


Abstract
Previous studies have shown the importance of the addition of albumin for characterization of hepatic glucuronidation in vitro; however, no reports exist on the effects of albumin on renal or intestinal microsomal glucuronidation assays. This study characterized glucuronidation clearance (CLint, UGT) in human kidney, liver, and intestinal microsomes in the presence and absence of bovine serum albumin (BSA) for seven drugs with differential UDP-glucuronosyltransferase (UGT) 1A9 and UGT2B7 specificity, namely, diclofenac, ezetimibe, gemfibrozil, mycophenolic acid, naloxone, propofol, and telmisartan. The impact of renal CLint, UGT on accuracy of in vitro-in vivo extrapolation (IVIVE) of glucuronidation clearance was investigated. Inclusion of 1% BSA for acidic drugs and 2% for bases/neutral drugs in incubations was found to be suitable for characterization of CLint, UGT in different tissues. Although BSA increased CLint, UGT in all tissues, the extent was tissue- and drug-dependent. Scaled CLint, UGT in the presence of BSA ranged from 2.22 to 207, 0.439 to 24.4, and 0.292 to 23.8 ml · min−1 · g tissue−1 in liver, kidney, and intestinal microsomes. Renal CLint, UGT (per gram of tissue) was up to 2-fold higher in comparison with that for liver for UGT1A9 substrates; in contrast, CLint, UGT for UGT2B7 substrates represented approximately one-third of hepatic estimates. Scaled renal CLint, UGT (in the presence of BSA) was up to 30-fold higher than intestinal glucuronidation for the drugs investigated. Use of in vitro data obtained in the presence of BSA and inclusion of renal clearance improved the IVIVE of glucuronidation clearance, with 50% of drugs predicted within 2-fold of observed values. Characterization and consideration of kidney CLint, UGT is particularly important for UGT1A9 substrates.
Introduction
Glucuronidation, via the UDP-glucuronosyltransferases (UGTs), is an important clearance mechanism for many xenobiotics and endogenous compounds (Tukey and Strassburg, 2000; Kiang et al., 2005). In vivo data indicate that the kidneys play an important role in the glucuronidation of certain drugs (Mazoit et al., 1990; Takizawa et al., 2005) and in the case of nonsteroidal anti-inflammatory drugs glucuronidation has also been linked to renal toxicity (Knights et al., 2009). Expression of certain UGT enzymes, e.g., 1A9 and 2B7, has been reported along the convoluted tubules, the loop of Henle, and collecting ducts and in the macula densa of the kidney (Gaganis et al., 2007; Lash et al., 2008); mRNA data suggest that expression of other UGTs is lower (Nishimura and Naito, 2006; Ohno and Nakajin, 2009). Considering the importance of UGT1A9 and UGT2B7 for drug clearance, renal glucuronidation may make a substantial contribution to systemic clearance, as illustrated in the case of propofol for which the kidney contributes up to one-third of total glucuronidation (Takizawa et al., 2005). In vitro characterization of renal glucuronidation and its importance relative to liver and intestinal glucuronidation has been relatively limited (Raoof et al., 1996; Bowalgaha and Miners, 2001; Soars et al., 2001).
Previous studies have shown that UGT1A9 and UGT2B7 are competitively inhibited by free fatty acids (FFAs) released during microsomal incubation; this effect is particularly evident for linoleic and arachidonic acids (Tsoutsikos et al., 2004). In contrast, FFAs were reported to have no effect on glucuronidation via UGT1A1 or UGT1A6 (Rowland et al., 2008). Bovine serum albumin (BSA) has been used to sequester FFAs in microsomal assays and prevent inhibition of glucuronidation (Rowland et al., 2007, 2008). Addition of BSA has resulted in up to 16-fold increases in glucuronidation intrinsic clearance (CLint, UGT) obtained either by depletion or metabolite formation in human liver microsomes (HLM), resulting in improved prediction accuracy when in vitro-in vivo extrapolation (IVIVE) of such data was performed (Rowland et al., 2008, 2009; Kilford et al., 2009; Manevski et al., 2011). Analogous to liver, arachidonic and linoleic acids represent two of the most abundant FFAs in renal and intestinal tissue (Soydan et al., 1996; Hoffmann et al., 2005; Rowland et al., 2007). However, to date there have been no studies assessing the effects of BSA on glucuronidation in human kidney (HKM) or intestinal (HIM) microsomes.
In vitro glucuronidation is influenced by assay conditions, and high intersubject variability has been observed for many UGTs, including UGT1A9 and UGT2B7 (Fisher et al., 2000; Soars et al., 2003; Court, 2010). UGT expression is not consistent throughout the kidney or intestine, and the region used for microsomal preparation will affect glucuronidation capacity (Gaganis et al., 2007; Lash et al., 2008; Bellemare et al., 2011). At present, absolute abundance data are available for only a few UGTs, primarily quantified in liver tissue and from a limited number of donors (Harbourt et al., 2012; Ohtsuki et al., 2012; Schaefer et al., 2012). In addition, UGT mRNA/protein expression data are often inconsistent among studies and are associated with high variability (Izukawa et al., 2009; Milne et al., 2011). IVIVE studies have concentrated on hepatic glucuronidation and have consistently underpredicted in vivo clearance (Boase and Miners, 2002; Miners et al., 2006; Cubitt et al., 2009; Kilford et al., 2009) but arguably no more than predictions of cytochrome P450 clearance (Hallifax et al., 2010). There have been few attempts to include extrahepatic glucuronidation in IVIVE (Soars et al., 2002; Al-Jahdari et al., 2006); lack of consideration of extrahepatic glucuronidation, differences in scaling factors, and in vitro assay conditions may contribute to the underprediction trend observed.
The aim of this study was to characterize and compare glucuronidation clearance in the kidney, liver, and intestine for seven selected drugs, namely diclofenac, ezetimibe, gemfibrozil, mycophenolic acid (MPA), naloxone, propofol, and telmisartan. The drugs selected covered a range of physicochemical properties, differing specificity for UGT1A9 and/or UGT2B7 and formed both acyl and phenolic glucuronides. Unbound intrinsic glucuronidation clearance (CLint, u, UGT) was characterized in alamethicin-activated HLM, HKM, and HIM under standardized in vitro conditions. For the first time, the effect of BSA on renal and intestinal glucuronidation has been assessed and compared with the effects observed in liver. In vitro data in the presence and absence of BSA were corrected for microsomal recovery to allow assessment of the relative contribution of each tissue to total glucuronidation clearance. These data were then scaled using the well stirred model to assess the impact of BSA on the prediction accuracy of IVIVE of glucuronidation clearance. Prediction accuracy of IVIVE determined using HLM data alone or using combined HLM and HKM data was also investigated.
Materials and Methods
Chemicals.
Diclofenac, gemfibrozil, MPA, naloxone, propofol, alamethicin (from Trichoderma viride), UDP-glucuronic acid, EDTA, BSA, and saccharic acid lactone were purchased from Sigma-Aldrich (Gillingham, Dorset, UK). Ezetimibe was purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). Telmisartan was purchased from Sequoia Research Products (Pangbourne, UK). All other reagents were of the highest grade available.
Source of the Microsomes.
Pooled HLM were purchased from BD Gentest (Woburn, MA). Pooled HKM and HIM were purchased from XenoTech (Tebu-Bio Ltd., Peterborough, Cambridgeshire, UK). Microsomes were stored at −80°C. HLM were pooled from 150 white donors, with 50% female and a mean age of 53 years (range, 18–79 years). HKM were pooled from 8 donors; with 88% white, 50% female, and a mean age of 61 years (range, 48–69 years). HIM were pooled from 18 donors; with 72% white, 39% female, and a mean age of 43 years (range, 21–67 years). Activity for pooled HLM was reported as 0.93, 0.85, 16.0, 2.00, and 0.72 nmol · min−1 · mg protein−1 for UGT1A1-, UGT1A4-, UGT1A6-, UGT1A9-, and UGT2B7-specific substrates. For HKM and HIM pools, activity was only reported for 4-methylumbelliferone (a substrate of multiple UGTs); with values of 125 and 8.44 nmol · min−1 · mg protein−1, respectively.
Microsome Experimental Conditions.
