


Abstract
Cryopreserved hepatocytes are often used as a convenient tool in studies of hepatic drug metabolism and disposition. In this study, the expression and activity of drug transporters in human and rat fresh and cryopreserved hepatocytes was investigated. In human cryopreserved hepatocytes, Western blot analysis indicated that protein expression of the drug uptake transporters [human Na+-taurocholate cotransporting polypeptide (NTCP), human organic anion transporting polypeptides (OATPs), human organic anion transporters, and human organic cation transporters (OCTs)] was considerably reduced compared with liver tissue. In rat cryopreserved cells, the same trend was observed but to a lesser extent. Several rat transporters were reduced as a result of both isolation and cryopreservation procedures. Immunofluorescence showed that a large portion of remaining human OATP1B1 and OATP1B3 transporters were internalized in human cryopreserved hepatocytes. Measuring uptake activity using known substrates of OATPs, OCTs, and NTCP showed decreased activity in cryopreserved as compared with fresh hepatocytes in both species. The reduced uptake in cryopreserved hepatocytes limited the in vitro metabolism of several AstraZeneca compounds. A retrospective analysis of clearance predictions of AstraZeneca compounds suggested systematic lower clearance predicted using metabolic stability data from human cryopreserved hepatocytes compared with human liver microsomes. This observation is consistent with a loss of drug uptake transporters in cryopreserved hepatocytes. In contrast, the predicted metabolic clearance from fresh rat hepatocytes was consistently higher than those predicted from liver microsomes, consistent with retention of uptake transporters. The uptake transporters, which are decreased in cryopreserved hepatocytes, may be rate-limiting for the metabolism of the compounds and thus be one explanation for underpredictions of in vivo metabolic clearance from cryopreserved hepatocytes.
Introduction
Isolated hepatocytes are often used to determine in vitro pharmacokinetic characteristics of drug candidate compounds (Soars et al., 2007b, 2009; Chiba et al., 2009). To provide useful information for in vivo pharmacokinetic predictions, the isolated hepatocyte should accurately reflect the cell's functions in liver tissue. This includes drug transporter expression and localization, activity of drug-metabolizing enzymes, gene regulatory mechanisms, and cofactor concentrations (Sohlenius-Sternbeck and Schmidt, 2005; Chiba et al., 2009; Soars et al., 2009).
In intact liver tissue, hepatocytes express a number of drug uptake transporters of the solute carrier (SLC) superfamily on their basolateral, blood-facing, sinusoidal membranes. The major families of hepatic drug uptake transporters are organic anion transporters (OATs), organic anion transporting polypeptides (OATPs), organic cation transporters (OCTs), and the Na+-taurocholate cotransporting polypeptide (NTCP). These transporters facilitate uptake of drugs and other xenobiotics from the blood into the hepatocyte (Klaassen and Aleksunes, 2010). In hepatocytes, drug molecules can be metabolized by a number of enzymes [such as cytochrome P450s (P450s) and UDP-glucuronosyltransferases (UGTs)] (Soars et al., 2007b; Chiba et al., 2009). Drugs and drug metabolites can be eliminated by biliary secretion mediated by drug efflux transporters belonging to the ATP-binding cassette transporter superfamily located on the bile-facing, canalicular hepatocyte membrane.
Uptake transporters may be rate-determining for drug clearance (CL), and it has been shown that intrahepatocyte accumulation by active uptake of a compound can influence both its metabolism and its P450 inhibitory effect (Grime et al., 2008; Kusuhara and Sugiyama, 2009; Watanabe et al., 2009; Brown et al., 2010). The International Transporter Consortium has recently published guidelines for the application of drug uptake transporter kinetics in pharmacokinetics and strongly encourages screening for interactions with the major drug uptake transporters during drug development (in the liver: OAT2, OATP1B1, OATP1B3, OCT1, and OCT3) (Giacomini et al., 2010; Zamek-Gliszczynski et al., 2013).
Freshly isolated hepatocytes are easily available from preclinical animal model species. However, because access to freshly isolated human hepatocytes is erratic, many drug development laboratories are using cryopreserved hepatocytes (Chiba et al., 2009; Soars et al., 2009). Therefore, human cryopreserved hepatocytes have been extensively characterized, focusing mainly on drug metabolic capacity (Stringer et al., 2008; Floby et al., 2009).
Most studies on uptake transporter activity in cryopreserved human hepatocytes have used the uptake of selective marker substrates to investigate a certain class of uptake transporters in the cells (Houle et al., 2003; Shitara et al., 2003; Lu et al., 2006; Soars et al., 2009; Badolo et al., 2011). Previous studies have reported partly conflicting results, ranging from no change to significant losses of either OATP or OCT uptake activity in cryopreserved hepatocytes as compared with freshly isolated cells. Recently, absolute quantification of transporter expression has indicated that OATP1B1, OATP1B3, and OATP2B1 protein levels were lower in human cryopreserved hepatocytes compared with liver tissue (Kimoto et al., 2012). The amount of NTCP seemed unaffected by cryopreservation in human hepatocytes while displaying some decline in rat cryopreserved hepatocytes (Qiu et al., 2013). As hepatocyte measurements were performed on a single, unmatched batch of cryopreserved cells from each species, any conclusions about NTCP expression in these cells are equivocal. A loss of drug transporters in cryopreserved hepatocytes, resulting in reduced membrane permeability for some drugs, may explain why in vitro intrinsic CL (CLint) obtained from cryopreserved hepatocytes often leads to an underprediction of in vivo clearance, progressively larger for higher clearances (Foster et al., 2011; Hallifax et al., 2012).
The aim of this study was to investigate whether underpredictions of in vivo CL based on in vitro CLint measurements from cryopreserved hepatocytes can be due to loss of drug uptake transporters in cryopreserved hepatocytes. We studied the protein expression, protein localization, and activity of several of the most important hepatic drug uptake transporters, including OATs, OATPs, OCTs, and NTCP. We compared activity and expression of the transporters in freshly isolated and cryopreserved hepatocytes from human and rat. The expression levels in hepatocytes were also compared with liver tissue levels from both species. Our study showed that cryopreserved hepatocytes from both human and rat exhibited large reductions in drug uptake transporter expression (including OATPs, OATs, and OCTs in both species, as well as NTCP in human cells) and activity compared with both fresh hepatocytes and intact liver tissue. This loss of uptake transporters may be one important factor explaining the underpredictions when scaling hepatic clearance from cryopreserved hepatocytes.
Materials and Methods
Nomenclature.
