


Abstract
Telmisartan, a nonpeptide angiotensin II receptor antagonist, is selectively distributed to liver. In the present study, we have characterized the contribution of organic anion transporting polypeptide (OATP) isoforms to the hepatic uptake of telmisartan by isolated rat hepatocytes, human cryopreserved hepatocytes, and human transporter-expressing cells. Because it is difficult to evaluate the transport activity of telmisartan because of its extensive adsorption to cells and culture materials, we performed the uptake study in the presence of human serum albumin. The saturable uptake of telmisartan into isolated rat hepatocytes took place in a Na+-independent manner and was inhibited by pravastatin, taurocholate, and digoxin, which are Oatp substrates and inhibitors, but not by organic cation, tetraethylammonium, indicating the involvement of Oatp isoforms in its uptake into rat hepatocytes. To identify which human OATP transporters are important for the hepatic uptake of telmisartan, the uptake assay was carried out using OATP1B1- and OATP1B3-expressing human embryonic kidney 293 cells and cryopreserved human hepatocytes. The uptake of telmisartan by OATP1B3-expressing cells was saturable (Km = 0.81 μM) and significantly higher than that by vector-transfected cells. In contrast, no significant uptake was observed in OATP1B1-expressing cells. We also observed the saturable uptake of telmisartan by human hepatocytes. Thirty micromolar estrone-3-sulfate, which can selectively inhibit OATP1B1-mediated uptake compared with OATP1B3, did not inhibit the uptake of telmisartan in human hepatocytes, whereas it could inhibit the uptake of estradiol 17β-d-glucuronide mediated by OATP1B1. These results suggest that OATP1B3 is predominantly involved in the hepatic uptake of telmisartan in humans.
The renin-angiotensin-aldosterone system plays a central role in blood pressure regulation (Hedner, 1999). This system leads to the production of the hormone angiotensin I, which is converted to the active hormone angiotensin II by angiotensin-converting enzyme. Angiotensin II receptor antagonists prevent angiotensin II from exerting its vasoconstrictive effects on blood vessels (Oliverio and Coffman, 1997). Five nonpeptide angiotensin receptor antagonists, losartan, candesartan cilexetil, valsartan, telmisartan, and olmesartan medoxomil, are commercially available in Japan. Losartan, candesartan cilexetil, and olmesartan medoxomil are prodrugs, whereas valsartan and telmisartan are themselves pharmacologically active. All of them are mainly excreted into feces from the liver.
Telmisartan (Fig. 1) is a lipophilic compound with a log P value of 3.2, and it exists in anionic form at neutral pH (Wienen et al., 2000). Telmisartan is metabolized to an inactive acylglucuronide conjugate by UDP-glucuronosyltransferases in the intestinal wall and liver (Stangier et al., 2000c). The acylglucuronide is rapidly excreted into the bile and accounts for 10% of the circulating drug-related material 1 h after p.o. administration of telmisartan (Stangier et al., 2000a,c). Telmisartan is selectively distributed to the liver in rats with a liver/plasma concentration ratio of more than 40 (Wienen et al., 2000). After a single p.o. and i.v. administration in humans, more than 98% of the total radioactivity was recovered in feces as a parent drug, and less than 1% of the radioactivity was recovered in urine (Stangier et al., 2000a). Telmisartan shows a large interindividual variability in its plasma concentrations, and both the maximum concentration in plasma (Cmax) and area under the plasma concentration-time curve (AUC) increased in a slightly more than the dose-proportional manner after p.o. administration (Stangier et al., 2000a,c). Because liver is a major clearance organ of telmisartan, it is essential to assess the uptake mechanism of telmisartan by human hepatocytes to gain an insight into the mechanism for its nonlinear pharmacokinetics and large interindividual variability.
Several transporters, such as Na+-taurocholate cotransporting polypeptide and organic anion transporting polypeptide (OATP) 1B1 (previously called OATP-C/OATP2/LST-1), OATP1B3 (previously called OATP8/LST-2), OATP2B1 (previously called OATP-B), organic anion transporter 2, and organic cation transporter 1, are expressed on the sinusoidal membrane of hepatocytes and are thought to be involved in the transport of a wide variety of compounds including clinically used drugs, such as 3-hydroxy-3-methylglutaryl CoA reductase inhibitors (statins), from blood into hepatocytes (Abe et al., 1999; Hsiang et al., 1999; Kok et al., 2000; Konig et al., 2000a,b; Tamai et al., 2000; Faber et al., 2003; Hirano et al., 2004). In particular, OATP1B1 and OATP1B3 are mainly expressed in human liver (Hagenbuch and Meier, 2003), and the substrate specificity of OATP1B3 commonly overlaps that of OATP1B1, so several compounds can be bisubstrates of both OATP1B1 and OATP1B3, such as estradiol 17β-d-glucuronide (E217βG), pitavastatin, and rifampicin (Vavricka et al., 2002; Hirano et al., 2004). Hirano et al. (2004) have recently established methods for estimating the contribution of OATP1B1 and OATP1B3 to the hepatic uptake of a number of compounds. They have shown that pitavastatin and E217βG are taken up in human hepatocytes mainly by OATP1B1. On the other hand, the uptake of fexofenadine, an H1 receptor antagonist, was mainly mediated by OATP1B3 rather than OATP1B1 (Shimizu et al., 2005). This kind of information is helpful for predicting the effect of changes in expression level and function of certain transporters caused by genetic polymorphisms, pathophysiological conditions, and transporter-mediated drug-drug interactions on the overall hepatic uptake clearance and subsequent pharmacokinetics of drugs. Moreover, if compounds can selectively inhibit OATP1B1- or OATP1B3-mediated transport, we can also easily calculate the contribution of each transporter to the uptake of particular compounds in human hepatocytes by estimating the fraction of their uptake that can be inhibited by transporter-selective inhibitors. However, our preliminary study and a previous report showed that cholecystokinin octapeptide (CCK-8) could also inhibit OATP1B1-mediated transport (Nozawa et al., 2003), indicating that it cannot be used as an OATP1B3-selective inhibitor, although it is selectively transported by OATP1B3 (Ismair et al., 2001). On the other hand, there is little information about selective inhibitors against OATP1B1.