Incubations were performed in duplicate using a Thermomixer (Eppendorf, Hamburg, Germany). The final substrate concentration used was 1 μM, with the exception of propofol for which 5 μM was used. Substrate concentrations were >5-fold below the reported Km values, when these data were available (Kiang et al., 2005); therefore, the enzyme kinetics assessed are expected to be in the linear range. Microsomal protein concentrations ranged from 0.25 to 1.0 mg/ml; details for individual drugs and tissues are provided in Supplemental Table 1. Microsomes were activated by incubation with alamethicin (50 μg/mg protein) on ice for 15 min (Fisher et al., 2000; Cubitt et al., 2009; Kilford et al., 2009). Substrates were preincubated with activated microsomes and 0.1 M phosphate buffer, pH 7.1, containing 3.45 mM magnesium chloride, 1.15 mM EDTA, and 115 μM saccharic acid lactone, for 5 min at 37°C shaken at 900 rpm (Fisher et al., 2001; Cubitt et al., 2009; Kilford et al., 2009). The reaction was initiated by addition of glucuronic acid (5 mM in incubation) to give a final incubation volume of 1 ml (Cubitt et al., 2009; Kilford et al., 2009). Stability of acyl glucuronides at pH 7.1 was tested using gemfibrozil as a representative example. Gemfibrozil acyl glucuronide was stable at pH 7.1 over the incubation time, and, therefore, this pH was considered to be suitable for microsomal assays of drugs that form this type of glucuronide in the current study. Concentration of the organic solvent used (methanol) was <0.6% of the incubation media. Control incubations were performed for each drug with no cofactor present to account for any potential cofactor-independent loss of the drug over the incubation time. The total length of the incubations ranged from 30 to 60 min (Supplemental Table 1). Samples of 60 to 100 μl of the incubation were removed at each time point and added to an equal volume of ice-cold methanol containing the internal standard as specified in Table 1 to terminate the reaction. Samples were frozen at −20°C for at least 1 h and then thawed and centrifuged (MSE Mistral 3000i centrifuge; MSE, London, UK) at 4°C and 2500 rpm for 30 min. An aliquot of the supernatant (10 or 20 μl) was analyzed by LC-MS/MS for parent drug concentration. Experiments were repeated on three separate occasions with the exception of those with HIM because of limited availability of the microsomal batch investigated.
Incubation conditions for experiments including BSA were comparable to those for experiments without BSA. In preliminary experiments, minimal depletion was observed for the acidic drugs when 2% BSA was used, and, therefore, CLint, UGT could not be determined. This is most likely because acids bind strongly to BSA (Rowland et al., 2009), and the remaining free drug for depletion is minimal. Nonspecific binding of gemfibrozil and diclofenac at BSA concentrations ranging from 0.5 to 2% were determined (data not shown); nonspecific binding was reduced at lower BSA concentrations, but the resultant fraction unbound from protein in the incubation (fu, inc) values remained <0.1. Previous research showed a plateau in the increase of CLint, u, UGT results at 1% BSA (Rowland et al., 2007, 2009). Therefore, to allow quantifiable depletion and adequate sequestration of free fatty acids, a concentration of 1% BSA was used for the acidic drugs (diclofenac, gemfibrozil, MPA, and telmisartan), and 2% BSA was found to be suitable for bases/neutral drugs (ezetimibe, naloxone, and propofol). BSA concentration, microsomal protein concentrations, and the total length of the incubations are provided in Supplemental Table 1. The reaction was terminated by adding samples of the incubation to a double volume of ice-cold acetonitrile containing the internal standard. Samples were refrigerated for at least 10 min and then centrifuged (Mini Spin; Eppendorf) at 13,400 rpm and room temperature for 5 min. An aliquot of the supernatant (10 or 20 μl) was analyzed by LC-MS/MS for parent drug concentration. Experiments were repeated 3 times with the exception of those for HIM for which limited availability prevented repeat analysis.
LC-MS/MS.
All compounds were analyzed on a Waters 2790 with a Micromass Quattro Ultima triple quadruple mass spectrometer in negative mode with the exception of naloxone and telmisartan, which were analyzed on a Waters 2790 with a Micromass Quattro Micro triple quadruple mass spectrometer in positive mode. Source temperature was 125°C, desolvation temperature was 350°C, desolvation gas rate was 600 l/h, and cone gas rate was 150 l/h, with the exception of MPA and ezetimibe for which cone gas was 50 l/h. The capillary voltage was 3.25 kV for the Micromass Quattro Ultima and 3.5 kV for the Micromass Quattro Micro. Propofol was analyzed using single ion recording owing to its limited fragmentation. Analytes were separated using a Luna C18 (3 μm, 50 × 4.6 mm) column (Phenomenex, Macclesfield, UK) with the exception of naloxone, for which a Luna Phenyl Hexyl (3 μm, 50 × 4.6 mm) column (Phenomenex) was used. Four mobile phases were used, with varying gradients for each drug: A, 90% water, 10% methanol, and 0.05% formic acid; B, 10% water, 90% methanol, and 0.05% formic acid; C, 90% water, 10% methanol, and 1 mM ammonium acetate; and D, 10% water, 90% methanol, and 1 mM ammonium acetate. The flow rate was 1 ml/min, splitting to 0.25 ml/min before entry into the mass spectrometer. Mass spectrometric conditions for each drug are detailed in Table 1.
LC-MS/MS conditions used for assessing glucuronidation depletion over time of seven drugs in kidney, liver, and intestinal microsomes
Correction for Nonspecific Protein Binding.
The high throughput dialysis method (Gertz et al., 2008) was used to determine the fu, inc values for all drugs at 0, 0.1, 0.25, 0.5, and 1.0 mg/ml HLM protein concentrations in the presence and absence of 1 or 2% BSA. Al-Jahdari et al. (2006) reported similar fu, inc values for propofol in HLM and HKM; therefore, nonspecific binding was assumed to be comparable across systems at the same protein concentration. At the low substrate concentrations used herein, nonspecific binding is expected to be below the saturation limit and therefore independent of substrate concentration. This assumption is supported by previous data showing that nonspecific binding of propofol (in the presence and absence of BSA) and MPA (in the absence of BSA) is independent of substrate concentration when determined over a range of concentrations exceeding those used in the current study (Bowalgaha and Miners, 2001; Rowland et al., 2008, 2009). Dialysis membranes (12–14 kDa molecular mass cutoff) were purchased from HTDialysis, LLC (Gales Ferry, CT). Phosphate buffer (0.1 M, pH 7.1), containing 3.45 mM magnesium chloride, 1.15 mM EDTA, 115 μM saccharic acid lactone, and, where applicable, 1 or 2% BSA was added to the acceptor side of the membrane, and the relevant concentration of HLM in buffer was added to the donor side. Each HLM protein concentration was tested in triplicate. A 1 μM concentration of drug (5 μM for propofol) was spiked into each side of the membrane. The plate was left to equilibrate for 6 h on a plate shaker (450 rpm) at 37°C. Then samples from both the acceptor and donor sides of the membrane were transferred to Eppendorf tubes containing an equal volume of methanol with the relevant internal standard for non-BSA samples or a double volume of acetonitrile with internal standard for BSA samples. Sample preparation and LC-MS/MS methods were the same as those for microsomal incubations.
Recovery from equilibrium dialysis was greater than 70% for all drugs with the exception of propofol. Although recovery of propofol was low (<30%), fu, inc values determined both in the presence and absence of BSA were comparable to values reported by Rowland et al. (2008), and so the experimental data have been used. The fu, inc values for the other drugs investigated (both in the presence and absence of BSA) were comparable to values reported in the literature (where available) (Bowalgaha and Miners, 2001; Al-Jahdari et al., 2006; Gertz et al., 2008; Cubitt et al., 2009; Kilford et al., 2009). From preliminary experiments, the volume shift between the donor and acceptor side of the membrane was generally less than 5%, and therefore correction for volume shift had minimal impact on the experimental fu, inc.
Nonspecific binding data were analyzed across the range of protein concentrations in GraFit 5 (Erithacus Software, Horley, UK) to determine the binding constant (Ka) for each drug. The Ka was then used to calculate the fu, inc at the protein concentrations used for the glucuronidation depletion assays (eq. 1). The experimentally determined fu, inc values in the presence of BSA were constant over the range of microsomal protein concentrations and, therefore, Ka could not be calculated. In these cases, the mean of the experimental fu, inc values was used for all protein concentrations with BSA. The CLint, UGT values from HLM, HKM, and HIM were corrected for fu, inc (CLint, UGT/fu, inc) to generate the unbound intrinsic clearance (CLint, u, UGT) (microliters per minute per milligram of protein).
 where C is the microsomal protein concentration.
where C is the microsomal protein concentration.