As recommended by Hagenbuch and Stieger (2013), this article follows the gene and protein nomenclature guidelines of the HUGO Gene Nomenclature Committee (Gray et al., 2013) and the International Committee for Standardized Genetic Nomenclature in Mice (http://www.informatics.jax.org/mgihome/nomen/2009_gene.shtml) (2009). Human gene names are written in capital letters (e.g., SLCO2B1), while rodent genes are given in upper and lower case (e.g., Slco2b1). Protein names for both human and rodent proteins are written in capital letters (e.g., OATP2B1). In the interest of clarity, human protein names are prefixed with "h" and rat proteins are prefixed with "r" (e.g., hOATP2B1, rOATP2B1). Protein names without prefix refer to the protein in general, not belonging to any particular species (e.g., an OATP2B1 inhibitor).
Chemicals.
Chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. A series of AstraZeneca (AZ) project compounds, including microsomal prostaglandin E synthase-1 (mPGES-1) inhibitors [chemical structures are presented in Bylund et al. (2013)], was obtained from AstraZeneca’s compound collection.
Animals.
Sprague-Dawley rats were acquired from Harlan Laboratories B.V. (Venray, The Netherlands). Rats were housed up to three per cage in transparent plastic cages. All animals were acclimatized for 2 weeks prior to studies. The room was lit in a 12-hour light/dark cycle. Bedding material consisted of aspen wood chips; plastic tunnels and aspen chewing blocks were provided. The rats had free access to food and water. All animal experiments were approved by the local animal ethics committee (Stockholms Södra djurförsöksetiska nämnd) and were conducted in compliance with national guidelines for the care and use of laboratory animals.
Preparation of Fresh and Cryopreserved Hepatocytes.
Rat hepatocytes were isolated according to a modified collagenase perfusion method previously described (Bissell and Guzelian, 1980) from 7- to 8-week-old male Sprague-Dawley rats, anesthetized with isoflurane (Abbott Scandinavia AB, Kista, Sweden). Shortly, the liver was perfused in situ using a peristaltic pump, first with 37°C calcium-free buffer containing 0.5 M EGTA, followed by prewarmed William’s E medium, supplemented with 10 mM Hepes (Gibco, Life Technologies, Paisley, UK), pH 7.4 (WE). To the buffer was added 0.16 mg/ml of collagenase type XI. After completed perfusion, the organ was cut from its ligaments and transferred to a prewarmed collagenase solution. Released from connective tissue, the hepatocytes were washed to obtain a homogenized cell suspension without debris from extracellular matrix. The suspension was filtered through gauze, repeatedly centrifuged at 50g for 2 minutes, and resuspended in new Krebs-Henseleit buffer supplemented with 10 mM Hepes (Gibco), pH 7.4 (KHL). Cell number and viability were determined using a Bürker counting chamber and the trypan blue exclusion method. Viability typically exceeded 90%. Cell preparations with viability of <80% were discarded.
Liver tissue samples were obtained from donors undergoing surgical liver resection in Uppsala University Hospital (Uppsala, Sweden), as approved by Uppsala Regional Ethical Review Board. Small biopsies from histologically normal areas of liver resections were snap frozen in ethane cooled by liquid nitrogen in the operating theater immediately after the surgical procedure (n = 3). Frozen biopsies were stored at −80°C until use. Fresh human hepatocytes were prepared from liver resections as described elsewhere (LeCluyse and Alexandre, 2010). Fresh human hepatocytes were not available for Western blotting experiments or for studies on AZ project compounds.
Human cryopreserved hepatocytes comprised lots Hu4035, Hu4037, and Hu4152 (CellzDirect, Life Technologies), all of plateable quality, and lots OZL, SQJ, and REL (Celsis In Vitro Technologies, Baltimore, MD). The lots represented a mix of genders and displayed average P450 metabolism. Rat cryopreserved hepatocytes were derived from male Sprague-Dawley rats, 7–8 weeks of age (CellzDirect); only two lots were available. Cryopreserved hepatocytes were thawed in a 37°C water bath immediately after removal from the −150°C freezer and transferred to a 50-ml vial of WE at 37°C. Centrifugation was done according to the manufacturer’s instructions, and the cell pellet was resuspended in a small volume of KHL. Cell counting and viability were determined as described for rat hepatocytes.
Donor information for liver tissue and fresh and cryopreserved hepatocytes is given in Supplemental Table 1.
Preparation of Hepatocyte Protein.
From liver tissue: Rat liver was handled in WE in a Petri dish, cut into smaller pieces, and homogenized with a glass pestle homogenizer containing cracking buffer, consisting of 5 mM Hepes, 0.5 mM EDTA, Protease Inhibitor Cocktail (Sigma) according to the manufacturer’s instruction, pH 7.2. The resulting suspension was transferred to a tube and homogenized thoroughly with a small-gauge syringe before centrifugation to remove unhomogenized tissue. Small frozen samples from human liver biopsies were handled in the same manner.
From fresh hepatocytes: Aliquots of 1 million fresh rat hepatocytes collected from the preparation technique described above were snap frozen after isolation. These were later thawed and suspended in 200 μl cracking buffer. Homogenization was performed as described for liver tissue.
From cryopreserved hepatocytes: Rat and human cryopreserved hepatocytes (CellzDirect, Celsis In Vitro Technologies), stored at −150°C, were thawed and pelleted by centrifugation at 90 rpm for 3 minutes. Homogenization was performed as described for liver tissue.
The preparations were supplemented with 50 mM mannitol and stored at −20°C until use. Determination of protein concentration was done according to BCA protein assay kit (Pierce Biotechnology, Thermo Fischer Scientific Inc., Rockford, IL) according to the manufacturer’s instructions.
Determination of Transporter Expression by Western Blot.
Ten to 40 micrograms of hepatocyte or liver protein was diluted in distilled H2O and loading buffer [0.5 ml Laemlli buffer, 50 μl β-mercaptoethanol, and 2.5 μl 1 M dithiothreitol, all from Bio-Rad (Hercules, CA)] and separated by SDS-PAGE on 9% gels followed by transfer of protein to a polyvinylidene fluoride membrane (PerkinElmer, Waltham, MA) at 200 mA for 2 hours. Membranes were washed 3× in Tris-buffered saline Tween 20 [0.1% Tween 20 in a buffer consisting of 150 mM NaCl and 25 mM Tris-HCl, pH 7.4 (TBST)] before continuing with blocking for 1 hour in blocking buffer (consisting of 5% nonfat dry milk in TBST, pH 7.2). Membranes were incubated with primary antibody (Supplemental Table 2), diluted 1:500 or 1:1000, at 4°C overnight followed by washing in TBST. Reblocking was performed during 1 hour followed by incubation with 1:1000 diluted polyclonal goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) (Abcam, Cambridge, UK). To detect glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (internal standard), an HRP-conjugated primary antibody was used (Santa Cruz Biotechnology, Houston, TX) Incubations were followed by a new washing step.