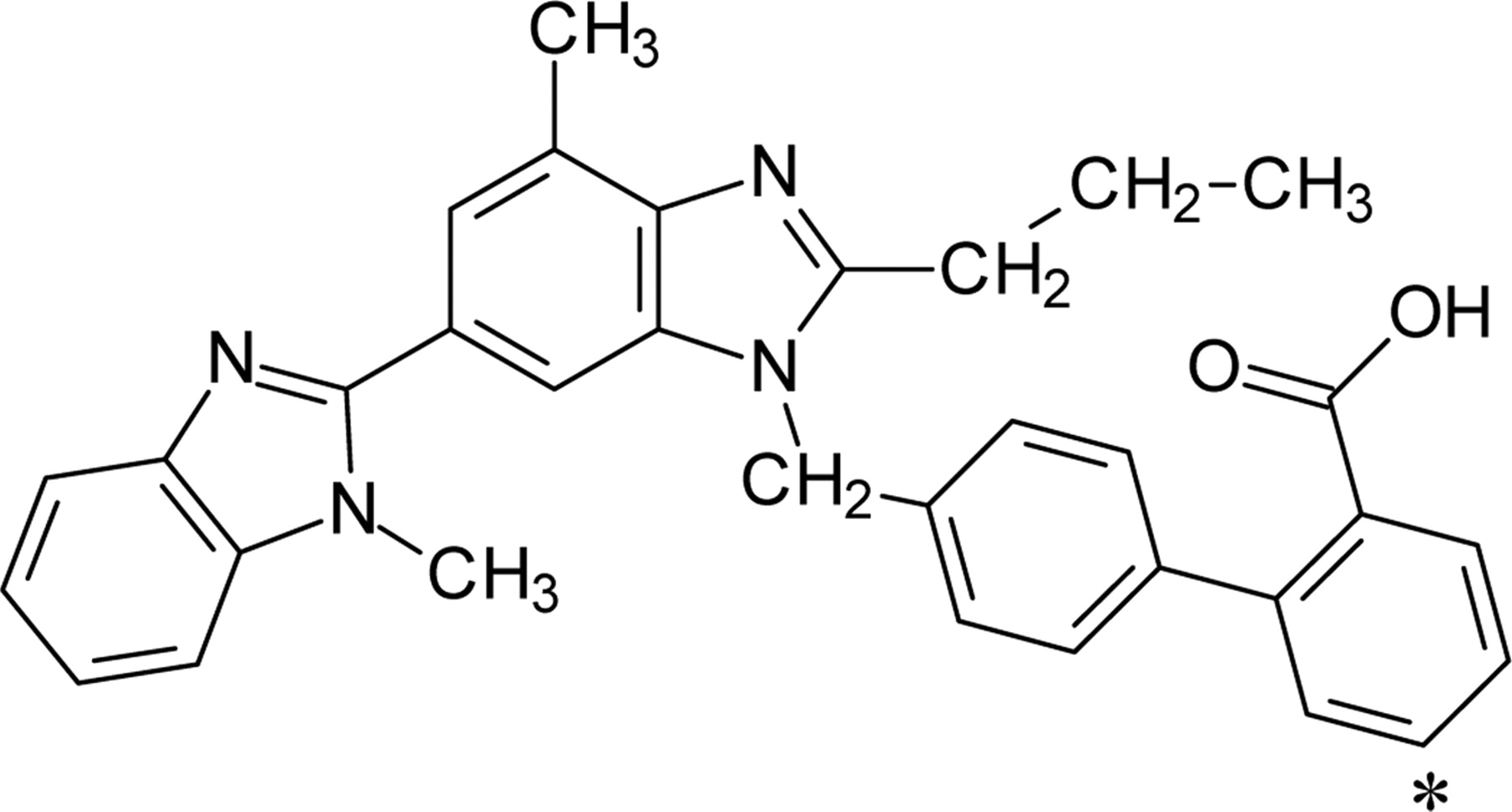
Chemical structure of [3H]telmisartan. The asterisk denotes the position of the 3H-label.
Therefore, the aim of our study is to show the involvement of OATP family transporters in the hepatic uptake process of telmisartan and estimate the contribution of OATP1B1 and OATP1B3 to its uptake in human hepatocytes by a newly developed estimation method using the OATP1B1-selective inhibitor estrone-3-sulfate (E-sul).
Materials and Methods
Chemicals. [3H]Telmisartan (762 GBq/mmol, radiochemical purity >98%), 4′-[(1,4′-dimethyl-2′-propyl[2,6′-bi-1H-benzimidazol]-1′-yl)methyl]-[1,1′-biphenyl]-2-carboxylic acid, and unlabeled telmisartan were synthesized by Boehringer Ingelheim Pharma KG (Biberach, Germany) (Ries et al., 1993). [3H]E217βG, [3H]E-sul, and [3H]taurocholate were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). [3H]CCK-8 was purchased from Amersham Biosciences UK Ltd. (Little Chalfont, Bucking-hamshire, UK). Unlabeled E217βG, E-sul, taurocholate, CCK-8, and digoxin were purchased from Sigma-Aldrich (St. Louis, MO). Pravastatin and tetra-ethylammonium (TEA) were obtained from Wako Pure Chemicals (Kyoto, Japan). All the other chemicals and reagents were commercial products of reagent grade.
Cell Culture. OATP1B1-, OATP1B3-, and OATP2B1-expressing or vector-transfected human embryonic kidney (HEK) 293 cells were established previously (Hirano et al., 2004; Shimizu et al., 2005). HEK293 cells were grown in Dulbecco's modified Eagle's medium low glucose (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B at 37°C with 5% CO2 and 95% humidity. Cells were then seeded in 12-well plates [coated with 50 mg/l poly(l-lysine) and 50 mg/l poly(l-ornithine); Sigma] at a density of 1.5 × 105 cells/well. For the transport study, the cell culture medium was replaced with culture medium supplemented with 5 mM sodium butyrate for 24 h before transport assay to induce the expression of transporters.