Data Analysis.
The mean concentration of the duplicate samples at each time point was analyzed using GraFit 5 to determine the elimination rate constant (k). A nonlinear single exponential fit was used, with the exception of diclofenac in HLM without BSA for which a double exponential fit was used. The rate constant was used to calculate the CLint, UGT (eq. 2) and corrected for nonspecific binding to give CLint, u, UGT (microliters per minute per milligram of protein).

To compare CLint, u, UGT values from different tissues, CLint, u, UGT was expressed per gram of tissue by correcting the values for the microsomal protein yield, resulting in scaled CLint, u, UGT. Microsomal protein yields of 40, 12.8, and 20.6 mg of protein/g tissue were used for hepatic, renal, and intestinal data, respectively (Al-Jahdari et al., 2006; Barter et al., 2007; Cubitt et al., 2009). In contrast with hepatic microsomal recovery, only one estimate of microsomal recovery for HKMs could be found, based on data from only five donors and an unspecified kidney region (Al-Jahdari et al., 2006). Data from rats support this report; relative microsomal recovery for kidney is 30% of that for liver. The mean scaled CLint, u, UGT and S.D. have been presented for each drug. The mean scaled clearance values were compared using ratios.
Prediction of Intravenous Glucuronidation Clearance.
Ezetimibe cannot be dosed intravenously, and the lack of human fraction absorbed and fraction escaping gut metabolism data prevent the conversion of oral plasma clearance data to CLint, UGT; therefore, ezetimibe was not included in IVIVE. In vivo pharmacokinetic parameters including intravenous plasma clearance (CLi.v.), plasma binding (fu, p), blood/plasma ratio (RB), and renal clearance were collated from the literature for the remaining six drugs. In cases for which multiple clinical studies were available, the mean was calculated, weighted for the number of subjects in each study; references for the in vivo parameters and values from individual studies are shown in Supplemental Tables 2 to 6. Clinical data from Japanese, Korean, and Chinese populations, disease populations, subjects receiving concomitant medication, elderly or obese populations, extended release formulations, and cases in which the area under the curve was reported over an insufficient time period were excluded. No nonlinearity issues were observed across multiple dose levels, so all data were included. Only plasma clearance data were used, with the exception of propofol for which blood data were reported. CLi.v. data for each drug were collated from up to 12 clinical studies (up to 121 subjects in total) (Supplemental Table 2). Data for renal clearance were more limited and for the majority of drugs were available from only one report in the literature. For many of the drugs RB values were determined in vitro and intersubject variability in this parameter was not assessed. The fu, p data were more readily available in the literature, and values for individual drugs were collated from up to four studies (<59 subjects in total per drug). Intersubject variability for CLi.v. and fu, p was generally <30% within individual studies; values varied to a similar extent between studies, with the exception of fu, p for mycophenolic acid for which values from different studies varied by 48%. The ranges of values from individual studies collated for each parameter are shown elsewhere (Supplemental Tables 2–6).
Plasma CLi.v. of all drugs was corrected for RB and renal clearance to give blood metabolic clearance (CLH) (Ito and Houston, 2005; Gertz et al., 2010). Clearance via metabolism was corrected for the fraction metabolized by glucuronidation (fm, UGT) to give the apparent in vivo glucuronidation clearance (CLUGT). Where available, data were corrected using in vitro fm, UGT from HLM data in the presence or absence of BSA (from in-house data), which were generated in a pool of HLM with UGT and cytochrome P450 characterization similar to that used for the current work. In the case of MPA and telmisartan, no in vitro fm, UGT data were available; however, for these two drugs in vivo data suggest almost complete glucuronidation (fm, UGT = 1 for telmisartan or 0.95 for MPA), and this assumption was applied in IVIVE.
Currently, there are no kidney-specific models available for IVIVE; therefore, perfusion-limited kinetics and assumptions of the well stirred model were applied for the kidney, which may have biased the IVIVE results presented herein. In vitro HLM and HKM CLint, u, UGT data (per gram of tissue) were scaled to give CLint, u, UGT (per kilogram body weight) using average organ weights of 21.4 g of tissue/kg (Ito and Houston, 2005) and 4.5 g tissue/kg for liver and kidney, respectively. The average kidney weight (range from 4.1 to 5.0 g tissue/kg) was estimated from the collated literature reports (Supplemental Table 7). These data were then scaled up using the well stirred model to give apparent glucuronidation clearance for the respective organ (eq. 3) (Houston, 1994). The intestine was expected to have minimal impact on glucuronidation clearance after intravenous drug administration; therefore, HIM CLint, u, UGT data were not included during IVIVE.
 where fu, B represents fraction unbound in blood, Qh is hepatic blood flow (Qh = 20.7 ml · min−1 · kg−1), and CLUGT represents apparent in vivo glucuronidation clearance. An analogous model was used for the kidney where Qh was replaced with Qr (average renal blood flow of 16.4 ml · min−1 · kg−1) and in vitro HKM data were used. The values reported for renal blood flow, gender differences, and methods used to estimate this parameters were collated from >50 studies reported in the literature (Supplemental Table 8).
where fu, B represents fraction unbound in blood, Qh is hepatic blood flow (Qh = 20.7 ml · min−1 · kg−1), and CLUGT represents apparent in vivo glucuronidation clearance. An analogous model was used for the kidney where Qh was replaced with Qr (average renal blood flow of 16.4 ml · min−1 · kg−1) and in vitro HKM data were used. The values reported for renal blood flow, gender differences, and methods used to estimate this parameters were collated from >50 studies reported in the literature (Supplemental Table 8).
For IVIVE using both the liver and kidney data, combined CLUGT was calculated as the sum of scaled hepatic and renal CLUGT. Predicted CLUGT values from HLM data alone or in combination with HKM data were compared with the observed CLUGT values from in vivo data. Both observed CLUGT and predicted CLUGT values were calculated using fm, UGT and in vitro scaled CLint, u, UGT data determined in the presence of BSA (where available). Similar comparisons were made using data in the absence of BSA. Prediction bias and precision were assessed using geometric fold error and root mean squared error, respectively, as described previously (Gertz et al., 2010).
Results
CLint, UGT was determined for seven drugs in pooled HKM, HLM, and HIM in the presence and absence of BSA using the substrate depletion approach. The fu, inc values were experimentally determined in HLM in the presence and absence of BSA and were used to correct the corresponding CLint, UGT values for nonspecific binding (Table 2).
CLint, u, UGT of seven drugs in human kidney, liver, and intestinal microsomes in the presence and absence of bovine serum albumin
Data represent the mean (±S.D.) of n = 3 for human kidney and liver microsomes or n = 1 for human intestinal microsomes. Experiments were performed in duplicate.
Glucuronidation Depletion in Human Kidney, Liver, and Intestinal Microsomes in the Absence of Bovine Serum Albumin.