Antibodies were detected by chemiluminescence (ECL Plus Western blotting reagents; GE Healthcare, Piscataway, NJ), a substrate for HRP, according to the manufacturer’s instruction. Molecular weights were determined using the Broad Range Molecular Weight Marker (Bio-Rad). The blot was photographed with Amersham Hyperfilm ECL (GE Healthcare), developed, and fixed. Several exposures were made to ensure that quantification was performed on film that had not been overexposed. Protein bands were scanned and the background was subtracted. To normalize band intensities, the ratio between the transporter band and the band from the housekeeping protein GAPDH was used.
Immunofluorescence.
Human cryopreserved hepatocytes were seeded on collagen type I–covered cover slips (BD Biosciences, Franklin Lakes, NJ) for 1 hour in WE in a cell incubator at 37°C and 5% CO2. After washing in KHL, adherent cells were fixed in ice-cold 50% acetone for 2 minutes, followed by a 5-minute fixation in 100% acetone. Cover slips were then dried and stored at −20°C until use.
Cover slips were washed 4 × 5 minutes in PBS and blocked with 10% fetal calf serum (FCS) (Gibco) in PBS for 30 minutes at room temperature. They were then washed again and incubated with primary antibody in 1% FCS in PBS for 1 hour at room temperature. Primary antibodies against human OATP1B1 and OATP1B3 (both rabbit polyclonals kindly provided by Dr. Bruno Stieger, Department of Clinical Pharmacology, University of Zürich, Zürich, Switzerland) were used at a dilution of 1:100. Cells were washed again and blocked with 10% goat serum (Life Technologies) in PBS for 30 minutes at room temperature. Slides were then incubated with goat anti-rabbit Alexa Fluor conjugated fluorescent secondary antibodies (Life Technologies). Secondary antibodies were used at a dilution of 1:1000 in PBS with 10% goat serum. Cover slips were washed one final time for 4 × 5 minutes in PBS and mounted with Prolong Gold anti-fade mounting media (Life Technologies). Stained cells were inspected and photographed using a confocal microscope (DMIRE 2; Leica Microsystems, Wetzlar, Germany).
Human liver biopsies were cut into 8-μm-thick sections on a cryotome and dried down on slides. Liver sections were fixed, stained, and visualized as described for hepatocyte samples above.
Uptake and Metabolic Stability Measurements in Hepatocytes.
Uptake measurements in suspended cells were performed using the media loss technique described by Soars et al. (2007a). Briefly, hepatocytes (106/ml) were incubated with test compound in KHL at a concentration of 1 μM and a final dimethylsulfoxide (DMSO) concentration of 0.1%. Experiments were completed within 2 hours of initial thawing or isolation of the hepatocytes. To distinguish media loss due to uptake or binding, test compound was incubated under the same conditions with cells that had been subjected to repeated freeze-thawing. Trypan blue staining revealed these cells to be intact but totally permeabilized. Uptake was followed for 30–45 minutes; the initial uptake phases used to determine CLint,uptake were 5–15 minutes for different compounds, the longest time used to quantify the slow uptake of fexofenadine. Aliquots were removed to a glass tube loaded into a bench-top centrifuge, and hepatocytes were pelleted by centrifugation. A sample of the buffer supernatant was removed and mixed with two volumes of an ice-cold stop solution of acetonitrile (ACN) containing 200 nM warfarin (internal standard). Plates containing samples were incubated on ice for at least 10 minutes to ensure complete precipitation of protein. The plate contents were shaken for a few minutes prior to centrifugation. An aliquot of 200 μl supernatant from each well was transferred to a 96-well analysis plate. Before analysis by liquid chromatography–tandem mass spectrometry (LC-MS/MS), each fraction was diluted in KHL to contain 25% ACN. Compound binding was measured in glass vials and the binding was found to amount to less than 1% of initial compound concentrations.
Hepatocytes in suspension (106/ml) were used to determine the metabolic stability of test compounds. Hepatocytes were incubated with test compound in KHL with 0.1% DMSO for 0–90 minutes at 37°C. Samples were taken at intervals, stopped and precipitated with two volumes of stop solution, and processed as described above. The activities of phase I and II drug-metabolizing enzymes in rat fresh and cryopreserved hepatocytes in suspension were compared using a cocktail containing highly permeable substrates for P450 and phase II enzymes (Floby et al., 2009): 7-hydroxycoumarin (phase II, UGT), bufuralol (CYP2D2), diazepam (CYP2C11), diclofenac (CYP2C6), midazolam (CYP3A), and phenacetin (CYP1A2), each at 2 μM final concentration. P450 isoform specificities were from Kobayashi et al. (2002) and Sakai and Ishizuka (2009).
Uptake experiments with plated fresh or cryopreserved hepatocytes were performed essentially as described (Menochet et al., 2012). Briefly, hepatocytes were seeded in 24-well collagen type I–covered plates (BD Biocoat; BD Biosciences) in WE supplemented with 10% FCS, 2 mM glutamine, and 0.01% insulin-transferrin-selenium A solution (Gibco) according to the provider’s instructions. Cells were left to attach for 2 hours at 37°C in 5% CO2. After washing and removal of unattached cells, leaving a semiconfluent cell monolayer (>90% confluent), uptakes were performed at 1 μM substrate concentration in KHL. The activities of uptake transporters were tested with several well characterized substrates: rosuvastatin (AstraZeneca), OATPs (Ho et al., 2006); fexofenadine, OATPs (Cvetkovic et al., 1999); ipratropium, OCTs (Nakanishi et al., 2011); and taurocholate, NTCP (Ozawa et al., 2004). Rosuvastatin is also an NTCP substrate in human but not rat cells (Ho et al., 2006). After uptake completion, cells were washed three times with ice-cold KHL, lysed with ACN stop solution, processed, and analyzed as described above. Uptake rates were quantified after 2 minutes of uptake, well within the linear phase of uptake. All experiments with plated hepatocytes were completed within 4 hours of initial thawing or isolation of the hepatocytes, including preparation and plating. Compound binding was measured in empty plates and subtracted from uptake measurements.
Metabolic Stability Measurements with Human and Rat Microsomes.
