly demonstrated the involvement of PON1 by using its recombinant proteins. Furthermore, we compared the enzyme characteristics of the two plasma esterases reported to hydrolyze OM, namely, PON1 and albumin, and estimated their contributions to the overall OM bioactivation in human blood circulation.
Materials and Methods
Materials.
OM, olmesartan, and 2-butyl-4-(1-hydroxy-1-methylethyl)-1-[2′-(1H-tetrazole-5-yl)-4-biphenylyl]-1H-imidazole-5-carboxylic acid (RNH-6272; a structural analog of olmesartan) used as the internal standard for olmesartan determination, were synthesized at Daiichi Sankyo (Tokyo, Japan). Phenyl acetate and paraoxon were purchased from Sigma-Aldrich Japan (Tokyo, Japan). Benzoyl choline was purchased from Nakalai Tesque (Kyoto, Japan). Human plasma or serum was prepared from blood collected from healthy subjects by using a protocol approved by the institutional human ethical committee of Daiichi Sankyo. Animal plasma was prepared from blood collected in-house from Wister-Imamichi rats, ddY mice, Japanese white rabbits, cynomolgus monkeys, and beagle dogs, in accordance with the guidelines of the institutional animal care and use committee of Daiichi Sankyo. Purified human serum albumin and anti-human albumin rabbit polyclonal IgG were purchased from Sigma-Aldrich.
Hydrolase Activity Measurement.
OM-hydrolyzing activity was determined as follows. Animal and human plasma for the species difference examination and each eluent fraction obtained from successive column chromatography for purification of our target hydrolase were appropriately diluted (final dilutions, 50–400-fold and 10–100-fold, respectively) with 10 mM potassium phosphate buffer. The diluted protein solutions were incubated at 37°C for 5 to 10 min with OM as a substrate (final solvent concentration, 2% acetonitrile). After reaction termination through addition of ice-cold acetonitrile, the concentration of the active metabolite olmesartan was determined with a high-performance liquid chromatography system (SLC-10A system; Shimadzu, Kyoto, Japan). Olmesartan was separated from the OM peak with a reverse-phase C18 column (YMC-Pack ODS-A A-312, C18, 5 μm, 6.0 i.d. × 150 mm; YMC, Kyoto, Japan) and a mobile phase of 40% acetonitrile containing 2% paired-ion chromatography reagent A (Waters, Milford, MA), at a flow rate of 1.0 ml/min, and was detected with UV detection at 254 nm. The lower limit of quantitation was set at 0.2 μM. In the other in vitro experiments, the OM-hydrolyzing activities of diluted human plasma (final dilution, 500-fold), recombinant PON1 proteins (final protein concentration, 0.01 mg/ml), and purified human serum albumin (final protein concentration, 5 mg/ml) were measured at 37°C with OM as a substrate, in 100 mM Tris-HCl buffer (pH 7.5) containing 1 mM CaCl2 (final solvent concentration, 2.5% acetonitrile). After incubation at 37°C for a designated reaction time, the reaction was terminated through addition of ice-cold acetonitrile containing RNH-6272, as an internal standard for determination of the metabolite concentration, and 0.25% trifluoroacetic acid, for prevention of the nonenzymatic degradation of OM. After filtration and addition of 50% methanol containing 1% formic acid, the concentration of the active metabolite olmesartan was determined with a liquid chromatography-tandem mass spectrometry system consisting of a Prominence LC-20A system (Shimadzu) and an API 3200 mass spectrometer (AB Sciex, Foster City, CA). Olmesartan was separated with a reverse-phase C18 column (Atlantis T3, 5 μm, 2.1 mm i.d. × 150 mm; Waters) and a mobile phase of 64% methanol containing 0.2% formic acid, at a flow rate of 0.2 ml/min, and was determined through monitoring of the ion transition of m/z 447 to m/z 207 with multiple-reaction monitoring in the positive electrospray ionization mode. The lower limit of quantitation was set at 20 nM. The enzymatic activity was expressed as a metabolite formation rate (in nanomoles per minute per milligram of protein) on the basis of the production of olmesartan for the reaction by the recombinant protein, which was subtracted from that in the buffer control (nonenzymatic hydrolysis).
Phenyl acetate-hydrolyzing activity was determined as follows. Over the process of protein purification from human serum, 20 to 40 μl of each fraction was incubated at 37°C for 10 to 20 min with 0.2 ml of 10 mM phenyl acetate solution in 50 mM Tris-HCl buffer (pH 8). After reaction termination through addition of 1 ml of 0.04% 4-aminoantipyrine in acetonitrile and then color development with addition of 2 ml of 0.08% potassium ferricyanide with 50 mM Tris-HCl buffer (pH 8) in acetonitrile, the absorbance at 510 nm was measured. After column purification, the activity of the purified protein was measured as reported previously (Gan et al., 1991), with slight modification. Twenty microliters of the purified esterase solution were incubated at room temperature with 1.5 ml of 1 mM phenyl acetate solution in 50 mM Tris-HCl buffer (pH 8) containing 1 mM CaCl2. Formation of the metabolite phenol was determined through monitoring of absorbance changes at 270 nm. The activity was expressed with units of change in absorbance at 270 nm per minute per milligram of protein.