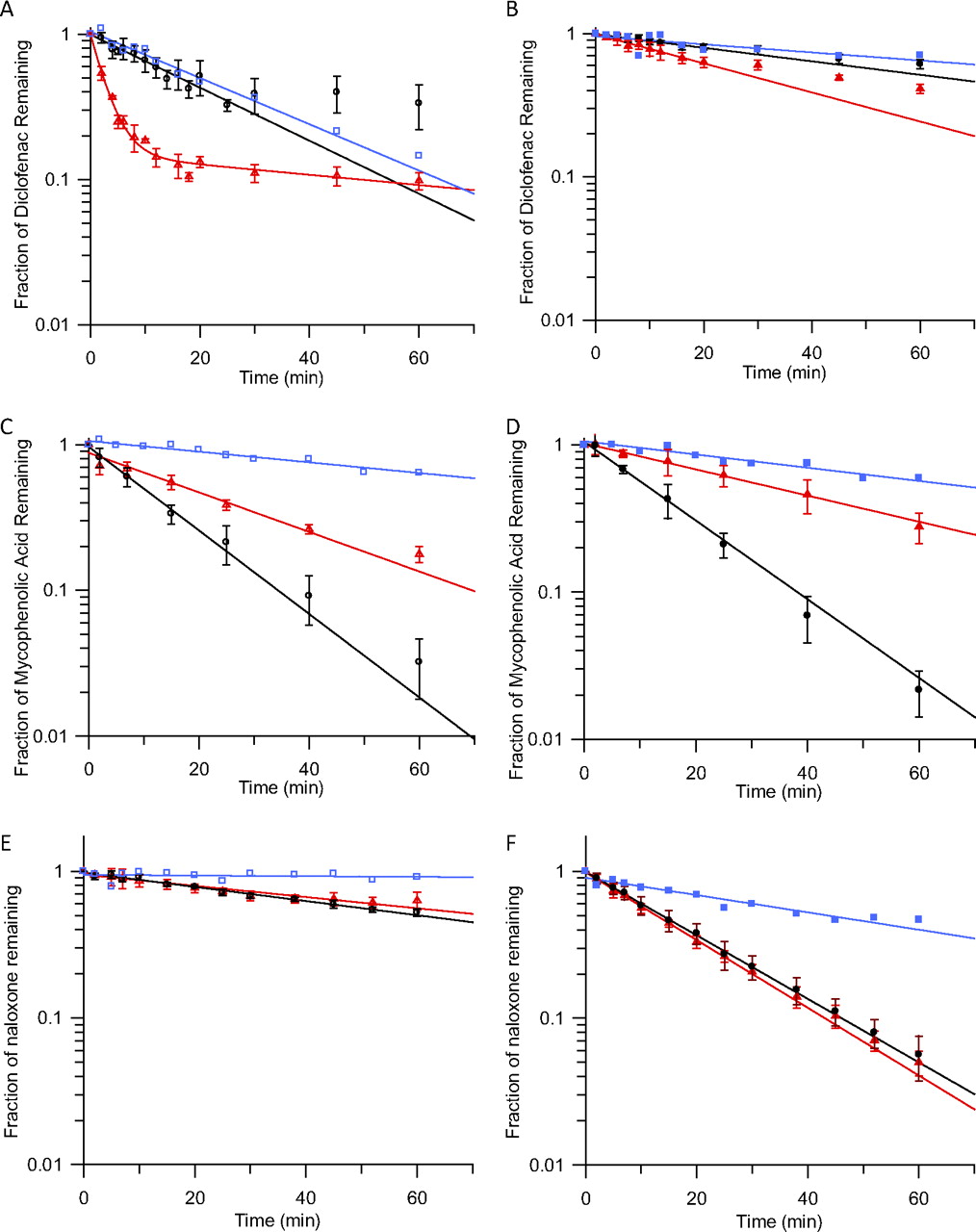
Figure 1 illustrates representative examples of depletion rates in different tissues and the effects of BSA on depletion of diclofenac, MPA, and naloxone; individual examples are discussed below in the text. The profiles for the remaining four drugs are shown in Supplemental Fig. 1. Depletion was linear over time for six of the seven drugs; however, for diclofenac a biphasic decline was observed (Fig. 1A). The initial linear decline in diclofenac concentration was used to calculate the CLint, UGT. Biphasic depletion of several drugs has been observed both in-house and in literature reports (Cubitt et al., 2009; Kilford et al., 2009) for microsomal and hepatocyte assays (data not shown); however, no trends regarding the assay conditions, type of glucuronide formed (acyl or phenol), substrate physicochemical properties, or UGT specificity could be identified. Reduced microsomal activity over time or end product inhibition may contribute to the observed biphasic decline. Diclofenac assays were performed over 60 min with the initial rate of depletion ending within 10 to 15 min. Assays for other drugs were performed up to 60 min with no apparent decline in depletion rate. Therefore, it is unlikely that the microsomes were losing activity over the incubation. The end of the initial phase occurred after 60 to 80% depletion of diclofenac. For other drugs, no reduction in depletion rate was observed after up to 99% depletion, suggesting that insufficient substrate or cofactor was not the cause of the decrease in diclofenac depletion rate.

Mean fraction of drug remaining (±S.D.) over time in human kidney, liver, and intestinal microsomes in the presence (B, D, and F) and absence (A, C, and E) of bovine serum albumin. ○, ▵, and □, depletion in human kidney, liver, and intestinal microsomes without BSA, respectively; ●, ▴, and ■, depletion in human kidney, liver, and intestinal microsomes with BSA, respectively. A and B, diclofenac. C and D, mycophenolic acid. E and F, naloxone.
For MPA and propofol, the rate of depletion was faster in HKM than in HLM, whereas for diclofenac, ezetimibe, and telmisartan the opposite was observed; for the remaining drugs the depletion rate was similar (Fig. 1, A, C, and E). The rate of depletion in HIM was slower than that in HLM for all drugs and only exceeded that in HKM for ezetimibe and telmisartan (Fig. 1, A, C, and E). The fu, inc in the absence of BSA ranged from 0.18 to 1.00 for ezetimibe and naloxone, respectively and was ≥0.65 for six of the seven drugs.
CLint, u, UGT in HKM without BSA ranged from 9.72 to 295 μl · min−1 · mg protein−1 for telmisartan and ezetimibe, respectively (Table 2). Glucuronidation clearance in HLM without BSA covered a 10-fold larger range than that in HKM (CLint, u, UGT up to 2930 μl · min−1 · mg protein−1 for ezetimibe). However, the largest range of CLint, u, UGT for the drugs investigated was observed for HIM (>500-fold), with the lowest value being 0.502 μl · min−1 · mg protein−1 for naloxone. Ezetimibe had the highest CLint, u, UGT in all tissues, whereas telmisartan or naloxone had the lowest glucuronidation clearance. However, the rank order of clearance values for the other drugs differed among the tissues.
Effect of Albumin on Glucuronidation Depletion by Human Kidney, Liver, and Intestinal Microsomes.
All the drugs studied bind strongly to BSA (fu, inc in the presence of BSA = 0.01–0.25) with the exception of naloxone (fu, inc = 0.96) (Table 2); binding to BSA was constant over the 0 to 1 mg/ml HLM protein concentration range studied. For four of seven drugs investigated, fu, inc under these conditions was <0.1 (Table 2). Because of the reduced free drug available for glucuronidation, the rate of depletion decreased for most drugs upon inclusion of BSA. To allow quantifiable depletion, the microsomal protein concentration in the incubation was increased to 1 mg/ml, and the incubation length was increased to 60 min for diclofenac, gemfibrozil, and telmisartan (Supplemental Table 1). Even with the increase in microsomal protein concentration and incubation length, inclusion of BSA (1% for acids and 2% for bases/neutral drugs) reduced the observed rate of depletion for diclofenac, ezetimibe, and gemfibrozil (Fig. 1, A and B). The biphasic nature of diclofenac depletion observed without BSA was not apparent in the presence of BSA (Fig. 1, A and B). In contrast, inclusion of BSA increased the rate of depletion for naloxone, MPA, and propofol in HKM (Fig. 1, C–F); these drugs also had the highest fu, inc in the presence of BSA of 0.96, 0.18, and 0.25, respectively. BSA had minimal effect on the rate of depletion for telmisartan or for MPA and propofol in HLM (Fig. 1, C and D). Depletion of telmisartan in HKM in the presence of 1% BSA resulted on average in <10% depletion from the initial concentration over 60 min, most likely as a result of the high binding to BSA, despite the lower BSA concentration used. Depletion from the initial concentration in individual replicates ranged from 0 to 21% at 60 min and also varied by up to 20% at other time points. Clearance estimates for individual incubations of telmisartan in HKM with BSA were consequently highly variable (coefficient of variation = 50%). Data in the absence of BSA showed depletion of 20%, and variability between replicates was low (<20%).