A pool of microsomes from 33 donors (28 male and 5 female) was purchased from BD Gentest (Woburn, MA). Rat microsomes were derived from an in-house preparation. Microsomes were incubated at a concentration of 0.5 mg/ml in 96-well microplates in the presence of 1 μM drug compound at a final DMSO concentration of 0.1%. The reaction was initiated by the addition of NADPH (final concentration, 1.5 mM). The final volume was 100 μl, and the incubations were performed in duplicate for 5, 15, 30, and 60 minutes. The reactions were stopped by the addition of 100 μl ACN, after which the samples were mixed and centrifuged and the supernatant thus obtained was analyzed. Control incubations (without microsomes or with microsomes but without NADPH) were performed for all compounds. Samples were processed and analyzed by LC-MS/MS as described above.
Determination of Plasma Protein Binding.
Plasma protein binding was determined by equilibrium dialysis. Human plasma was obtained from AstraZeneca's internal blood tapping at Clinical Pharmacology & DMPK. Plasma from three individuals was pooled and mixed with the compound at a concentration of 10 μM (in 0.1% DMSO). A dialysis membrane (Spectra/Por MWCO 6-8000; Spectrum Laboratories, Inc., Rancho Dominguez, CA) was soaked in distilled water and thereafter placed between two dialysis plate halves with 48 wells. A volume of 180 μl of 0.122 M phosphate buffer, pH 7.4, with 75 mM NaCl was added to each well on one side of the dialysis plate side, and 180 μl of the plasma/compound mix to the opposite side. After incubation on an orbital shaker (4 mm in diameter and 100 rpm) at 37°C for 18 hours, the samples on both sides of the membrane were analyzed as described below. The fraction unbound (fu) in plasma was calculated from the ratio of the mass spectrometry (MS) area of the compound in the buffer to the MS area of the compound in the plasma. Recovery was measured from the ratio of the sum of the MS peak areas in buffer and sample to the MS peak areas in the plasma/compound mix at zero time. All compounds were found to be stable during the 18-hour incubation. The volume change over time was negligible (<10%).
Plasma Clearance of Test Compound.
Blood samples were drawn from male Sprague-Dawley rats, at an age of ∼8 weeks, just before and at 1 minute, 5 minutes, 20 minutes, 40 minutes, 1 hour, 1.5 hours, 3 hours, 6 hours, and 24 hours after intravenous bolus administration of test compound (in 0.3 M N-methyl-d-glucamine). A dose of 3 μmol/kg was given for all compounds. Standards, diluted in 50% ACN with distilled H2O, were subsequently diluted 1:10 with blank plasma. Fifty microliters of each standard or analytic sample together with 150 μl cold stop solution was mixed in a 96-deep-well working plate and centrifuged at 4°C and 4000 rpm for 20 minutes. An aliquot of 120 μl supernatant was transferred to a 96-deep-well analysis plate and diluted with 300 μl of the liquid chromatography mobile phase A and analyzed as described below.
Analysis of Study Compounds.
Analysis of parent compounds and metabolites were performed by LC-MS/MS using a Micromass Quattro Micro triple quadrupole (Micromass, Manchester, UK) coupled to a gradient pump composed of two Shimadzu LC-10AD VP isocratic pumps (Shimadzu Corporation, Kyoto, Japan) and a CTC HTS PAL autosampler (CTC Analytics, Zwingen, Switzerland). The software MassLynx (Waters Corporation, Milford, MA) (controlling the LC system and mass spectrometer), which includes QuanLynx (quantification) and QuanOptimize (MS/MS optimization), was used. A High Purity C18 5-μm 30 × 2.1 mm analytical column (Thermo Electron Corporation, Waltham, MA) was used. Chromatography was performed using a generic gradient at a flow rate of 0.4 ml/min. The mobile phase consisted of the solvents 2% acetonitrile in 0.1% (v/v) acetic acid in water (A) and 80% acetonitrile in 0.1% (v/v) acetic acid in water (B). The injection volume was 20 μl, and warfarin was used as internal standard. Other source parameters (e.g., collision energy, cone voltage, ion mode, molecular weight of parent and daughter) were individually optimized for each compound (Supplemental Table 3). Quantification of each compound was achieved by comparison of the analyte/internal standard peak area ratios. A standard curve was included for each compound analyzed spanning a concentration range from twice the initial experimental concentration to below the limit of quantification (LOQ). LOQ, signal-to-noise ratio for detection, was set to 5 times.
Pharmacokinetic Calculations.
In vitro–in vivo extrapolation (IVIVE) was performed for a number of selective uptake transporter substrates to compare predictions deriving from uptake clearance measurements (CLint,uptake) in freshly isolated and cryopreserved hepatocytes.
In a retrospective study, scaled in vivo CLint (scaled CLint,in vivo; see eq. 6) was calculated for 83 chemically diverse AstraZeneca project compounds. In vitro data on rat and human metabolic CLint (CLint,met) using fresh and cryopreserved hepatocytes, respectively; CLint,met using liver microsomes; fu in plasma (fuplasma); and blood-to-plasma concentration ratio (Cblood/Cplasma) were derived from AstraZeneca in-house databases. All compounds derived from the CNS and Pain therapeutic area and had been synthesized at the AstraZeneca R&D facility in Södertälje, Sweden. The study included all compounds that had the in vitro data listed above available for both rat and human determined using the same protocols as presented in this study.
Determination of Intrinsic Clearance.
The elimination rate constant for metabolic stability or uptake experiments was calculated from the first-order elimination equation
 (1)
(1)where [S] is the concentration of substrate at a given time point, t, after the incubation concentration of the substrate, [S0]; and k is the elimination rate constant determined by nonlinear regression within a time point interval considered to represent the initial elimination rate (Soars et al., 2007a). In vitro intrinsic clearance (CLint,in vitro) for the elimination phase could then be calculated from:
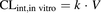 (2)
(2)where V represents the volume of the incubation.
Fraction Unbound in the Incubations.
The fraction unbound in the hepatocyte incubation was predicted according to Kilford et al. (2008):
 (3)
(3)where VR is the ratio between the cell volume and the incubation volume, with a value of 0.005 at the cell concentration of 106 cells/ml.
The fraction unbound in the microsome incubation was predicted according to Hallifax and Houston (2006):
 (4)
(4)where P is the microsomal protein concentration (0.5 mg/ml). In both eqs. 3 and 4, log P is used for bases and log D for other ion classes. Predicted log P and log D values were generated using a commercial package from Advanced Chemistry Development (Toronto, ON, Canada).
Calculations of Fraction Unbound in the Blood.