Paraoxon-hydrolyzing activity was measured as reported previously (Gan et al., 1991), with slight modification. Twenty microliters of the appropriately diluted purified protein solution were incubated at room temperature with 0.8 ml of paraoxon solution (1 mM) in 50 mM Tris-HCl buffer (pH 8) containing 1 mM CaCl2 and 1 M NaCl. Formation of the metabolite p-nitrophenol was determined through monitoring of absorbance changes at 412 nm. The activity was expressed with units of change in absorbance at 412 nm per minute per milligram of protein.
Benzoyl choline-hydrolyzing activity was measured as follows. One hundred twenty microliters of the appropriately diluted purified protein solution were incubated at 37°C for 10 min with benzoyl choline (final concentration, 1 mM). After reaction termination through addition of ice-cold acetonitrile, the concentration of the metabolite benzoic acid was determined with a high-performance liquid chromatography system (SLC-10A system; Shimadzu). The metabolite was separated from the benzoyl choline peak with a reverse-phase C18 column (YMC-Pack ODS-A A-312, C18, 5 μm, 6.0 i.d. × 150 mm; YMC) and a mobile phase of 55% acetonitrile containing phosphoric acid for pH adjustment (pH 3.0), at a flow rate of 1.0 ml/min. UV detection was performed at 225 nm. The activity was expressed with units of nanomoles per minute per milligram of protein.
Purification of OM Hydrolase from Human Plasma.
Because there were findings of species differences in plasma OM-hydrolyzing activities (Fig. 2) and chemical inhibition properties were reported previously (Ishizuka et al., 2010) that resembled the enzymatic characteristics of human PON1, all column purification steps were performed according to the plasma PON1 purification method reported previously (Gan et al., 1991), with slight modification. The peak fraction was determined on the basis of phenylacetate-hydrolyzing activity with absorbance at 510 nm, because OM-hydrolyzing activity and phenylacetate-hydrolyzing activity were confirmed to behave quite similarly in the preliminary column chromatography. Over the purification process, each fraction was analyzed with SDS-PAGE performed according to the method described by Laemmli (1970), with 8% or 10% SDS-polyacrylamide gels (Bio-Rad Laboratories, Hercules, CA). Human plasma was added to 2 volumes of column buffer A (50 mM Tris-HCl buffer, pH 8, containing 1 mM CaCl2, 5 μM EDTA, and 5% glycerol) containing 3 M NaCl. The mixture was allowed to stand for ∼1 h. After centrifugation, the supernatant was loaded onto a Blue Sepharose column (5 × 7 cm; GE Healthcare Japan, Tokyo, Japan). The column was pre-equilibrated with column buffer A containing 3 M NaCl, washed with column buffer A containing 3 M NaCl and then with the same buffer without NaCl, and then eluted with column buffer A containing 0.1% sodium deoxycholate. The active fractions were dialyzed against column buffer A, concentrated, and loaded onto an ion-exchange column (DEAE-Sephacel column, 2.5 × 8 cm; GE Healthcare Japan), which had been pre-equilibrated with 25 mM Tris-HCl buffer (pH 8) containing 1 mM CaCl2, 5 μM EDTA, and 5% glycerol (referred to as column buffer B), was washed with column buffer B containing 1% Emulgen 911 (Kao Corp., Tokyo, Japan) and 5% dimethylacetamide and then buffer containing 0.1% Emulgen, and then was eluted with a linear gradient of 0 to 350 mM NaCl in column buffer B. The active fractions were again loaded onto a DEAE-Sephacel column, which was washed with column buffer B containing 1% Emulgen and 5% dimethylacetamide, buffer containing 0.1% Emulgen, and buffer containing 100 mM NaCl and then was eluted with a linear gradient of 100 to 350 mM NaCl in column buffer B. The resultant active fractions were dialyzed against column buffer B containing 0.1% Emulgen, concentrated, and stored frozen at −80°C until use as the final purified esterase, which was subsequently determined to be human PON1. A portion of the active fraction from each column purification step was loaded onto a SDS-polyacrylamide gel (8% gel; Bio-Rad Laboratories) according to the method described by Laemmli (1970), and the gel was stained with Coomassie Brilliant Blue. The protein concentration was determined by using a detergent-compatible protein assay (Bio-Rad Laboratories), with bovine serum albumin as the standard.