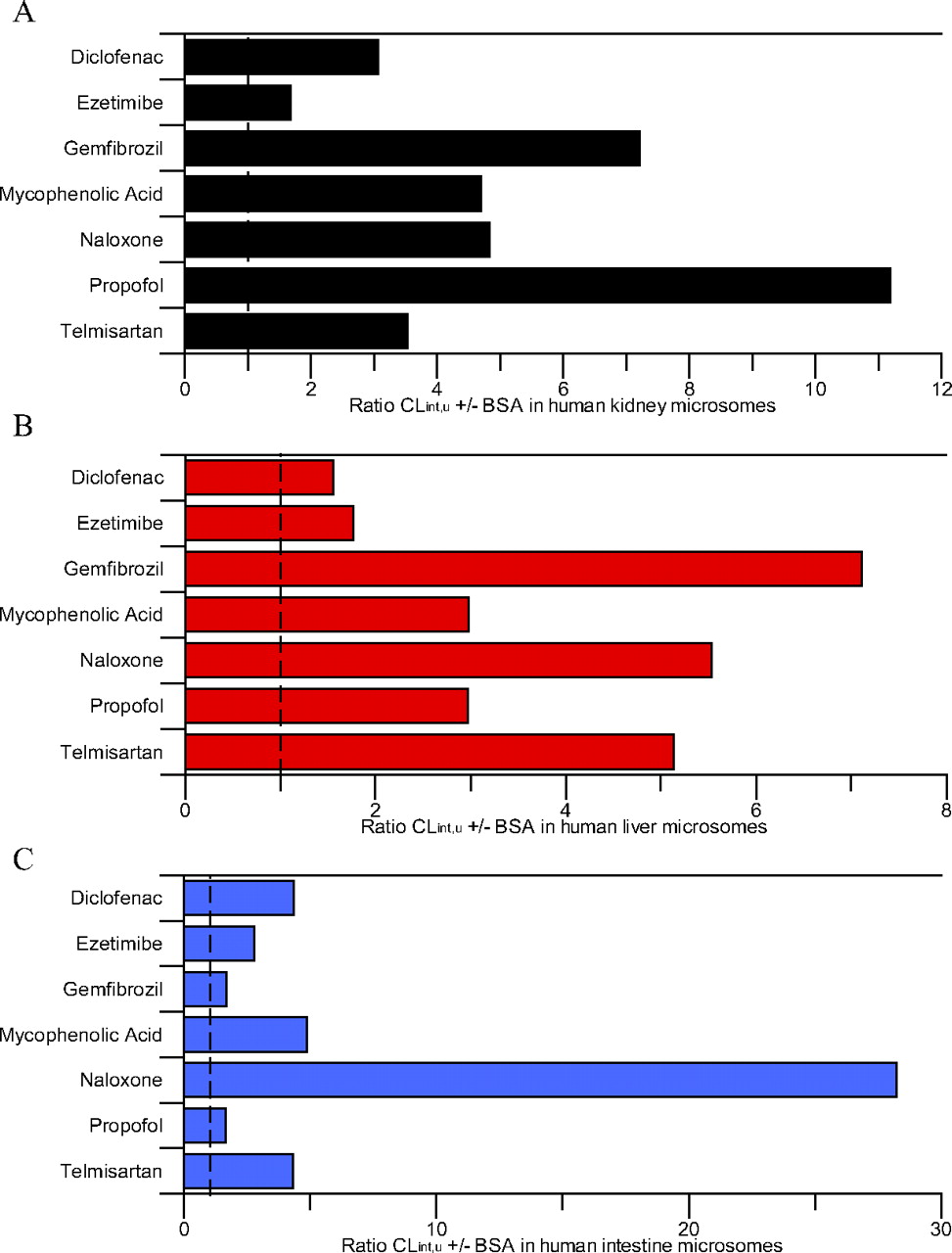
Inclusion of BSA in the incubation resulted in a 56-fold range of renal glucuronidation clearance; telmisartan CLint, u, UGT was the lowest, consistent with the data in the absence of BSA (Table 2). A larger range of CLint, u, UGT was again observed for HLM (CLint, u, UGT up to 5180 μl · min−1 · mg protein−1 for ezetimibe) (Table 2). Comparable to HLM, an 82-fold range in CLint, u, UGT was observed for HIM in the presence of BSA. Although the CLint, u, UGT for ezetimibe remained the highest and that for naloxone remained the lowest in HLM and HIM, the rank order of CLint, u, UGT for the other drugs changed in all tissues after the inclusion of BSA.
Inclusion of BSA increased CLint, u, UGT by 1.6- to 11-fold in all tissues, with the exception of naloxone in the intestine for which CLint, u, UGT increased by 28-fold (Fig. 2). No consistent trends in the increase of CLint, u, UGT resulting from BSA inclusion were observed between the three tissues (Fig. 2). For HLM and HKM, ezetimibe had one of the lowest increases in clearance (1.8- and 1.7-fold, respectively), whereas for HIM similarly low increases in CLint, u, UGT were observed for gemfibrozil and propofol. Similar increases in CLint, u, UGT were observed between HLM and HKM for gemfibrozil and naloxone, whereas the increase in CLint, u, UGT was higher in HKM than in HLM for diclofenac, MPA, and propofol. The increase in intestinal CLint, u, UGT was greater than in liver and kidney for diclofenac, ezetimibe, and naloxone, but lower for gemfibrozil and propofol. The increase in CLint, u, UGT was comparable in HIM and HKM for MPA (4.7- and 4.9-fold, respectively). Upon inclusion of BSA, increases in CLint, u, UGT in this study were generally lower than those reported previously using both the substrate depletion and metabolite formation methods, most noticeably for propofol for which the increases in CLint, u, UGT values were on average 3-fold lower than reported previously (Rowland et al., 2008; Kilford et al., 2009).
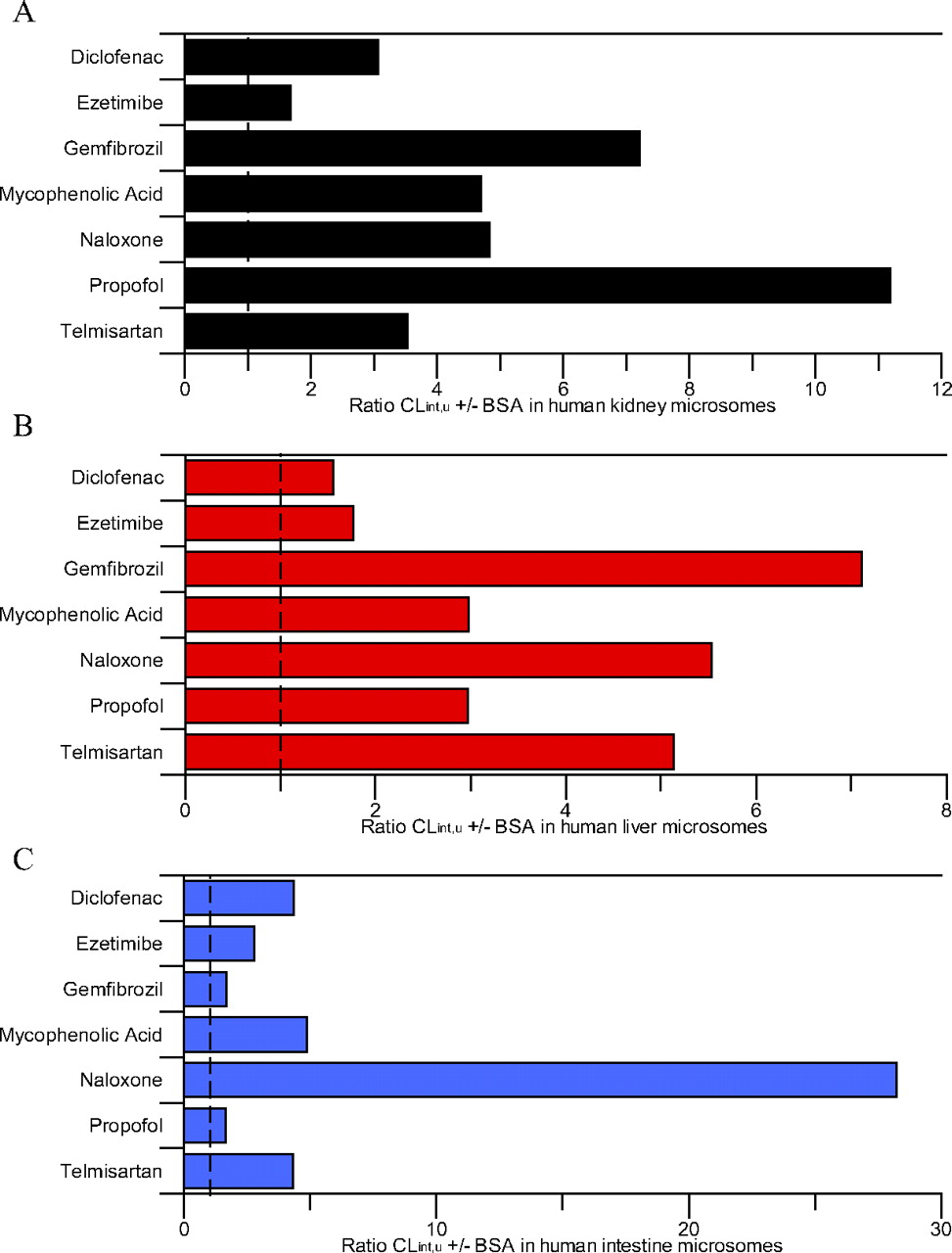
Ratio of mean glucuronidation CLint, u, UGT ± bovine serum albumin in human kidney, liver, and intestinal microsomes. A, human kidney microsomes. B, human liver microsomes. C, human intestine microsomes. Dashed line represents the line of unity.