Transport Study Using Transporter Expression Systems. The transport study was carried out as described previously (Hirano et al., 2004). Uptake was initiated by adding Krebs-Henseleit buffer (118 mM NaCl, 23.8 mM NaHCO3, 4.8 mM KCl, 1.0 mM KH2PO4, 1.2 mM MgSO4, 12.5 mM HEPES, 5.0 mM glucose, and 1.5 mM CaCl2 adjusted to pH 7.4) containing radiolabeled and unlabeled substrates after cells had been washed twice and preincubated with Krebs-Henseleit buffer at 37°C for 15 min. The uptake was terminated at designated times by adding ice-cold Krebs-Henseleit buffer after removal of the incubation buffer. Then, cells were washed twice with 1 ml of ice-cold Krebs-Henseleit buffer, solubilized in 1 N NaOH, and kept for 1 h at 37°C. Aliquots were transferred to scintillation vials after adding a half volume of 2 N HCl. The radioactivity associated with the cells and incubation buffer was measured in a liquid scintillation counter (TRI-CARB 2500TR, PerkinElmer) after adding 2 ml of scintillation fluid. The remaining 50 μl of cell lysate was used to determine the protein concentration by the method of Lowry with bovine serum albumin as a standard.
Preparation of Rat and Human Hepatocytes. Isolated rat hepatocytes were prepared from Sprague-Dawley rats weighing 200 to 300 g by the collagenase perfusion method described previously (Yamazaki et al., 1993). Isolated hepatocytes (viability >80%) were suspended in Krebs-Henseleit buffer, adjusted to 2.0 × 106 cells/ml, and stored on ice before the uptake experiment. Cryopreserved human hepatocytes (lot. OCF, MYO, and 094) were purchased from In Vitro Technologies, Inc. (Baltimore, MD). The preparation of hepatocytes was performed as described previously (Shitara et al., 2003). The cryopreserved human hepatocytes were resuspended in Krebs-Henseleit buffer to give a final cell density of 1.0 × 106 viable cells/ml for the uptake study. The number of viable cells was determined by trypan blue staining. To measure the uptake in the absence of Na+, sodium chloride and sodium bicarbonate in Krebs-Henseleit buffer were replaced with choline chloride and choline bicarbonate.
Transport Study Using Hepatocytes. Before the uptake studies, the cell suspensions were prewarmed at 37°C for 3 min. The uptake studies were initiated by adding an equal volume of buffer (120–200 μl) containing labeled and unlabeled substrates to the cell suspension. After incubation at 37°C for 0.5, 2, or 5 min, the reaction was terminated by separating the cells from the substrate solution. For this purpose, an aliquot of 80 μl of incubation mixture was collected and placed in a centrifuge tube (450 μl) containing 50 μl of 2 N NaOH under a layer of 100 μl of oil mixture (density, 1.015, a mixture of silicone oil and mineral oil; Sigma-Aldrich), and subsequently the sample tube was centrifuged for 20 s using a standard centrifuge (17,500g, MX-100, TOMY). During this process, hepatocytes passed through the oil layer into the alkaline solution. After an overnight incubation in alkali to dissolve the hepatocytes and the centrifuge tube was frozen in liquid nitrogen, the centrifuge tube was cut, and each compartment was transferred to a scintillation vial. The compartment containing the dissolved cells was neutralized with 50 μl of 2 N HCl. The aliquots were mixed with scintillation mixture, and the radioactivity was measured in a liquid scintillation counter.
Estimation of Protein Unbound Concentration of Telmisartan in the Presence of Human Serum Albumin. The unbound concentration of 1 μM telmisartan in the presence of human serum albumin (HSA) (0, 0.1, 0.3, 1, 3, and 5%) was determined after a 2-h incubation at 37°C by equilibrium dialysis (DIANORM, Dainippon Pharmaceutical Ltd., Osaka, Japan).
Kinetic Analyses. Ligand uptake was expressed as the uptake volume (microliters per milligram protein), given as the radioactivity associated with the cells (disintegrations per milligram protein) divided by its concentration in the incubation media (disintegrations per microliter). Specific uptake was obtained by subtracting the uptake into vector-transfected cells from the uptake into cDNA-transfected cells. Kinetic parameters were obtained using eq. 1: 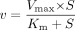 where v is the uptake velocity of the substrate (picomoles per minute per milligram protein), S is the substrate concentration in the medium (micromolar), Km is the Michaelis constant (micromolar), and Vmax is the maximum uptake rate (picomoles per minute per milligram protein). Fitting was performed by the nonlinear least-squares method using a MULTI program (Yamaoka et al., 1981). The half-inhibitory concentration (IC50) of inhibitors was obtained by examining their inhibitory effects on the uptake of CCK-8, E217βG, and telmisartan based on eq. 2:
where v is the uptake velocity of the substrate (picomoles per minute per milligram protein), S is the substrate concentration in the medium (micromolar), Km is the Michaelis constant (micromolar), and Vmax is the maximum uptake rate (picomoles per minute per milligram protein). Fitting was performed by the nonlinear least-squares method using a MULTI program (Yamaoka et al., 1981). The half-inhibitory concentration (IC50) of inhibitors was obtained by examining their inhibitory effects on the uptake of CCK-8, E217βG, and telmisartan based on eq. 2: 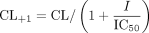 where CL and CL+I represent the uptake clearance in the absence and presence of inhibitor, respectively, and I is the concentration of inhibitor. IC50 values were estimated by the nonlinear least-squares method using a MULTI program (Yamaoka et al., 1981). To determine the saturable hepatic uptake clearance in rat and human hepatocytes, we first determined the hepatic uptake clearance [CL(2 min-0.5 min)] (microliters per minute per 106 cells) by calculating the slope of the uptake volume (Vd) (microliters per 106 cells) between 0.5 and 2 min (eq. 3). The hepatic uptake clearance was fitted to eq. 1 by means of nonlinear least-squares regression analysis using a MULTI program (Yamaoka et al., 1981). The saturable hepatic uptake clearance (CLhep) was determined by subtracting CL(2 min-0.5 min) in the presence of an excess of unlabeled substrate (excess) from that in the absence of unlabeled substrate (tracer) (eq. 4).