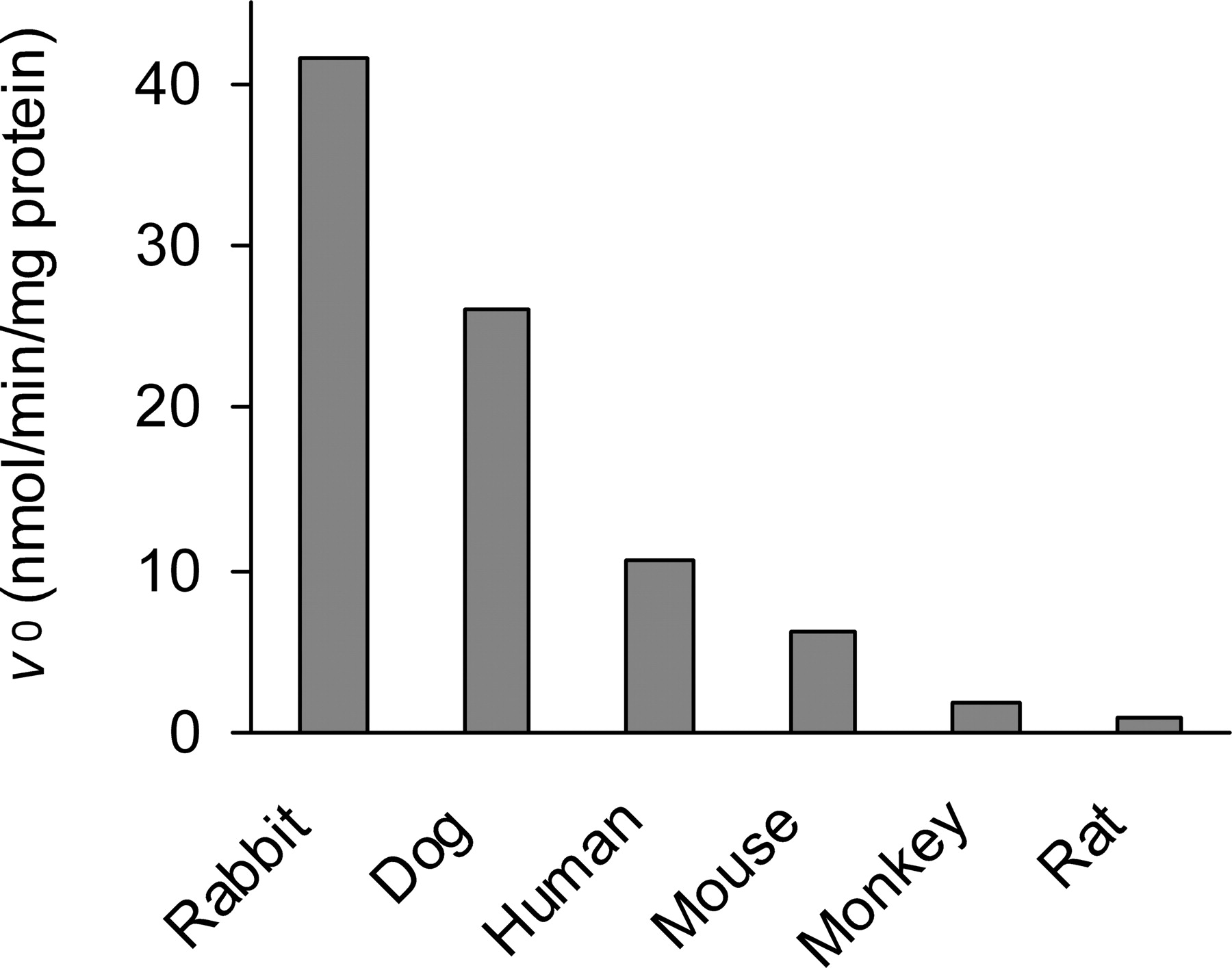
Cross-species differences in the activity of OM hydrolysis in human and animal plasma. Data represent the results of single determinations with pooled plasma from three individuals.
N-Terminal Amino Acid Sequencing.
The N-terminal sequence of the target protein was determined from the purified protein transferred electrophoretically onto a PVDF membrane (Immun-Blot PVDF membrane; Bio-Rad Laboratories) after SDS-PAGE. Amino acids were sequenced through automated Edman degradation by using a gas-phase protein sequencer (model PPSQ-10; Shimadzu), according to the manufacturer's procedure.
Immunoblotting and Immunoprecipitation Analysis.
Specific antiserum against the purified protein, which was later identified as human PON1, was raised in female Japanese white rabbits. The animals received three boosts with equal volumes of the purified protein as an antigen in complete Freund's adjuvant, with 2-week intervals. The antiserum was obtained from the animals, and the IgG was purified from the antiserum by using 50% saturated ammonium sulfate precipitation, followed by DEAE-Sephacel column chromatography. The IgG was further purified with an Econo-Pac serum IgG purification column kit (Bio-Rad Laboratories), to remove components with OM-hydrolyzing activity.
The plasma proteins in each purification step that were separated with SDS-PAGE and then blotted on a PVDF membrane were detected with the purified anti-PON1 IgG described above, followed by enhanced chemiluminescence, horseradish peroxidase-linked, donkey anti-rabbit IgG (GE Healthcare, Little Chalfont, UK), as primary and secondary antibodies, respectively. These immunoblots were observed through chemiluminescence with an enhanced chemiluminescence detecting reagent (GE Healthcare).
Inhibitory effects of the purified IgG against PON1 and albumin on the OM-hydrolyzing activity in human plasma were investigated to estimate the contribution of each protein. Diluted human plasma at an appropriate dilution ratio was incubated overnight at 4°C with the respective IgG fractions at various IgG fraction/plasma volume ratios. After separation of the antigen-antibody complex through centrifugation, the supernatant was used as an enzyme source for OM-hydrolyzing activity measurements.