The fraction unbound in the blood (fublood) was calculated using the following equation:
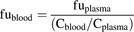 (5)
(5)When Cblood/Cplasma was unavailable, a value of 1 was assumed for neutral and basic compounds and 0.55 for acidic compounds.
Scaled In Vivo CLint.
CLint,in vitro from microsomes or hepatocytes was scaled to an in vivo CLint:
 (6)
(6)The scaling factors were as follows: for human hepatocytes, (120 × 106 cells/g liver) × (1500 g liver/70 kg body weight); for human microsomes, (45 mg protein/g liver) × (1500 g liver/70 kg body weight) (Bayliss et al., 1990; Obach et al., 1997; Sohlenius-Sternbeck, 2006). In the rat, liver and body weight was 10 g and 250 g, respectively, and the hepatocellularity and microsome protein content per gram of liver the same as for human.
Prediction of In Vivo Hepatic Blood CL.
Predictions were based on the well stirred model (Yang et al., 2007).
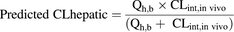 (7)
(7)Hepatic blood flow, Qh,b, was set to 72 and 20 ml/min/kg for rat and human, respectively (Brown, et al., 1997; Delp et al., 1998).
Determination of In Vivo Clearance.
In vivo blood clearance was calculated according to
 (8)
(8)where
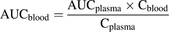 (9)
(9)The blood CLhepatic was calculated from the CLblood and the hepatic fraction, fhepatic, given in the literature (listed in Table 1).
IVIVE of fexofenadine, ipratropium, and rosuvastatin in vivo CLhepatic
Statistical Analysis.
Data were analyzed using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA). Data are given as mean ± S.D. Statistical significances were tested by the two-tailed Student’s t test. Probability values (P values) are symbolized by: * = P < 0.05, ** = P < 0.01, *** = P < 0.001; P values of <0.05 were considered statistically significant.
Results
Uptake Transporter Expression in Human and Rat Hepatocytes.
Western blotting was used to determine total protein expression levels in liver cells and liver tissue of several major drug uptake transporters—hOATP1B1, hOATP1B3, hOATP2B1, hOAT2, hOCT1, hOCT3, and hNTCP—as well as transporters such as human peptide transporter 1 (hPEPT1) and human monocarboxylate transporter 1 (hMCT1) involved in uptake of endogenous substrates. The expression levels of transporter in human liver tissue and freshly thawed cryopreserved hepatocytes measured by densitometry of Western blots indicated that the drug uptake transporters were significantly lower in the cryopreserved hepatocytes (Fig. 1A). Liver tissue and cryopreserved hepatocytes derived from different donors. The hOATP family of transporters was the most affected, showing of 80%–95% lower levels in human cryopreserved hepatocytes as compared with liver tissue. hOAT2 as well as hOCT1 and hOCT3 were 70%–80% lower in cryopreserved hepatocytes as compared with liver tissue, whereas hNTCP showed 50% lower levels in the cryopreserved hepatocytes. The transporters hMCT1 and hPEPT1 displayed no reductions in protein levels after cryopreservation as compared with the levels in liver tissue (Fig. 1A). No significant differences in transporter expression were found between cryopreserved hepatocytes from different vendors (data not shown). Figure 1B depicts Western blotting of hOATP1B1, hOATP1B3, and hOAT2 clearly showing the loss of the transporter proteins in human cryopreserved hepatocytes as compared with liver tissue expression levels (Fig. 1B).

(A) Comparison of expression levels of SLC uptake transporters in human cryopreserved hepatocytes and human liver tissue. Human liver tissue was acquired from patients undergoing hepatectomy due to different forms of liver cancer, while cryopreserved hepatocytes were acquired from commercial sources. Transporter protein levels were normalized against GAPDH; expression levels in liver tissue were set to 1. Data represent mean ± S.D.; n = 3. *P < 0.05; **P < 0.01; ***P < 0.001. (B) Representative Western blots depicting the levels of hOATP1B1, hOATP1B3, and hOAT2 transporters in three human cryopreserved hepatocyte preparations and three human liver samples. hOATP1B1 and hOATP1B3 were seen at approximately 75 kDa and hOAT2 at 70 kDa. All proteins could be detected in all human samples. Lower panels depict the internal standard GAPDH, detected at 35 kDa. Black bars denote different sample groups.
Western blot analysis of uptake transporters in rat liver, freshly isolated hepatocytes, and cryopreserved hepatocytes indicated that rOATP2B1, rOCT3, and rOAT2 levels were significantly lower already after cell isolation (Fig. 2). The loss of these transporters then increased further after cryopreservation. rMCT1 showed little loss after hepatocyte isolation, but expression levels decreased more than 50% after cryopreservation. rOCT1, rNTCP, and rPEPT1 expression levels were unaffected by isolation and cryopreservation of rat hepatocytes. rOATP2B1 was the uptake transporter most affected in isolated and cryopreserved rat hepatocytes, which is similar to the results in human cells.

Comparison of expression levels of SLC uptake transporters in rat cryopreserved hepatocytes, freshly isolated rat hepatocytes, and rat liver tissue. Transporter protein levels were normalized against GAPDH; expression levels in liver tissue were set to 1. Data represent mean ± S.D.; n = 3. *P < 0.05; **P < 0.01 for liver and fresh hepatocytes. For cryopreserved hepatocytes, two individual measurements were performed; statistical significance of differences can therefore not be calculated for these samples. (B) Representative Western blot depicting the levels of rOAT2 transporters in rat liver (L), fresh hepatocytes (FH), and cryopreserved hepatocytes (CH). rOAT2 was detected at approximately 60 kDa. One animal seemed devoid of rOAT2 staining. Lower panels depict the internal standard rGAPDH, detected at 35 kDa. Black bars denote different sample groups.
Immunolocalization of hOATP1B1 and hOATP1B3 in Human Cryopreserved Hepatocytes and Liver.
Despite the large loss of transporters after cryopreservation, hOATP1B1 and hOATP1B3 could readily be detected by immunohistochemistry in human cryopreserved hepatocytes attached to collagen-covered cover slips (Fig. 3, A and B). Transporter proteins were detected on the plasma membranes but also seemed to have been internalized to a large extent. This internalization was particularly evident for hOATP1B3. In liver tissue, hOATP1B1 and hOATP1B3 were mainly localized to plasma membranes of the hepatocytes (Fig. 4, A and B). Negative control stainings of liver sections, omitting primary antibody, were devoid of signal (Fig. 4C).

Immunofluorescent detection of hOATP1B1 (A) and hOATP1B3 (B) in human cryopreserved cells attached to collagen type I–covered cover slips.