where CL and CL+I represent the uptake clearance in the absence and presence of inhibitor, respectively, and I is the concentration of inhibitor. IC50 values were estimated by the nonlinear least-squares method using a MULTI program (Yamaoka et al., 1981). To determine the saturable hepatic uptake clearance in rat and human hepatocytes, we first determined the hepatic uptake clearance [CL(2 min-0.5 min)] (microliters per minute per 106 cells) by calculating the slope of the uptake volume (Vd) (microliters per 106 cells) between 0.5 and 2 min (eq. 3). The hepatic uptake clearance was fitted to eq. 1 by means of nonlinear least-squares regression analysis using a MULTI program (Yamaoka et al., 1981). The saturable hepatic uptake clearance (CLhep) was determined by subtracting CL(2 min-0.5 min) in the presence of an excess of unlabeled substrate (excess) from that in the absence of unlabeled substrate (tracer) (eq. 4). 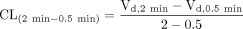
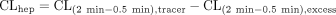
Estimation of the Maximum Unbound Concentration of Inhibitors at the Liver Inlet. The maximum unbound concentration at the liver inlet (Iin,max, u) was calculated from the following equation (eq. 5) as described previously (Ito et al., 1998).  where Cmax,blood and fu,blood are estimated by the reported values of the maximum blood concentration of drug after p.o. administration of the clinical dose (D = 160 mg) and the plasma protein unbound fraction (0.005) and blood-to-plasma concentration ratio (about 1.0) in humans (Stangier et al., 2000a,b). Qh is the hepatic blood flow rate (96.6 l/h). To avoid the false-negative prediction, ka is set to a theoretically maximum absorption rate constant (6/h), and Fa is set to one.
where Cmax,blood and fu,blood are estimated by the reported values of the maximum blood concentration of drug after p.o. administration of the clinical dose (D = 160 mg) and the plasma protein unbound fraction (0.005) and blood-to-plasma concentration ratio (about 1.0) in humans (Stangier et al., 2000a,b). Qh is the hepatic blood flow rate (96.6 l/h). To avoid the false-negative prediction, ka is set to a theoretically maximum absorption rate constant (6/h), and Fa is set to one.
Statistical Analysis. The two-tailed Dunnett test was used to assess the significance of differences between three sets of data. Differences were considered to be statistically significant when P < 0.05.
Results
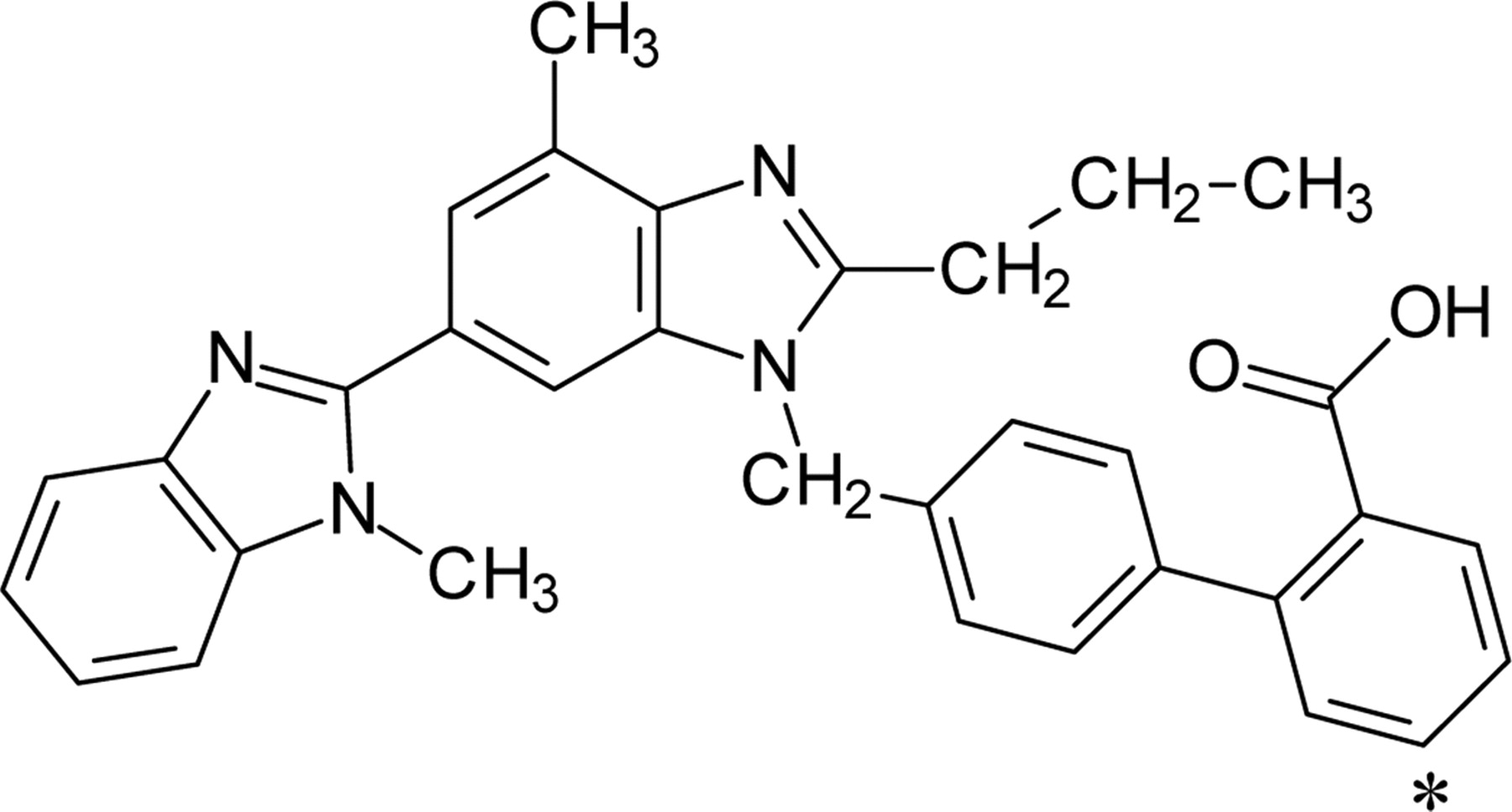
Uptake of Telmisartan into Isolated Rat Hepatocytes. Telmisartan was taken up into isolated rat hepatocytes in a time-dependent manner. Whereas saturation of the uptake of telmisartan by an excess of unlabeled telmisartan (40 μM) was not clearly observed in the incubation media with 0 and 0.1% HSA, it could be observed in the presence of 0.3 to 5% HSA (Fig. 2, A–F). Thus, we decided to evaluate the telmisartan uptake with more than 0.3% of HSA in the incubation media to prevent its extensive adsorption to the cells and culture materials. The protein unbound fraction of telmisartan in the incubation media with 0.3, 1, 3, and 5% HSA was 0.056, 0.018, 0.006, and 0.004, respectively. Both the uptake and the unbound fraction of telmisartan were reduced in parallel as the concentration of HSA was increased (Fig. 2G). In the presence of 