Expression of Human PON1 in Mammalian Cell Line.
The open reading frame of the full-length human PON1 (amino acids 1–355) and that of the Q192R mutant were subcloned into a vector plasmid (pcDNA6-myc-His; Invitrogen, Carlsbad, CA) providing a C-terminal myc-polyhistidine-epitope tag, confirmed with DNA sequencing, and expressed in mammalian FreeStyle 293-F cells (Invitrogen). The transfected cells were cultured for 7 days in FreeStyle 293 expression medium (Invitrogen), and the conditioned media from the cells overexpressing human PON1192QQ and PON1192RR were filtered with polyethersulfone membrane filters (0.45 μm; Thermo Fisher Scientific, Waltham, MA). After dialysis against 20 mM Tris-HCl buffer (pH 7.5), the overexpressed histidine-tagged proteins were purified with a two-step purification process involving anion chromatography with a HiTrap Q-XL column (GE Healthcare) and Ni-affinity chromatography with a HisTrap FF column (GE Healthcare). The eluates were collected and desalted with PD-10 columns (GE Healthcare), and then the resultant proteins were stored frozen at −80°C until use. The protein concentration was determined by using a micro-bicinchoninic acid Pierce protein assay (Thermo Fisher Scientific), with bovine serum albumin as the standard.
Kinetic Analysis.
The enzyme kinetics for OM hydrolysis by human plasma, recombinant PON1 proteins, and purified human serum albumin were evaluated with substrate concentrations ranging from 3.125 to 400 μM. For reactions with purified serum albumin, 100 mM potassium phosphate buffer (KPB) (pH 7.4) was used instead of 100 mM Tris-HCl buffer (pH 7.5) containing 1 mM CaCl2. Kinetic parameters, namely, the Michaelis constant (Km) and the maximal velocity (Vmax), were estimated from the data on substrate concentrations ([S]) and initial velocities (v) with WinNonlin Professional (version 5.2.1; Pharsight, Mountain View, CA), by using nonlinear least-squares regression analysis fitted to the Michaelis-Menten equation, v = Vmax × [S]/(Km + [S]).
Distribution of OM-Hydrolyzing Activity in Serum Lipoprotein Fractions.
The lipoprotein fractions, namely, very-low-density lipoprotein (VLDL) including chylomicron, low-density lipoprotein (LDL), and HDL, were separated from human serum through sequential ultracentrifugation in continuous-density gradients (Havel et al., 1955) (CS150GXL ultracentrifuge with S120AT2 rotor; Hitachi Koki, Tokyo, Japan), desalted (PD-10 desalting columns; GE Healthcare), and concentrated through centrifugation (Amicon Ultra filters; molecular weight cutoff, 10,000; Millipore Corporation, Billerica, MA). The lipoprotein-deficient serum (LPDS) fraction obtained after the lipoprotein separation, which was thought to include serum albumin, was also desalted with PD-10 columns and was used for activity measurements. The OM-hydrolyzing activity was measured at a substrate concentration of 10 μM, as described above. Incubations with the LPDS fraction in 100 mM KPB (pH 7.4) also were performed, because purified serum albumin showed higher OM-hydrolyzing activity in KPB than in Tris-HCl buffer containing CaCl2.
Other Methods.
Protein concentrations were determined by using the Bradford (Bradford, 1976) protein assay (Bio-Rad Laboratories), with bovine serum albumin as the standard, unless indicated otherwise.
Results
Characteristics of Plasma OM Hydrolase.
In plasma fractions from six different species including humans, OM was substantially hydrolyzed and converted into the active metabolite olmesartan. The OM-hydrolyzing activities in human and animal plasma are shown comparatively in Fig. 2. Rabbit plasma demonstrated the highest activity, followed by dog and human plasma. The hydrolysis in rat plasma was much slower than that in human plasma.
Purification of OM Hydrolase from Human Plasma.
The OM hydrolase was purified from human plasma through successive column chromatography. As shown in Table 1, the purification resulted in a 386-fold increase in specific activity of OM hydrolysis, in accordance with those of phenyl acetate hydrolase and paraoxon hydrolase activities, which were monitored as markers of PON1 activity, whereas the marker activity for choline esterase, the benzoyl choline-hydrolyzing activity, was completely removed during the purification process. The fractions containing the OM hydrolase were separated through SDS-PAGE and stained with Coomassie Brilliant Blue, which showed a highly purified enzyme preparation with a dominant protein band exhibiting an apparent molecular mass of 48.5 kDa (Fig. 3A, lane 5). After transfer onto the PVDF membrane, the band was excised from the membrane and subjected to the following amino acid sequencing.
Purification of OM-bioactivating hydrolase from human plasma and three marker activities of typical plasma esterases
Phenyl acetate hydrolysis and paraoxon hydrolysis activities, as markers of PON1 activity, and benzoyl choline hydrolysis activity, as a marker for choline esterase, were simultaneously monitored during the purification process.