Immunofluorescent detection of hOATP1B1 (A) and hOATP1B3 (B) in human liver sections. The OATP transporters were seen almost exclusively on hepatocyte membranes. In (C), a negative control section stained omitting the primary antibody is shown; neither unspecific staining nor autofluorescence is evident.
Uptake Transporter Activity in Fresh and Cryopreserved Human and Rat Hepatocytes.
The uptake of several well characterized substrates was tested in plated human fresh and cryopreserved hepatocytes. Rosuvastatin, an hNTCP and hOATP substrate; fexofenadine, an hOATP substrate; and ipratropium, an hOCT substrate, all showed significantly lower uptake rates by 75%, 90%, and 50%, respectively, in cryopreserved hepatocytes as compared with fresh cells (Fig. 5A). In agreement with Western blotting data, the hNTCP substrate taurocholate showed similar uptake rates in both fresh and cryopreserved hepatocytes.

Activity of drug uptake transporters in plated fresh and cryopreserved hepatocytes. (A) Uptake rates were determined for uptake substrates in plated fresh or cryopreserved human hepatocytes (fexofenadine is a substrate of hOATP transporters; rosuvastatin, hOATP and hNTCP transporters; ipratropium, hOCT transporters; taurocholate, hNTCP transporter). (B) The same experiment was performed using plated rat fresh and cryopreserved hepatocytes (fexofenadine is a substrate of rOATP transporters; rosuvastatin, rOATP but not rNTCP transporters; ipratropium, rOCT transporters; taurocholate, rNTCP transporter). Data represent mean ± S.D.; n = 3. *P < 0.05; **P < 0.01; ***P < 0.001.
In rat cryopreserved hepatocytes, the uptake of the rOATP substrate fexofenadine was reduced by >90% as compared with fresh hepatocytes (Fig. 5B). The OATP-mediated uptake of rosuvastatin was reduced by 80%. rOCT-mediated uptake of ipratropium was reduced by 40% in cryopreserved cells compared with fresh hepatocytes. As for NTCP activity in human cryopreserved hepatocytes, taurocholate uptake by rNTCP was unaffected by cryopreservation of rat hepatocytes.
Activity of Phase I and Phase II Drug-Metabolizing Enzymes in Fresh and Cryopreserved Rat Hepatocytes.
No significant differences in drug metabolism between fresh and cryopreserved cells were indicated when measuring the metabolism of a cocktail of highly permeable model substrates for P450 and UGT enzymes: bufuralol (CYP2D2), diazepam (CYP2C11), diclofenac (CYP2C6), midazolam (CYP3A), phenacetin (CYP1A2), and 7-hydroxycoumarin (phase II, UGT) (Fig. 6). Because these substances are highly permeable, transporter-mediated uptake is unlikely to be rate-determining for their metabolism (Benet et al., 2011).
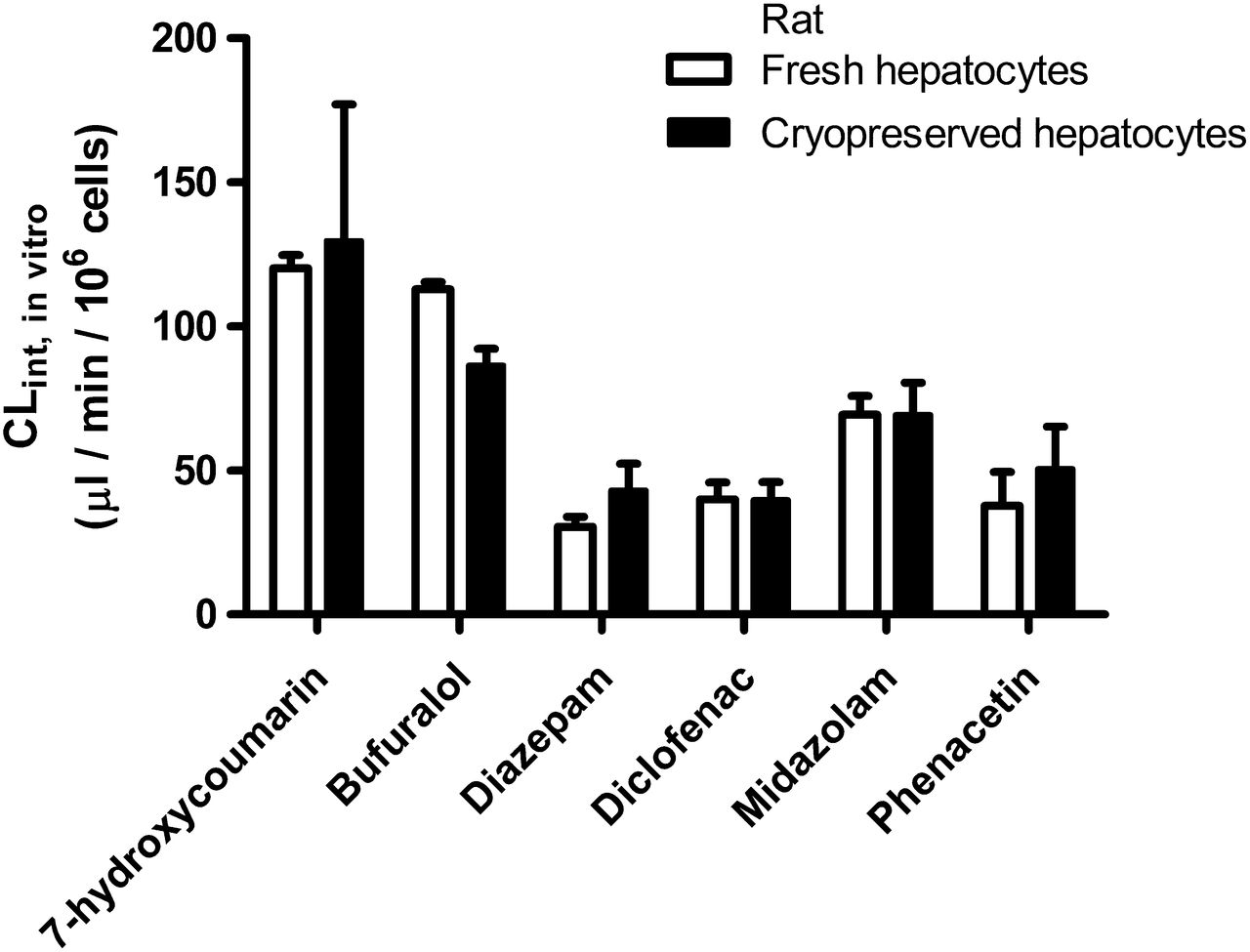
Drug metabolism in rat fresh and cryopreserved hepatocytes. Drug metabolism of P450 and phase II enzyme substrates was measured in rat fresh and cryopreserved hepatocytes in suspension. Enzyme specificities: 7-hydroxycoumarin, UGT; bufuralol, CYP2D2; diazepam, CYP2C11; diclofenac, CYP2C6; midazolam, CYP3A; and phenacetin, CYP1A2. Data represent mean ± S.D.; n = 3.