1% HSA, telmisartan was taken up into isolated rat hepatocytes linearly up to 5 min (Fig. 3A). The concentration dependence of the uptake of telmisartan was studied over concentration range of 0.1 to 40 μM in the presence of 1% HSA. Eadie-Hofstee plot showed one saturable component (Fig. 3B), and the apparent Km and Vmax values for telmisartan uptake in the presence of 1% HSA were 21.7 ± 4.4 μM and 371 ± 58 pmol/min/106 cells, respectively. Depletion of Na+ in the incubation media did not affect the uptake of telmisartan (Fig. 4A), and the uptake was inhibited by pravastatin, digoxin, and taurocholate, which are substrates and inhibitors of Oatp isoforms, with IC50 values of 58.6 ± 15.5, 45.3 ± 11.7, and 300 ± 99 μM, respectively. However, 1 mM TEA, a typical substrate of organic cation transporter, did not affect the uptake of telmisartan (Fig. 4B).
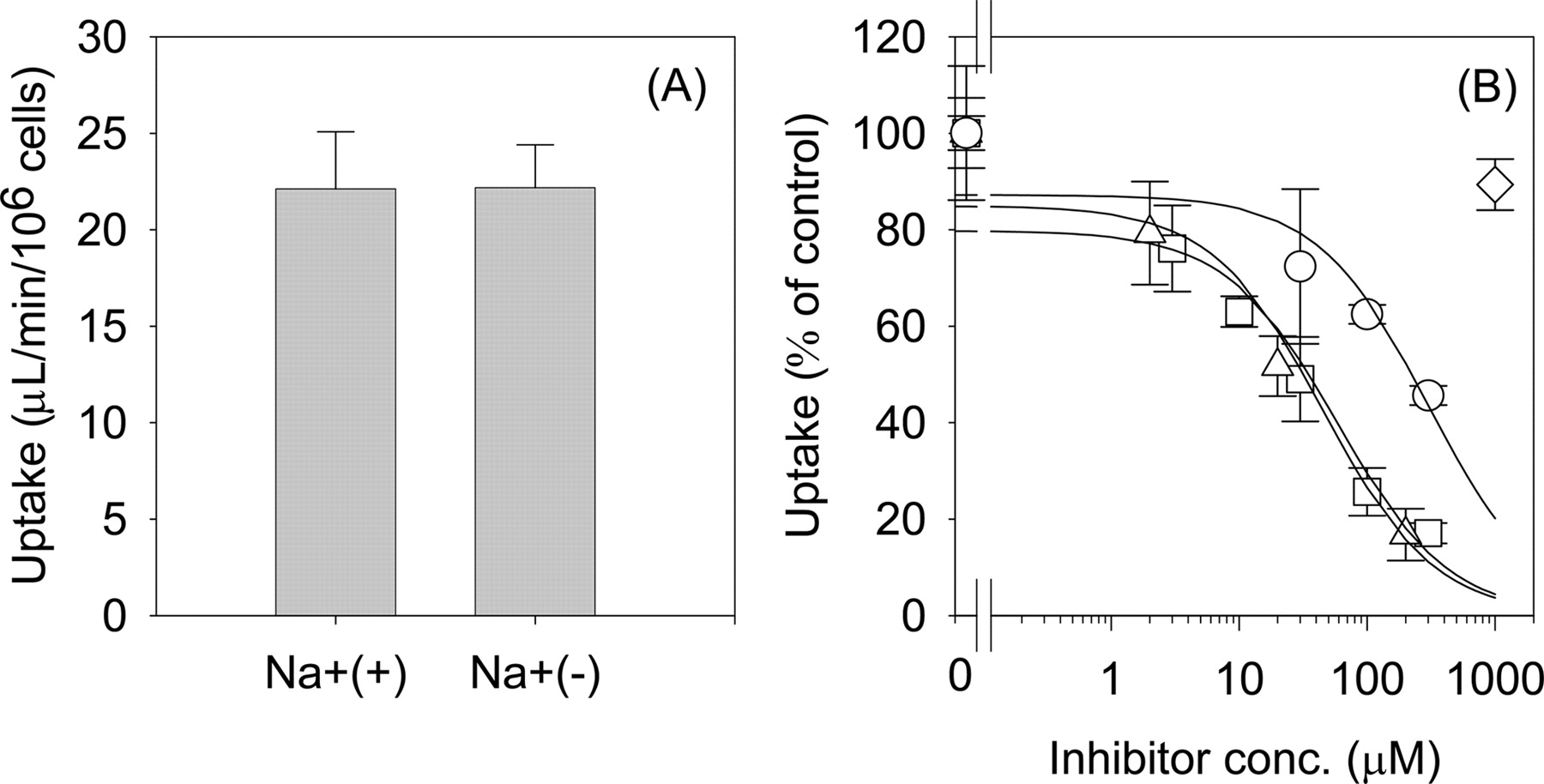
Uptake of Telmisartan by Transporter-Expressing HEK293 Cells. In the transport study using transporter expression systems, we reduced the HSA concentration from 1% to 0.3% in the incubation media because only a minimal transport activity of E217βG, which is used as the probe substrate for OATP1B1, was detected in OATP1B1-expressing cells in the presence of 1% HSA because of the significant decrease in its unbound concentration by binding to HSA (vector-transfected control cells, 2.32 ± 0.21 μl/2 min/mg protein; OATP1B1-expressing cells, 3.95 ± 0.21 μl/2 min/mg protein). To identify which transporters are important for the hepatic uptake of telmisartan in humans, the uptake assay was carried out using OATP1B1- and OATP1B3-expressing HEK293 cells in the presence of 0.3% HSA in the incubation media. Under these conditions, significant uptake of E217βG by OATP1B1- and OATP1B3-expressing HEK293 cells was observed (Fig. 5B). On the other hand, telmisartan was taken up by OATP1B3 but not by OATP1B1 (Fig. 5A). Because the difference in the degree of uptake between vector- and OATP1B3-transfected cells was too small to assess its saturation kinetics in the presence of 0.3% HSA, the concentration dependence of telmisartan uptake was evaluated over the concentration range of 0.05 to 10 μM in the absence of HSA at 5 min. The Km and Vmax values of telmisartan transport by OATP1B3 were calculated to be 0.81 ± 0.18 μM and 6.7 ± 0.91 pmol/min/mg protein, respectively (Fig. 6).
Inhibitory Effect of E-sul on OATP1B1- and OATP1B3-Mediated Uptake of Telmisartan in Transporter-Expression Systems. The inhibitory effect of E-sul on OATP1B1- and OATP1B3-mediated uptake of E217βG, CCK-8, and telmisartan was evaluated using OATP1B1- and OATP1B3-expressing HEK293 cells in the presence of 0.3% HSA. E-sul strongly inhibited OATP1B1-mediated E217βG uptake with an IC50 value of 0.79 ± 0.51 μM, whereas E-sul did not inhibit the OATP1B3-mediated CCK-8 uptake up to 30 μM (IC50 = 97.1 ± 37 μM) (Fig. 7A). In addition, the OATP1B3-mediated uptake of telmisartan was not inhibited by 30 μM E-sul (Fig. 7B).