Purification of PON1 from human plasma. The OM hydrolase was purified from human plasma through successive column chromatography. The SDS-PAGE gel stained with Coomassie Brilliant Blue (A) and the immunoblot membrane stained with anti-PON1 IgG (B) for the pooled active fractions of each purification step are shown. Lane 1, molecular mass marker; lane 2, human plasma; lane 3, eluate from Blue Sepharose column; lanes 4 and 5, first- and second-step eluates, respectively, from DEAE-Sephacel column. Arrowheads, 48.5-kDa bands (subjected to amino acid sequencing in A, lane 5).
N-Terminal Amino Acid Sequencing.
The automated Edman degradation procedure provided the N-terminal sequence of the first 20 amino acids of the column-purified OM hydrolase from human plasma. The following sequence was obtained: Ala-Lys-Leu-Ile-Ala-Leu-Thr-Leu-Leu-Gly-Met-Gly-Leu-Ala-Leu-Phe-Arg-Asn-His-Gln. A Basic Local Alignment Search Tool search of a human protein database (the National Center for Biotechnology Information RefSeq database) demonstrated that PON1, which was postulated to be our target protein in plasma, is the only human protein that shows a perfect match to the determined 20-amino acid sequence.
OM Hydrolysis by Recombinant PON1 Proteins.
To confirm the protein identification results from N-terminal amino acid sequencing, we overexpressed recombinant human PON1 in FreeStyle 293-F cells, a mammalian cell line derived from human embryonic kidney 293 cells, and the OM-hydrolyzing activity of the recombinant protein was measured. Because residue 192 is a well investigated polymorphic site of human PON1 that accounts for marked qualitative differences (Harel et al., 2004; Ginsberg et al., 2009), two types of allelic homozygotes at residue 192, PON1192QQ and PON1192RR, were generated. To confirm the recombinant proteins produced, we examined tryptic fragments by using mass spectrometry and achieved 78% and 95% amino acid sequence coverage of PON1192QQ and PON1192RR, respectively, with covering of the Q192R mutated sequence. Both recombinant PON1 allozymes rapidly hydrolyzed OM, converted it into the active metabolite olmesartan, and showed calcium ion dependence of the enzymatic activity (Table 2).
Metal ion requirement for OM hydrolysis by various proteins in human plasma
Percentages were determined as the percentage activity in comparison with values measured in the presence of Ca2+ ions. The substrate concentration was 10 μM.
Immunoblotting of Plasma OM Hydrolase.
Specific immunoreactivity of the IgG fraction against the PON1 protein was confirmed with Western blotting (Fig. 3B). The anti-PON1 IgG immunostained the targeted protein in active fractions of each column purification step as a single band with the same migration point, whose intensity increased in accordance with the proceeding purification step.
Metal Ion Requirements.
In Table 2, the OM-hydrolyzing activities of several plasma esterases in incubation buffer containing Ca2+ ions are compared with those in buffer in which Ca2+ ions were replaced by either Zn2+ or Mg2+ ions. The enzymatic activities of the diluted plasma and recombinant PON1 proteins were greatly reduced in the absence of Ca2+ ions. In contrast, the purified serum albumin hydrolyzed OM equally in all incubations, regardless of the metal ions contained.
Kinetic Analysis.
The OM hydrolysis activity in human plasma, the recombinant proteins PON1192QQ and PON1192RR, and purified serum albumin exhibited single-enzyme Michaelis-Menten kinetics, as shown in Fig. 4. The parameters of enzyme kinetics are summarized in Table 3. Higher estimated Km values for the recombinant proteins PON1192QQ and PON1192RR (157 and 102 μM, respectively) than for plasma (Km = 6.71 μM) indicate a meaningful lower affinity of the substrate OM for the recombinant PON1 proteins than the natural protein in plasma. A comparison of Vmax/Km, representing enzymatic efficiency, between the two recombinant PON1 proteins showed that PON1192RR was more active with OM as a substrate than was PON1192QQ. The Vmax/Km value for serum albumin was considerably lower than those of the recombinant PON1 proteins.

Enzyme kinetics for OM hydrolysis catalyzed by human plasma esterases. Enzyme kinetics of the recombinant proteins PON1192QQ (A) and PON1192RR (B), diluted human plasma (C), and purified serum albumin (D) were investigated. Data represent the mean values of duplicate determinations. Solid lines, best fit to the Michaelis-Menten equation with nonlinear least-squares regression. Insets, Eadie-Hofstee plots.
Kinetic parameters for OM-hydrolyzing activities of various proteins in human plasma
Data generated in duplicate determinations were fitted to the single-enzyme Michaelis-Menten model by using nonlinear least-squares regression.
Immunoprecipitation of Plasma OM Hydrolase.
The contribution of PON1 and albumin to OM bioactivation in human plasma was estimated through immunoprecipitation with specific antibodies against the respective proteins. As shown in Fig. 5A, the IgG against human PON1 showed concentration-dependent inhibition of the OM-hydrolyzing activity in human plasma and completely abolished the activity at a 4-fold IgG fraction/plasma ratio. In contrast, the IgG against human serum albumin displayed only incomplete inhibition of the plasma OM-hydrolyzing activity even at the highest IgG fraction/plasma ratio (Fig. 5B), despite showing complete inhibition of the activity in purified albumin with the same volume of IgG fraction added (data not shown). The maximal inhibition magnitude, which was observed at IgG fraction/plasma ratios of more than 2-fold, was less than 30% of the control activity.