Correction of CLint, u, UGT for Microsomal Recovery.
In vitro data were corrected for microsomal recovery resulting in scaled CLint, u, UGT values (Tables 3 and 4). After correction, scaled CLint, u, UGT values for HIM without BSA ranged from 0.01 to 8.61 ml · min−1 · g tissue−1 for naloxone and ezetimibe, respectively. Scaled CLint, u, UGT values for HKM without BSA covered a similar range (0.12–3.78 ml · min−1 · g tissue−1 for telmisartan and ezetimibe, respectively). In the absence of BSA, the lowest and highest scaled CLint, u, UGT values for HLM were approximately an order of magnitude greater than those for HIM or HKM (Table 3). In the presence of BSA, the extent of the range of scaled CLint, u, UGT values was similar in all three tissues, but again HLM scaled CLint, u, UGT values were approximately an order of magnitude higher than those obtained in the kidney and intestine (Table 4).
Scaled CLint, u, UGT of seven drugs from human kidney, liver, and intestinal microsomal data obtained in the absence of albumin
Data represent the mean (±S.D.) of n = 3 for human kidney and liver microsomes or n = 1 for human intestine microsomes. Experiments were performed in duplicate.
Scaled CLint, u, UGT of seven drugs from human kidney, liver, and intestinal microsomal data obtained in the presence of 1 or 2% bovine serum albumin
Data represent the mean (±S.D.) of n = 3 for human kidney and liver microsomes or n = 1 for human intestine microsomes. Experiments were performed in duplicate.
Comparison of In Vitro Glucuronidation Clearance in Kidney Relative to That in Liver.
In the absence of BSA, kidney/liver scaled CLint, u, UGT ratios ranged from 0.03 to 1.2 for ezetimibe and MPA, respectively (Table 3). MPA was the only drug for which scaled CLint, u, UGT values obtained in the absence of BSA were greater in kidney than in liver microsomes (Fig. 3A). Scaled glucuronidation clearance by kidney microsomes represented 32 to 43% of that in liver for gemfibrozil, naloxone, and propofol and for the remaining drugs the renal scaled CLint, u, UGT was <8% of that in the liver (Fig. 3A). Inclusion of BSA gave a similar range of kidney/liver ratios (Table 4); however, in addition to MPA, scaled CLint, u, UGT for propofol was higher in the kidney than in the liver (Fig. 3B). Comparison of kidney and liver scaled CLint, u, UGT values remained similar for the other drugs after addition of BSA (Table 4). No trends between the scaled CLint, u, UGT values in liver and the ratio of kidney/liver scaled clearance were observed.
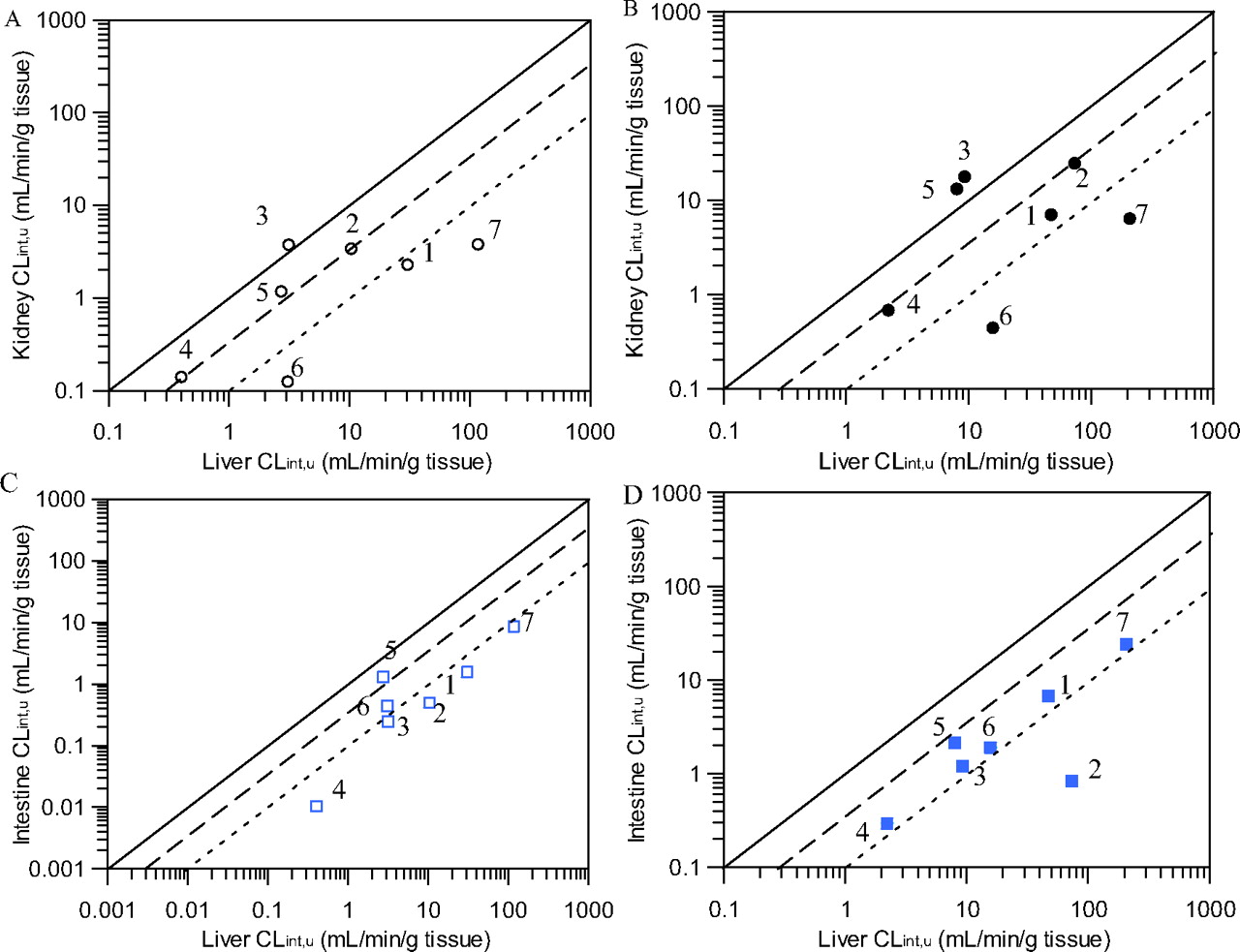
Comparison of mean scaled CLint, u, UGT in human kidney and intestinal microsomes compared with liver scaled CLint, u, UGT in the presence and absence of bovine serum albumin. A and B, represent kidney versus liver scaled CLint, u, UGT. C and D, represent intestine versus liver scaled CLint, u, UGT. ○ and □, represent CLint, u, UGT data obtained in the absence of BSA; ● and ■, represent CLint, u, UGT data generated in the presence of BSA. ——, line of unity; – – –, 3-fold difference; - - -, 10-fold difference. 1, diclofenac; 2, gemfibrozil; 3, mycophenolic acid; 4, naloxone; 5, propofol; 6, telmisartan; 7, ezetimibe.
Comparison of In Vitro Glucuronidation Clearance in Intestine Relative to That in Liver.
Intestinal scaled CLint, u, UGT in the absence of BSA represented <15% of that in the liver for all drugs, with the exception of propofol for which the intestinal scaled CLint, u, UGT was 48% of that in the liver (Fig. 3C). Addition of BSA increased intestine/liver scaled clearance ratios by approximately 2-fold for four of seven drugs. In the case of telmisartan, the clearance ratio between intestine and liver remained unchanged, whereas it decreased for propofol and gemfibrozil, in the latter case to 1% of the liver (Fig. 3D). Analogous to renal glucuronidation, no trends between the scaled clearance in liver and the intestine/liver scaled CLint, u, UGT ratios were observed.
Comparison of In Vitro Glucuronidation Clearance in Kidney Relative to That in Intestine.