Uptake of Telmisartan into Cryopreserved Human Hepatocytes. The uptake of 1 μM E217βG and 0.1 μM telmisartan by three different batches of cryopreserved human hepatocytes (lot. OCF, 094, and MYO) in the presence of 0.3% HSA was increased from 0.5 to 2 min [uptake of E217βG and telmisartan by cryopreserved human hepatocytes (OCF): 6.3 ± 1.0 and 56.7 ± 3.6 μl/min/106 cells, respectively], and their uptake was reduced in the presence of an excess of unlabeled E217βG (200 μM) and telmisartan (40 μM) to 0.6 ± 1.1 and 8.2 ± 4.7 μl/min/106 cells, respectively. The uptake of E217βG into human hepatocytes was inhibited by more than half at 30 μM E-sul, whereas that of telmisartan was not significantly inhibited by 30 μM E-sul (Table 1).
Effect of E-sul on the uptake of telmisartan and E217β G by cryopreserved human hepatocytes in the presence of 0.3% HSA
The substrate concentration used was 0.1 and 1 μM for telmisartan and E217β G. Saturable uptake of telmisartan and E217β G into cryopreserved human hepatocytes was determined after the subtraction of nonsaturable uptake (evaluated as the uptake clearance of the respective compounds in the presence of 40 μM telmisartan and 200 μM E217β G). In parentheses is the percentage of the saturable uptake of telmisartan and E217β G in the absence of inhibitor.

Effect of various concentrations of HSA on the uptake of telmisartan in isolated rat hepatocytes and the unbound fraction of telmisartan. Uptake of telmisartan was measured by incubating cells with 0.1 μM (closed circle) and 40 μM (open circle) telmisartan, and saturable uptake of telmisartan by isolated rat hepatocytes was determined using eq. 3 and eq. 4. HSA concentrations used were 0 (A), 0.1 (B), 0.3 (C), 1 (D), 3 (E), and 5% (F). G, triangles and circles represent the uptake of telmisartan into isolated rat hepatocytes (μl/min/106 cells) and unbound fraction of telmisartan, respectively. Each point represents the mean ± S.E. of three separate determinations.

Time profile (A) and Eadie-Hofstee plot (B) of the uptake of telmisartan by isolated rat hepatocytes in the presence of 1% HSA. A, substrate concentrations used were 0.1 (closed circles) and 40 μM (open circles). B, the uptake of telmisartan in isolated rat hepatocytes was measured at a concentration between 0.1 and 40 μM telmisartan. The initial uptake rate of telmisartan in isolated rat hepatocytes was determined using eq. 3. The solid line represents the fitted curve. Each point represents the mean ± S.E. of three separate determinations.
Discussion
In the present study, we have shown that telmisartan is likely to be taken up into rat and human hepatocytes by OATP family transporters because the uptake was Na+-independent and inhibited by some OATP substrates/inhibitors. We also have suggested that OATP1B3 predominantly contributes to the hepatic uptake of telmisartan in human hepatocytes.
Because it is difficult to evaluate the transport of telmisartan in the absence of HSA because of the extensive adsorption of lipophilic telmisartan to cells and/or culture materials, we examined the effect of different concentrations of HSA (0.1–5%) in the incubation media on the uptake of telmisartan by isolated rat hepatocytes (Fig. 2, A–F). In 0.3 to 5% HSA, saturable time-dependent uptake of telmisartan was clearly observed. The uptake of telmisartan into isolated rat hepatocytes was almost proportional to the protein unbound concentration of telmisartan in the incubation media, suggesting that the uptake of telmisartan followed the “free” hypothesis, in which only unbound ligand can be recognized by transporters (Fig. 2G).