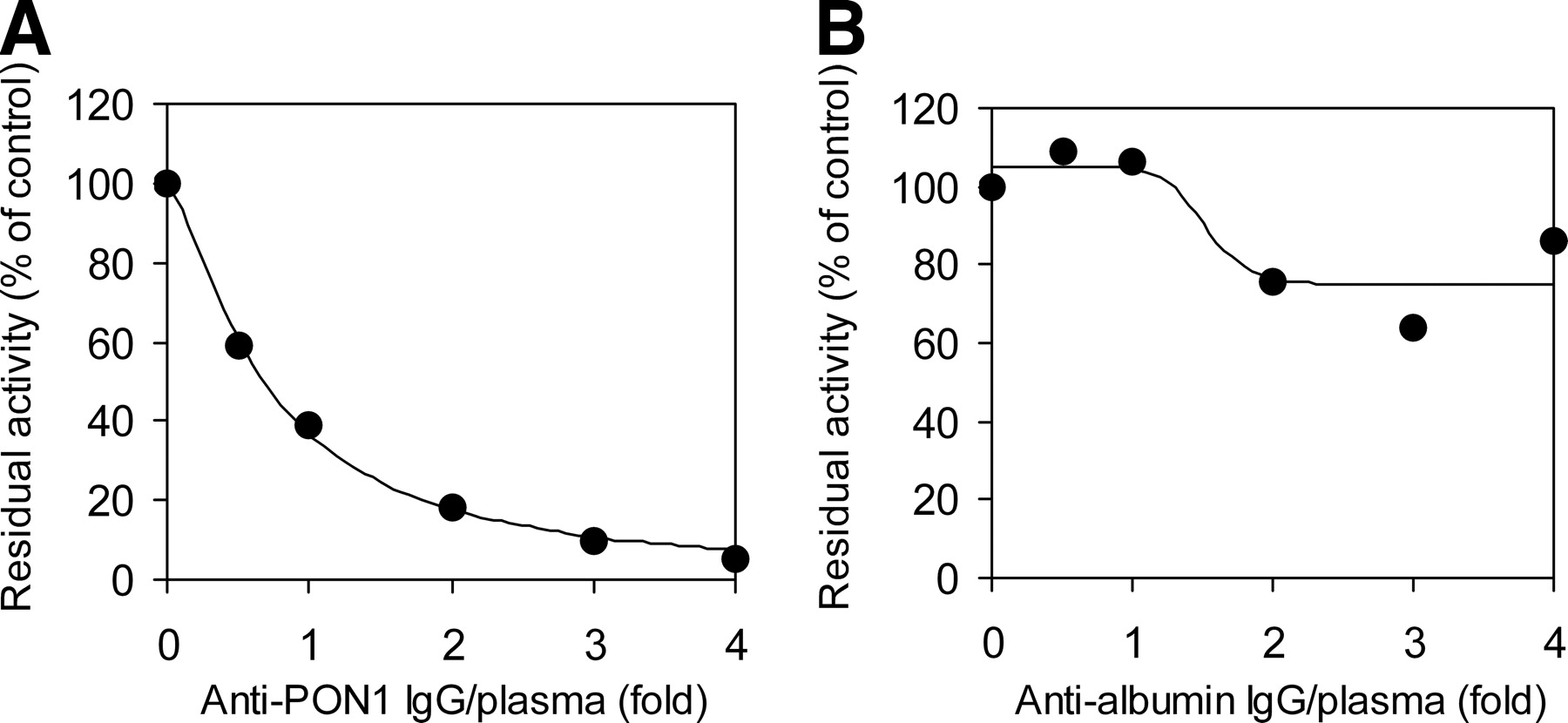
Immunoprecipitation analysis of OM-hydrolyzing activity in human plasma. Inhibitory effects of rabbit IgG raised against two human serum proteins, PON1 (A) and albumin (B), on the OM-hydrolyzing activity in human plasma are shown. Diluted human plasma was incubated overnight at 4°C with the two purified IgG fractions at various IgG fraction/plasma ratios. Solid lines, best fit to the sigmoidal Emax model of inhibitory effects with nonlinear least-squares regression.
Distribution of OM-Hydrolyzing Activity in Human Serum Lipoprotein Fractions.
The distribution of the OM-hydrolyzing activity in lipoprotein fractions (VLDL including chylomicron, LDL, and HDL) and LPDS separated from human serum is summarized in Table 4. Most of the activity (more than 93%) was located in the HDL fraction, whereas fairly low enzymatic activity was observed in LPDS, which is thought to include serum albumin.
Distribution of OM-hydrolyzing activity in lipoprotein fractions and LPDS separated from human serum
Human serum was fractionated into three lipoprotein fractions (VLDL including chylomicron, LDL, and HDL) and LPDS with a sequential ultracentrifugation method. The substrate concentration was 10 μM. The activity in the LPDS was tested in KPB and Tris-HCl buffer containing Ca2+ ions, because serum albumin previously showed higher activity for OM hydrolysis in KPB than in Tris-HCl buffer.