IVIVE of Clearance for Uptake Transporter Substrates in Human and Rat.
In Table 1, the IVIVE of in vivo CLhepatic using the well stirred model (eqs. 6 and 7) and in vitro uptake data for fexofenadine, rosuvastatin, and ipratropium from human and rat fresh as well as cryopreserved hepatocytes are shown. Uptake rates were measured both in hepatocyte suspensions and with plated cells. In vivo CLblood was calculated according to eqs. 8 and 9 for rat or given in the literature for human CLblood (Supplemental Material). CLhepatic was consistently underpredicted when using CLint,uptake from cryopreserved hepatocytes, regardless of species or experimental method used. Using CLint,uptake from fresh human or rat hepatocytes resulted in significantly higher predictions of CLhepatic.
Effects of Transporter Loss on Metabolic and Uptake CLint, in vitro of a Chemical Series of AZ Compounds.
Measuring in vitro metabolic stability for four close chemical analogs from an AstraZeneca series of mPGES-1 inhibitors showed that they displayed similar in vitro metabolism patterns: rapid metabolism in metabolic stability assays by human microsomes but stable when incubated with human cryopreserved hepatocytes [a representative example is shown in Table 2 and Fig. 7; structures are published by Bylund et al. (2013)]. No active uptake was indicated in human cryopreserved hepatocytes using the media loss method, as the decrease in extracellular concentration seen when incubating cryopreserved cells and compound closely corresponded to the binding seen when incubating compound with dead hepatocytes (Fig. 7A). In contrast, rat liver microsomes metabolized the compounds rapidly, as did freshly isolated rat hepatocytes (Table 2). In fresh rat hepatocytes, the compounds showed a pronounced uptake, well above the binding in dead cells (Fig. 7B; Table 2). Rat cryopreserved hepatocytes showed both slower uptake and metabolism than fresh hepatocytes (Fig. 7C; Table 2). Metabolic CLint,in vitro (eq. 2) was 75% lower (P < 0.001) and the uptake CLint,in vitro was 50% lower (P < 0.01) in cryopreserved hepatocytes as compared with fresh cells (Table 2). This suggests that metabolism of the compound is rate-limited by active uptake since metabolic capacity in rat freshly isolated and cryopreserved isolated hepatocytes is similar as presented above (Fig. 6).
Metabolism and uptake CLint,in vitro determinations for an AZ project compound

The uptake and metabolism of an AZ project compound is shown in human cryopreserved hepatocytes (A), rat fresh hepatocytes (B), and rat cryopreserved hepatocytes (C). Graphs show the total loss of compound from the incubation (cells and media) by metabolism, or loss from the extracellular medium by uptake or binding (to dead cells) measured after removal of cells by centrifugation. In human cryopreserved hepatocytes, metabolism was slow and uptake was limited to binding to cells. In fresh rat hepatocytes, and to a lesser extent in rat cryopreserved hepatocytes, the compound was rapidly taken up and metabolized. Data represent mean ± S.D.; n = 3.
Scaling CLint,in vivo from Rat and Human Microsomes and Hepatocytes.
A retrospective investigation was performed to scale the in vivo CLint (eq. 6) for a set of 83 chemically diverse AstraZeneca new chemical entities synthesized at AstraZeneca R&D, Södertälje, Sweden, including the 4 mPGES-1 inhibitors mentioned above. Figure 8A shows the scaled in vivo CLint (calculated using eq. 6) from human microsomes versus human cryopreserved hepatocytes for the 83 compounds. Data from fresh human hepatocytes were not available. Scaling from cryopreserved human hepatocytes indicated higher values for 23% of the compounds than the values from liver microsomes. However, in rat fresh hepatocytes, 81% of the compounds showed higher scaled in vivo CLint values (eq. 6) than in microsomes (Fig. 8B). Acids, neutrals, and bases in the data set showed similar behaviors (data not shown).
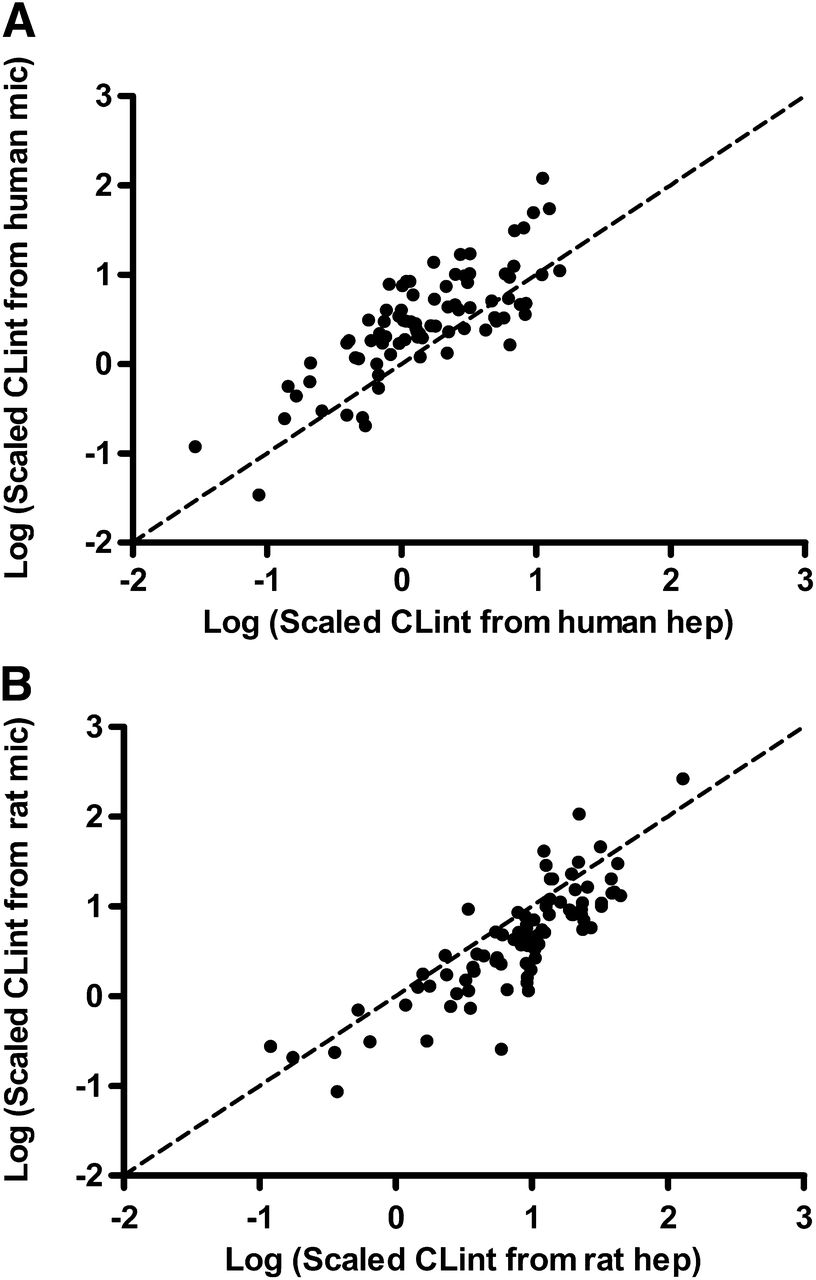
Comparison between the logarithms of scaled in vivo CLint (calculated using eq. 6) for human liver microsomes and human cryopreserved hepatocytes (A) and rat microsomes and rat fresh hepatocytes (B) for 83 AZ new chemical entities. Human liver microsomes tended to give higher scaled in vivo CLint than human cryopreserved hepatocytes. The reverse was true for rat experiments, where fresh rat hepatocytes tended to give higher scaled in vivo CLint than rat liver microsomes. The dashed line represents the line of unity.