Taking the balance between the absolute uptake amount and the avoidance of extensive adsorption of telmisartan to cells and/or culture materials by HSA into consideration, we decided to use 1 and 0.3% HSA in the incubation media for the further evaluation of telmisartan uptake by rat and human hepatocytes, respectively.
Initially, we characterized the transport property of telmisartan using isolated rat hepatocytes. Telmisartan was transported into isolated rat hepatocytes in a time- and concentration-dependent manner (Fig. 3). The Km and Vmax values of telmisartan uptake into isolated rat hepatocytes and the protein unbound fraction of telmisartan in the presence of 1% HSA were 21.7 μM, 371 pmol/min/106 cells, and 0.018, respectively. Then, the Km value normalized by the unbound concentration in the incubation media was estimated to be 0.4 μM. To evaluate the nonsaturable uptake of telmisartan, we defined 40 μM telmisartan as an excess concentration resulting from the limited solubility of telmisartan in the incubation media. The uptake of telmisartan into isolated rat hepatocytes was Na+-independent, indicating that telmisartan is not transported by Na+-taurocholate cotransporting polypeptide, the uptake by which is Na+-dependent (Fig. 4A). Furthermore, the uptake of telmisartan was inhibited by digoxin, pravastatin, and taurocholate with the IC50 value of 45.3, 58.6, and 300 μM, respectively. In contrast, a high concentration of TEA (1 mM) did not inhibit telmisartan uptake (Fig. 4B). Taurocholate, pravastatin, and digoxin are the substrates and inhibitors of Oatp1a1, 1a4, and 1b2 in rats (Noe et al., 1997; Kouzuki et al., 1999; Tokui et al., 1999; Cattori et al., 2000; Sasaki et al., 2004). It is reported that 100 μM digoxin completely inhibited the Oatp1a4 activity, but at most inhibited Oatp1a1-mediated uptake of digoxin by 70% (Shitara et al., 2002). Based on these results, it appears that telmisartan is taken up into rat hepatocytes by Oatp isoforms, and at least Oatp1a4 is involved in the uptake of telmisartan.

Effect of Na+ ion (A) and various compounds (B) on the uptake of telmisartan in isolated rat hepatocytes in the presence of 1% HSA. The substrate concentration used was 0.1 μM. Saturable uptake of telmisartan by isolated rat hepatocytes was determined using eq. 3 and eq. 4. B, data are shown as the percentage of the saturable uptake of telmisartan in the absence of inhibitors. Squares, triangles, circles, and diamonds represent the uptake of telmisartan in the presence of pravastatin, digoxin, taurocholate, and tetraethylammonium, respectively. Solid lines represent the fitted curves obtained by nonlinear regression analysis. Each bar and point represents the mean ± S.E. of three separate determinations.

Time profiles of the uptake of telmisartan (A) and E217βG (B) into transporter-expressing cells. The substrate concentrations of telmisartan and E217βG used were 0.1 μM. Squares, triangles, and circles represent the uptake by OATP1B3-, OATP1B1-, and vector-transfected cells. The uptake of telmisartan and E217βG was evaluated in the presence and absence of 0.3% HSA, respectively. *, a significant difference (P < 0.05) from the uptake by vector-transfected cells. Each point represents the mean ± S.E. of three separate determinations.

Time profile and Eadie-Hofstee plot of the uptake of telmisartan into transporter-expressing cells in the absence of HSA. Inset, the substrate concentration used was 0.1 μM. Squares, triangles, and circles represent the uptake by OATP1B3-, OATP1B1-, and vector-transfected cells. The uptake of telmisartan by transporter-expressing cells was measured at a concentration between 0.05 and 10 μM telmisartan in the absence of HSA. The OATP1B3-mediated telmisartan transport was obtained by subtracting the uptake in vector-transfected cells from that in OATP1B3-expressing cells for 5 min. *, a significant difference (P < 0.05) from the uptake by vector-transfected cells. Each point represents the mean ± S.E. of three separate determinations.

Inhibitory effect of E-sul on OATP1B1-mediated E217βG uptake (A, circle), OATP1B3-mediated CCK-8 uptake (A, triangle), and OATP1B3-mediated telmisartan uptake (B) in the presence of 0.3% HSA. The substrate concentration used was 0.1 μM for all the compounds. The OATP1B1- and OATP1B3-mediated transport was obtained by subtracting the uptake in vector-transfected cells from that in OATP1B1- or OATP1B3-expressing cells for 2 min for E217βG or 5 min for CCK-8 and telmisartan. Data are shown as the percentage of the OATP1B1- and OATP1B3-mediated substrate uptake in the absence of E-sul. The solid lines in A represent the fitted curves obtained by nonlinear regression analysis. Each bar and point represents the mean ± S.E. (n = 3).