Discussion
Although several reports stated that some human plasma esterases are capable of bioactivation of the prodrug OM (Laeis et al., 2001; Ma et al., 2005), we found no reports that presented evidence of the molecular identification of enzymes responsible for the hydrolytic reaction. In this report, we directly identified the OM hydrolase purified from human serum as human PON1 through N-terminal peptide sequencing and we demonstrated significant OM-hydrolyzing activities of recombinant PON1 proteins. In our previous work (Ishizuka et al., 2010), the OM-hydrolyzing activity in human plasma was strongly inhibited by p-chloromercuribenzoic acid (a free thiol modifier) and EDTA (a divalent cation chelator), which are both PON1 inhibitors, but was not sensitive to diisopropyl fluorophosphate (an organophosphate), which inhibits cholinesterases and carboxylesterases. The cross-species difference in plasma OM-hydrolyzing activities (abundant in rabbits and limited in rats, as shown in Fig. 2) also suggested the involvement of PON1 (Costa et al., 1990; Kuo and La Du, 1995) in OM bioactivation in plasma, rather than other plasma esterases. Therefore, we purified this OM-hydrolyzing activity according to the method historically used for PON1 purification (Gan et al., 1991). The N-terminal sequence (20 amino acids) of the extruded 48.5-kDa protein was, as we expected, identical to the N-terminal region spanning Ala2 to Gln21 of human PON1 (EC 3.1.8.1./EC 3.1.1.2).
In an attempt to confirm the OM-hydrolyzing activity of human PON1, we constructed recombinant human PON1 proteins, PON1192QQ and PON1192RR, in a mammalian cell line. We compared their enzyme characteristics with those of diluted human plasma and purified serum albumin, which was reported as another OM-hydrolyzing enzyme in human plasma. Consistent with the well known feature of paraoxonases as calcium-dependent metalloenzymes, both recombinant PON1 allozymes and diluted plasma showed calcium ion-dependent OM-hydrolyzing activities, whereas the activity of purified serum albumin was hardly affected by the metal ion replacements (Table 2).
OM was hydrolyzed slightly faster by recombinant PON192RR than by PON192QQ (Table 2). Genetic variability in human PON1 activity has been of interest and has been widely studied over the years. PON1 has two common coding-region polymorphisms, M55L and Q192R; more attention has been paid to the latter because it accounts for marked qualitative differences between the two allozymes in their affinity for and catalytic activity with a number of substrates (Mackness et al., 1998a,b; Costa et al., 2003). Some ester substrates, such as phenyl acetate, are hydrolyzed by the PON1 192Q and 192R allozymes at approximately equivalent rates, whereas most organophosphates are hydrolyzed by them at different rates. Prulifloxacin, a prodrug-type antibacterial agent with a medoxomil moiety (like OM), was reported to be hydrolyzed mainly by PON1 and to form its active metabolite (Tougou et al., 1998). The authors showed that the prulifloxacin-hydrolyzing activity was positively correlated with the paraoxon-hydrolyzing activity (which is catalyzed faster by PON1192R), which suggests that the PON192R allozyme is more active than the PON192Q allozyme in prodrug activation, in the same manner as OM bioactivation. The interindividual variation in the prulifloxacin-hydrolyzing activity was reported to be only 2-fold, whereas the variation in the paraoxon-hydrolyzing activity was 9-fold (Tougou et al., 1998). When these results are considered together with the result regarding OM, the effects of the PON1 Q192R polymorphism on the bioactivation of prodrugs with a medoxomil moiety is not considered to be significant. As another example, the latest research on PON1 involvement in the hydrolysis of pilocarpine, which is used as a treatment for xerostomia, demonstrated higher activity of the R/R genotype, compared with the Q/R and Q/Q genotypes, by using 50 individual human plasma samples (Hioki et al., 2011). The analogy of this polymorphic phenomenon in the pilocarpine lactone ring-opening reaction to those of the medoxomil prodrugs described above seems to be attributable to the structural similarity in the hydrolyzed groups of these drugs.
Next, we evaluated the contribution of the two plasma hydrolases, PON1 and albumin, to OM bioactivation in human plasma. In a comparison of kinetic parameters between these two proteins (Table 3), a significantly lower Km and higher Vmax, resulting in a higher Vmax/Km, for recombinant PON1 demonstrated a larger capacity of PON1 for OM hydrolysis, compared with albumin. Because albumin is the most abundant protein in plasma, however, it is still possible that albumin plays a certain role in OM bioactivation in human plasma. Indeed, a several hundred-fold difference in PON1 (∼0.2 mg protein/ml) (Garin et al., 1997; Connelly et al., 2008) and albumin (∼40 mg protein/ml) concentrations in human plasma numerically offset the difference in the in vitro catalytic efficiencies of these purified proteins. Therefore, we directly examined the contributions of PON1 and albumin by using human plasma in immunoprecipitation assays with specific IgG fractions against these two proteins. The anti-PON1 IgG completely abolished the OM-hydrolyzing activity in human plasma, whereas maximal inhibition of only 30% was observed with the antialbumin IgG (Fig. 5), which suggests a predominant contribution of PON1 rather than albumin. Similar to the findings for OM, Tougou et al. (1998) reported that human serum albumin had prulifloxacin-hydrolyzing activity but the contribution of albumin to the total activity in human serum was <5%, and the authors concluded that PON1 is mainly responsible for the hydrolysis of prulifloxacin.