Scaled CLint, u, UGT without BSA was higher in the kidney than in the intestine for diclofenac, gemfibrozil, MPA, and naloxone (kidney/intestine ratios = 1.47–15.4). For the remaining three drugs, scaled clearance by the kidney represented 28 to 91% of that in the intestine. There were no consistent trends in the change in kidney/intestine ratios upon inclusion of BSA. Scaled CLint, u, UGT was higher in the kidney than that in the intestine for five of seven drugs in the presence of BSA with kidney/intestine scaled CLint, u, UGT ratios ranging from 0.23 to 29.7 for telmisartan and gemfibrozil, respectively (Table 4).
Prediction of Intravenous Glucuronidation Clearance.
In vitro hepatic and renal scaled CLint, u, UGT values for six drugs were further scaled up using the well stirred model and compared with observed in vivo intravenous CLUGT. Table 5 provides a summary of the in vivo and in vitro data used for IVIVE; full details of the in vivo parameters are presented elsewhere (Supplemental Tables 2–6). Prediction success for IVIVE of scaled CLint, u, UGT using HLM data alone or in combination with HKM data is shown in Fig. 4 (full details in Supplemental Table 9). Comparison of predicted to observed intravenous CLUGT from HLM data alone in the presence of BSA leads to a median underprediction of 66% with diclofenac, MPA, and naloxone being predicted within 2-fold of observed values (Fig. 4A). A 3-fold prediction bias was observed and precision (root mean squared error) was 6.60. Predicted clearance ranged from 13 to 102% of observed values for telmisartan and diclofenac, respectively, with the exception of gemfibrozil for which predicted CLUGT was 3.6-fold greater than the observed value. Use of hepatic microsomal data obtained in the absence of BSA resulted in reduced prediction success for most of the drugs investigated, with increased bias (15-fold) and only two of six drugs predicted within 2-fold of the observed CLUGT. The exceptions were propofol and gemfibrozil for which predicted clearance based on the data in the absence of BSA was in better agreement with observed values (Fig. 4). Predictions from HLM data alone in the absence of BSA performed slightly better than those reported previously; the mean predicted CLUGT was 39% of observed CLUGT versus 10 to 22% in previous reports (Soars et al., 2002; Cubitt et al., 2009; Kilford et al., 2009).
In vitro and in vivo parameters used for IVIVE of glucuronidation clearance in human liver and kidney microsomes in the absence and presence of 1 or 2% bovine serum albumin
In vivo parameter data represent weighted mean; details of variability between literature reports are given elsewhere (Supplemental Tables 2–6). Predicted CLUGT data represent the mean (±S.D.) of n = 3 for human kidney and liver microsomes.

Comparison of observed intravenous to predicted CLUGT calculated using the well stirred model and mean (n = 3) in vitro human liver microsomal (A) or combined human liver and kidney microsomal (B) data in the presence and absence of bovine serum albumin. Observed intravenous clearance data were corrected for RB, renal clearance, and fm, UGT. ▵ and ▴, human liver microsome data only; ○ and ●, combined human liver and kidney microsomal data. ▵ and ○, data obtained in the absence of BSA; ▴ and ●, data obtained in the presence of BSA. ——, line of unity; – – –, 2-fold difference. Vertical bars represent S.D. for in vitro data; horizontal bars represent minimum and maximum in vivo CLUGT values (calculated from minimum and maximum CLi.v. values; Supplemental Table 2). 1, diclofenac; 2, gemfibrozil; 3, mycophenolic acid; 4, naloxone; 5, propofol; 6, telmisartan.
Upon inclusion of renal CLUGT (in the presence of BSA) predictions of MPA, naloxone, and propofol were all improved compared with use of HLM data alone (percentage of predicted/observed CLUGT increased from 72 to 102, from 61 to 69, and from 18 to 24%, respectively) (Fig. 4B). However, a comparable increase in the overprediction of gemfibrozil CLUGT was observed, and therefore the overall prediction success remained similar to using HLM data alone. In the absence of BSA, inclusion of renal CLUGT showed marginal improvement in prediction accuracy compared with predictions based on HLM data in isolation (Fig. 4B). Soars et al. (2002) reported a more pronounced increase in prediction accuracy when scaled renal CLint, u, UGT (in the absence of BSA) was included in IVIVE. However, kidney and liver microsomal protein recoveries were assumed to be equivalent in Soars et al. (2002), in contrast to the current study in which lower protein recovery for kidney relative to liver was applied, leading to a potentially underestimated impact of renal glucuronidation.
Diclofenac, gemfibrozil, and telmisartan all had very low fu, inc in the presence of BSA (<0.07), and any errors associated with experimental determination may have a pronounced effect on the success of CLUGT prediction. However, the fu, inc value for gemfibrozil and telmisartan would have to differ substantially (up to 7-fold) for the predicted clearance to be within 2-fold of observed values. The fu, inc value for diclofenac in the presence of BSA was also in this range (<0.1), but its CLUGT was predicted well. In addition, all nonspecific binding experiments were conducted at five microsomal concentrations (three replicates of each). For gemfibrozil and telmisartan, recovery was high (88–100%), and low variability between the fu, inc values of individual replicates (coefficient of variation of 11 and 14%, respectively) was observed. It is therefore unlikely that nonspecific binding or any errors associated with low fu, inc estimates can in isolation rationalize the pronounced over/underpredictions of CLUGT for gemfibrozil and telmisartan. For many of the drugs studied, fu, p values were also very low (<0.02 for five of the six drugs), and the variability associated with this parameter was generally <30% for individual drugs in the data set (Supplemental Table 4). Varying fu, p values by this magnitude had a marginal impact on the prediction accuracy of IVIVE and could not in isolation rationalize the pronounced over/underpredictions of CLUGT for gemfibrozil, naloxone, propofol, and telmisartan.
Discussion
This study characterized and compared extrahepatic and hepatic glucuronidation in alamethicin-activated microsomes for diclofenac, ezetimibe, gemfibrozil, MPA, naloxone, propofol, and telmisartan using standardized assay conditions. The impact of BSA on microsomal assays has previously been investigated for hepatic glucuronidation (Rowland et al., 2008; Kilford et al., 2009), and its effects on extrahepatic glucuronidation are reported here. The impact of albumin on the relative ratios in CLint, u, UGT among tissues was investigated. IVIVE including both kidney and liver CLint, u, UGT was performed, and the impact of BSA on prediction accuracy was assessed.
Impact of BSA on Microsomal Glucuronidation Clearance in Three Tissues.
Inclusion of BSA in HLM incubations increased CLint, u, UGT up to 7-fold, which is within the range of previously reported data (Kilford et al., 2009; Rowland et al., 2009; Manevski et al., 2011). In addition, the presence of BSA in the incubations led to increases in HKM and HIM CLint, u, UGT values, supporting reports that linoleic and arachidonic acids (the most potent UGT inhibitors of FFAs) (Tsoutsikos et al., 2004) are abundant in all three tissues investigated (Soydan et al., 1996; Hoffmann et al., 2005; Rowland et al., 2007). Inclusion of BSA (1% for acids and 2% for bases/neutral drugs) decreased the fu, inc for all drugs (<0.1 for four of seven drugs); consequently, depletion was very low for some of the drugs investigated. For highly bound drugs, the accuracy of fu, inc may have a significant impact on CLint, u, UGT values and subsequent IVIVE. Consideration of alternatives (e.g., human intestinal fatty acid binding protein with reduced drug binding) as proposed by Rowland et al. (2009) might eliminate some of these issues, but unfortunately this protein was not available at the time of the current study.
In HLM, drugs cleared primarily by UGT1A9 and UGT2B7 showed similar increases in CLint, u, UGT upon inclusion of BSA. Ezetimibe is mainly cleared via UGT1A1 and had the smallest increase in HLM and HKM CLint, u, UGT in the presence of BSA. UGT1A9 substrates (propofol and MPA) showed a more pronounced increase in CLint, u, UGT in HKM than in HLM, which could potentially be attributed to higher expression of UGT1A9 in kidney tissue. Surprisingly, diclofenac (primarily cleared via UGT2B7) showed a low increase in HLM CLint, u, UGT; as described above, this increase was less pronounced than that for HKM CLint, u, UGT. Gemfibrozil and naloxone had comparable increases in CLint, u, UGT in kidney and liver, indicating similar effects of BSA on UGT2B7 in these tissues. Trends observed in HLM and HKM are in agreement with limited available UGT mRNA/protein expression data, indicating higher expression of UGT1A9 in the kidney and comparable UGT2B7 expression in kidney and liver (Nishimura and Naito, 2006; Ohno and Nakajin, 2009; Harbourt et al., 2012; Ohtsuki et al., 2012). Lower expression of these UGTs reported in the intestine is in contrast to the pronounced increases in CLint, u, UGT observed for diclofenac, ezetimibe, and naloxone in intestinal microsomes upon inclusion of BSA.