Next, to examine which human OATP transporters are involved in the uptake of telmisartan into human hepatocytes, we evaluated the transport properties of telmisartan using HEK293 cells expressing individual OATP and cryopreserved human hepatocytes. In this transport study, we reduced the HSA concentration from 1% to 0.3% in the incubation media because no significant uptake of E217βG, which is used as a probe substrate for OATP1B1, was detected in OATP1B1-expressing cells in the presence of 1% HSA. Telmisartan could be taken up by OATP1B3 but not by OATP1B1 (Fig. 5A). Because the transporter-mediated uptake of telmisartan in the presence of HSA was not high enough to evaluate the saturation kinetics, we investigated the saturable uptake of telmisartan in the absence of HSA. The Km and Vmax values of telmisartan uptake by OATP1B3 in the absence of HSA were 0.81 μM and 6.7 pmol/min/mg protein, respectively (Fig. 6). This Km value was almost comparable with that in isolated rat hepatocytes normalized by the unbound concentration of telmisartan in the incubation media. Telmisartan was taken up into cryopreserved human hepatocytes in a saturable manner. Considering that telmisartan was taken up by OATP1B3, but not by OATP1B1, the uptake of telmisartan seems to be mediated by OATP1B3. To confirm the minor contribution of OATP1B1 to the hepatic uptake of telmisartan, we planned to perform an inhibition study using OATP1B1- and OATP1B3-selective inhibitor. From our analyses, E-sul inhibited OATP1B1-mediated E217βG uptake with an IC50 value of 0.8 μM, whereas E-sul did not inhibit OATP1B3-mediated CCK-8 uptake up to 30 μM (Fig. 7A). These results confirmed that 30 μM E-sul can selectively inhibit the OATP1B1-mediated uptake. On the other hand, CCK-8 inhibited both OATP1B1- and OATP1B3-mediated E217βG uptake with IC50 value of 6.79 ± 0.59 and 14.6 ± 2.2 μM, respectively, indicating that CCK-8 cannot be used as a selective inhibitor for OATP1B3 as reported previously (Nozawa et al., 2003). The uptake of telmisartan into OATP1B3-expressing cells and cryopreserved human hepatocytes was not inhibited by 30 μM E-sul (Fig. 7B; Table 1). On the contrary, 30 μM E-sul inhibited more than half of E217βG uptake in all the batches of cryopreserved human hepatocytes (Table 1). Hirano et al. (2004) have reported that the uptake of E217βG in human hepatocytes is mediated mainly by OATP1B1. These results suggest that telmisartan is transported into cryopreserved human hepatocytes by OATP1B3 rather than OATP1B1.
In a previous study, the ratio of the relative expression level of OATP1B1 and OATP1B3 in cryopreserved human hepatocytes to that in expression systems, determined by Western blot analysis, was 1.79 and 0.96, respectively (Hirano et al., 2004). In human liver, OATP2B1 is also expressed on the basolateral membrane (Kullak-Ublick et al., 2001). We checked that telmisartan was significantly taken up into OATP1B3- and OATP2B1-expressing HEK293 cells (2.6 ± 0.4 and 1.7 ± 0.9 μl/5 min/mg protein, respectively). The ratio of the protein expression level of OATP2B1 in human hepatocytes to that in our expression systems was less than 0.2 (Hirano et al., 2006). Therefore, it is suggested that the contribution of OATP2B1 to the hepatic uptake of telmisartan into human hepatocytes is at most one-fifth that of OATP1B3.
In general, it is accepted that OATP1B1 is responsible for the hepatic uptake of several compounds. Hirano et al. (2004) have shown that pitavastatin and E217βG are taken up mainly via OATP1B1. However, a recent report suggested that fexofenadine, an H1-receptor antagonist, is transported by OATP1B3 rather than OATP1B1 (Shimizu et al., 2005). Moreover, in the case of valsartan, which is in the same therapeutic class as telmisartan, the contribution of OATP1B1 and OATP1B3 to its hepatic uptake is estimated to be almost similar (Yamashiro et al., 2006). Therefore, the relative contribution of OATP1B1 and OATP1B3 to the hepatic uptake of organic anions depends on the substrate properties and chemical structures, and we cannot a priori decide which transporters are responsible for hepatic uptake without using dedicated experiments for estimating the contribution of each proposed transporter.
The Cmax value of telmisartan increases disproportionately with the dose (10–160 mg). In clinical situations, 160/25 mg/day telmisartan/hydrochlorothiazide combination therapy is approved for the treatment of hypertension in the United States. The Cmax values of telmisartan after single and multiple 160-mg doses were 3.0 and 5.6 μM, respectively (Stangier et al., 2000b). Considering that 99.5% of the telmisartan in blood is bound to plasma proteins (Stangier et al., 2000b), the unbound concentration of telmisartan is estimated to be 0.015 and 0.028 μM. These values are more than 20 times lower than the Km value of telmisartan uptake by OATP1B3 obtained in this study. In addition, to avoid the false-negative prediction of the contribution of OATP1B3 to its nonlinear pharmacokinetics, we calculated the maximum unbound concentration of telmisartan at the inlet to the liver (Iin, max, u) to be 0.12 μM after multiple 160-mg doses using an established method (Ito et al., 1998). However, the Km value of telmisartan uptake by OATP1B3 is still more than 5 times higher than the Iin, max, u of telmisartan. If the conventional assumption applies, in which only the unbound drug can interact with OATP1B3, the saturation of OATP1B3-mediated telmisartan uptake seems to have a minor effect on the nonlinear increase of Cmax and AUC over the clinical dose range. Furthermore, a large interindividual variability in the plasma profile of telmisartan has been observed in clinical situations (Stangier et al., 2000a,c). Letschert et al. (2004) have reported two naturally occurring mutations in the SLCO1B3 gene that cause a substrate-dependent functional change in OATP1B3. The genetic polymorphisms in OATP1B3 may be one of the reasons for the interindividual variability of the pharmacokinetics of telmisartan. In addition, the glucuronidation process of telmisartan and hepatobiliary transport of telmisartan glucuronide may also affect its interindividual variability, and further quantitative analyses in each process will be needed.
In conclusion, we have shown that telmisartan is taken up into human hepatocytes by OATP1B3 rather than by OATP1B1. In addition, these findings support and further extend the important role of OATP1B3 in overall hepatic elimination of some drugs.
Footnotes
-
This work was supported in part by a Health and Labor Sciences Research Grants from Ministry of Health, Labor, and Welfare for the Research on Advanced Medical Technology and Grant-in Aid for Young Scientists (B) (17790113) from the Ministry of Education, Culture, Sports, Science, and Technology.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.009175.
-
ABBREVIATIONS: AUC, area under the plasma concentration-time curve; OATP, organic anion transporting polypeptide; E217βG, estradiol 17β-d-glucuronide; CCK-8, cholecystokinin octapeptide; E-sul, estrone-3-sulfate; TEA, tetraethylammonium; HEK, human embryonic kidney; HSA, human serum albumin.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}