Furthermore, the distribution pattern for the OM-hydrolyzing activity in serum lipoprotein fractions and LPDS, with most of the activity located in the HDL fraction (Table 4), brings us evidence showing the practically exclusive involvement of PON1 in plasma OM hydrolysis. PON1 is an enzyme secreted into the blood, where it resides on HDL particles. Studies showed that both the activity and stability of PON1 are highly dependent on the HDL components (La Du et al., 1993; Sorenson et al., 1999). Gaidukov and Tawfik (2005) demonstrated that the stability and lactonase activity of recombinant PON1 were dramatically stimulated by treatment with apolipoprotein A-1-containing HDL; in addition, the paraoxonase and arylesterase activities were stimulated with HDL particles regardless of the apolipoprotein content. The association of PON1 with human phosphate-binding protein, a HDL component with a molecular weight similar to that of PON1, is highlighted to be essential for preserving active conformations of the enzyme (Rochu et al., 2007; Renault et al., 2010). Lacking these HDL components as chaperones might explain the considerably lower affinity for OM of our recombinant PON1 proteins, compared with that of natural PON1 in diluted human plasma in the enzyme kinetic analysis (Table 3).
OM is an orally administered prodrug. After oral administration of the prodrug, first-pass bioactivation may occur in the intestine, followed by the portal blood and liver, before the prodrug reaches the systemic circulation. We discovered the involvement of an unknown human protein, CMBL, in OM bioactivation in the intestine and liver (Ishizuka et al., 2010). High metabolic clearance of intestinal CMBL suggests that the intestinal bioactivation firstly and predominantly contributes to the quick onset of drug action after oral administration of OM. For reference, the intestinal first-pass availability in the prodrug form was estimated to be several percent in QGut model predictions (Yang et al., 2007) using the in vitro clearance for intestinal CMBL and a permeability estimate for OM (see Supplemental Method 1). However, the plasma esterase PON1, presumably in the portal blood, may play a supplemental role to complete the bioactivation of prodrug molecules that escape hydrolysis by CMBL in the intestine. Although the transit time through portal blood is quite short, the possibility of a significant contribution of plasma PON1 was indicated in our previous publication (Kobayashi et al., 2000), which showed that OM hydrolysis proceeds in human plasma with a half-life of less than several seconds. This multiple-enzyme contribution at multiple sites is considered to effectuate the minimal risk of significant interindividual variation regardless of possible inhibition by concomitantly administered drugs or genetic polymorphisms in CMBL that may cause varied production of the pharmacologically active metabolite. No components other than the active metabolite olmesartan were detected in plasma after oral administration of radiolabeled OM to healthy volunteers (Laeis et al., 2001). The multiple bioactivating enzymes at multiple sites in humans in vivo are considered to achieve the rapid and complete drug action of the orally administered prodrug OM.
In conclusion, we reported for the first time the purification of the OM-bioactivating hydrolase in human plasma and its identification as PON1, on a molecular basis. Moreover, we clearly demonstrated that PON1 plays a major role in OM bioactivation in human plasma, through a comparison of enzyme characteristics of PON1 and albumin.
Authorship Contributions
Participated in research design: Ishizuka, Fujimori, Yoshigae, Kurihara, and Ikeda.
Conducted experiments: Ishizuka, Fujimori, Nishida, and Sakurai.
Performed data analysis: Ishizuka, Fujimori, and Sakurai.
Wrote or contributed to the writing of the manuscript: Ishizuka, Yoshigae, Nakahara, Kurihara, Ikeda, and Izumi.
Acknowledgments
We gratefully acknowledge Kazumi Abiko and Junko Kawaguchi for the N-terminal amino acid sequencing. We also express our appreciation to Drs. Shinji Yamaguchi and Kazuishi Kubota for expert advice on the lipoprotein fractionation and liquid chromatography-tandem mass spectrometry peptide mapping, respectively; Dr. Masakatsu Kotsuma for assistance with the in vitro experiments and writing of the manuscript; and Miho Kazui and Eiko Suzuki for many helpful discussions.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- PON1
- paraoxonase/arylesterase 1
- OM
- olmesartan medoxomil
- CMBL
- carboxymethylenebutenolidase homolog
- RNH-6272
- 2-butyl-4-(1-hydroxy-1-methylethyl)-1-[2′-(1H-tetrazole-5-yl)-4-biphenylyl]-1H-imidazole-5-carboxylic acid
- PVDF
- polyvinylidene difluoride
- PAGE
- polyacrylamide gel electrophoresis
- KPB
- potassium phosphate buffer
- VLDL
- very-low-density lipoprotein
- LDL
- low-density lipoprotein
- HDL
- high-density lipoprotein
- LPDS
- lipoprotein-deficient serum.

- Received June 30, 2011.
- Accepted November 15, 2011.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}