Telmisartan is minimally cleared via UGT1A9 but showed one of the largest increases in CLint, u, UGT in HLM and HIM. This result may suggest that other UGTs involved in its clearance (UGT1A3 > 1A1/1A7/1A8) may also be affected by FFAs. Arachidonic acid is cleared via UGT1A3 (Little et al., 2004) and, therefore, may cause competitive inhibition of this enzyme, which would rationalize the large increase in CLint, u, UGT observed for telmisartan. Gemfibrozil is also partially cleared via UGT1A3 and showed >6-fold increases in clearance in HLM and HKM. For naloxone (UGT1A8 and UGT2B7 substrate), CLint, u, UGT increased by 28-fold in HIM in the presence of albumin, whereas the increase in gemfibrozil CLint, u, UGT (mainly UGT2B7 substrate) was only 2-fold. These findings suggest that UGT1A8 may also be inhibited by FFAs; to date, the effect of FFAs and BSA on UGT1A3 and the intestinal specific UGTs has not been studied. The effect of BSA on the different tissues may also be influenced by differences in their FFA content.
Comparison of In Vitro Glucuronidation Clearance in Different Tissues.
Inclusion of BSA altered the ratios of scaled clearance between different tissues in a substrate-dependent manner. Scaled renal CLint, u, UGT (per gram of tissue) obtained in the presence of BSA was 3% of that in liver for substrates cleared primarily by UGT1A1 and UGT1A3 (ezetimibe and telmisartan). In contrast, for UGT1A9 substrates (propofol and MPA), scaled renal CLint, u, UGT was up to 2-fold higher than hepatic values, suggesting that expression of UGT1A1 and UGT1A3 is lower and that of UGT1A9 is higher in the kidney than in the liver. The kidney/liver scaled CLint, UGT ratios for drugs primarily cleared by UGT2B7 ranged from 0.15 to 0.33, potentially because of lower expression of UGT2B7 in the kidney than in the liver or higher expression of other contributing UGTs for these drugs in the liver.
Our data showed the highest intestine/liver ratio for propofol. If propofol is selectively glucuronidated by UGT1A9, the tissue ratios should not change after inclusion of BSA; however, this was not the case, indicating that propofol may also be cleared by other UGTs, in agreement with previous reports that UGT1A8 contributes to propofol glucuronidation (Cheng et al., 1999). For the drugs predominantly cleared by UGT1A1 and UGT1A3, the intestine/liver CLint, u, UGT ratios were low. The scaled renal CLint, u, UGT was lower than that in intestine for these drugs, suggesting that UGT1A1 and UGT1A3 are most highly expressed in liver followed by intestine. These functional activity data are compatible with currently available UGT mRNA/protein expression data (Nishimura and Naito, 2006; Ohno and Nakajin, 2009; Harbourt et al., 2012; Ohtsuki et al., 2012).
For non-BSA data, the kidney/liver scaled CLint, u, UGT ratios were generally lower than those calculated from previously published data, with the exception of MPA (Bowalgaha and Miners, 2001; Shipkova et al., 2001; Bernard and Guillemette, 2004). Intestine/liver scaled CLint, u, UGT ratios for diclofenac, gemfibrozil, and naloxone were also lower than those calculated from data in Cubitt et al. (2009), with the exception of propofol (Raoof et al., 1996). Differences in the tissue scaled CLint, u, UGT ratios between studies could arise from the use of microsomes from different donors (Court, 2010), use of microsomes prepared from different sections of the kidney or intestine (generally not reported), inconsistencies in assay conditions and scaling factors applied.
Prediction of Intravenous Glucuronidation Clearance.
Prediction of intravenous CLUGT from HLM data alone in the presence of BSA resulted in underestimation of in vivo clearance, with three of the six drugs predicted within 2-fold of observed values. Similar to data reported by Kilford et al. (2009), gemfibrozil CLUGT was overpredicted. In agreement with previous reports, IVIVE of in vitro data in the absence of BSA resulted in reduced prediction success for most of the drugs investigated (Rowland et al., 2007, 2008, 2009; Kilford et al., 2009).
Inclusion of renal CLUGT increased prediction success for several drugs with half of the drugs being predicted well. The largest impact was observed for MPA and propofol, which is not surprising, considering that these drugs had a higher scaled renal CLint, u, UGT than liver CLint, u, UGT. However, inclusion of renal CLUGT also increased the overprediction observed for gemfibrozil. Further analysis of a larger set of drugs and development of more physiologically based kidney models will allow more conclusive assessment of the impact of renal CLUGT in IVIVE. Robust scaling factors for extrahepatic glucuronidation are yet to be substantiated, and interpretation of CLint, UGT differences between tissues is problematic. Scaling of CLint, u, UGT values using absolute abundance of individual UGTs may alter the tissue CLint, u, UGT ratios reported here. Abundance data from a large cohort of individuals (as in the case of cytochromes P450), information on regional expression differences across tissues of interest, and appropriate covariate analysis are required to allow further refinement of IVIVE of glucuronidation clearance and associated variability.
Errors in the accuracy of plasma or nonspecific binding estimates in isolation could not explain the poor predictions for gemfibrozil, propofol, naloxone, and telmisartan. However, variability in fu, p or other pharmacokinetic parameters collated from the literature combined with any errors in fu, inc estimation may have contributed to the poor IVIVE accuracy observed for some drugs. In addition, renal CLint, u, UGT was determined from a very limited pool of donors in comparison with liver CLint, u, UGT, and, therefore, any variability due to the age and/or health of these donors could not be accounted for in IVIVE. Use of more dynamic modeling techniques, e.g., physiologically based pharmacokinetic models, may improve predictions of CLUGT for drugs such as telmisartan, for which transporters play an important role in drug disposition (Ishiguro et al., 2006; Menochet et al., 2011). For nontransporter substrates, the use of physiologically based models may provide limited improvement as issues associated with the UGT scaling factors will affect their prediction accuracy, analogous to the current IVIVE strategy.
In conclusion, this is the first publication assessing the effect of BSA on characterization and comparison of glucuronidation in kidney, intestine, and liver. Use of 1% BSA for acids and 2% for bases/neutral drugs was found to be suitable for determination of CLint, UGT in microsomes from all three tissues. Inclusion of BSA in microsomal glucuronidation assays increased CLint, u, UGT in a tissue- and drug-dependent manner, altered the ratios of clearance between the tissues, and resulted in improved overall prediction accuracy of clearance. The potential for intestinal specific UGTs and UGT1A3 to be inhibited by FFAs, highlighted by the current findings, requires further investigation. For four of the seven drugs, the scaled renal CLint, u, UGT (with BSA) represented >30% of that in the liver, being higher than that in the liver for UGT1A9 substrates. Inclusion of renal CLUGT in IVIVE further improved predictions for some drugs, most noticeably for substrates of UGT1A9, and, therefore, should be characterized in the future for drugs cleared by this enzyme.
Authorship Contributions
Participated in research design: Gill, Houston, and Galetin.
Conducted experiments: Gill.
Performed data analysis: Gill.
Wrote or contributed to the writing of the manuscript: Gill, Houston, and Galetin.
Acknowledgments
We thank Sue Murby and Dr. David Hallifax (University of Manchester) for valuable assistance with the LC-MS/MS, Dr. Michael Gertz (University of Manchester) for useful discussions, and Sofie Andersson for assistance with the intestinal depletion assays.
Footnotes
The work was funded by a consortium of pharmaceutical companies (GlaxoSmithKline, Lilly, Pfizer, and Servier) within the Centre for Applied Pharmacokinetic Research at the University of Manchester. K.L.G. is a recipient of a Ph.D. studentship from Biotechnology and Biological Sciences Research Council.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.ABBREVIATIONS:
- UGT
- UDP-glucuronosyltransferase
- FFA
- free fatty acid
- BSA
- bovine serum albumin
- HLM
- human liver microsomes
- IVIVE
- in vitro-in vivo extrapolation
- HKM
- human kidney microsomes
- HIM
- human intestinal microsomes
- MPA
- mycophenolic acid
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry.

- Received November 29, 2011.
- Accepted January 23, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}