Discussion
In the present study we could clearly show that both human and rat cryopreserved hepatocytes have considerably lower levels of uptake transporter expression of especially OATPs, OAT2, and to a somewhat lesser extent OCTs and NTCP than in freshly isolated cells and in liver tissue. PEPT1 and MCT1 showed smaller reductions in protein levels. In addition to lower levels in cryopreserved hepatocytes, hOATP1B1 and hOATP1B3 also showed some internalization. This internalization may be a step in the breakdown of the transporters translocating the proteins from the cell surface to the lysosome, a process that might be induced by the isolation and cryopreservation procedure. Such internalization has earlier been described in the downregulation of ATP-binding cassette transporters in plated rat hepatocytes and hOATP1B1 and hOATP1B3 in plated human hepatocytes (Bow et al., 2008; Ulvestad et al., 2011).
In rat cells, rOAT2, rOATP2B1, and rOCT3 levels were decreased already in freshly isolated cells, suggesting that the isolation procedure itself initiates the loss of uptake transporters, which are then further decreased by cryopreservation. Houle et al. (2003) reported that cryopreservation of rat hepatocytes did not affect NTCP activities while OATP activity was only slightly reduced after cryopreservation. In the study by Houle et al. (2003), cells had been cryopreserved quickly after isolation using an optimized protocol, suggesting that the cryopreservation method may be important for the retention of functional uptake transporters. Loss of uptake transporters compared with liver levels was, however, not investigated.
There is now mounting evidence that cryopreserved human hepatocytes lose some or most of their drug uptake transporters. Recently it was demonstrated by LC-MS/MS protein quantification that hOATP1B1, hOATP1B3, and hOATP2B1 were significantly lower in human cryopreserved hepatocytes compared with expression levels in human liver, supporting the results presented in this study (Kimoto et al., 2012). In contrast, hNTCP expression was found to be retained in human cryopreserved hepatocytes and slightly downregulated in rat cryopreserved cells compared with liver levels (Qiu et al., 2013). Expression levels in cryopreserved cells were measured in one single batch, unrelated to the liver tissue samples, from each species. A small decrease in hNTCP expression as seen in the present study may thus have been masked by individual variations in the study by Qiu et al. (2013). In a study by Soars et al. (2009), freshly isolated human hepatocytes and cryopreserved cells were shown to exhibit similar levels of OATP activity while OCT activity was reduced in cells after cryopreservation. Badolo et al. (2011) have reported a loss of OCT activity in human cryopreserved hepatocytes, while OATP activity was similar in fresh and cryopreserved human hepatocytes in their study. The present study is to our knowledge the first study on uptake transporters in human cryopreserved hepatocytes combining measurements of protein expression levels of OATPs, OCTs, and NTCP with activity measurements.
Commercially available human cryopreserved cells often derive from liver resections or transplantation organs that have not found a recipient. The tissue may have been handled and shipped to laboratory for a significant amount of time before isolation of hepatocytes can be initiated (Richert et al., 2004). Ischemia and resulting hypoxia is always a consequence of hepatic surgery needed to isolate hepatocytes, regardless of species, and has been described to be critical for the quality of the isolated cells (Richert et al., 2004; Berendsen et al., 2011). Downregulation of drug uptake transporters in hepatocytes has been described to be a result of the time in ischemia and hypoxia as well as liver injury and cholestasis, starting within 30 minutes of liver injury in rat (Gartung et al., 1996; Gerloff et al., 1999; Vos et al., 1999; Donner et al., 2013). The downregulation of drug uptake transporters is likely a physiologic stress response protecting the hepatocyte from toxic levels of bile salts, bilirubin, or other endotoxic compounds upon loss of circulation (Vos et al., 1999; Donner et al., 2013). The downregulation of hepatic uptake transporters is initiated by proinflammatory cytokines released upon tissue damage, and the signaling pathways involved are being elucidated (Donner et al., 2013; Li et al., 2013). There are also indications that this downregulation of uptake transporters can be minimized, for example, by limiting the time of tissue ischemia during cell isolation or by induction of heme oxygenase-1 (Donner et al., 2013). In earlier studies where uptake transporter activity in human cryopreserved hepatocytes seemed to be retained, livers had been stored for extended periods before cells were isolated. In a study by Shitara et al. (2003), the livers had been stored in the cold for up to 24 hours before cell isolation and cryopreservation. It is possible that loss of uptake transporters had already taken place during the storage of livers, before initial cell isolation. In their study, most batches of cryopreserved hepatocytes showed some loss of NTCP and OATP activities compared with freshly isolated hepatocytes, while some batches exhibited no change. No comparison was made to liver tissue expression levels. Freshly isolated human hepatocytes used in the present study were isolated within 3–4 hours of hepatic surgery, which is close to the minimum elapsed time possible and only achievable if the laboratory isolating the hepatocytes is located in close proximity to the hospital performing the surgery. Information on the donors of liver tissue and fresh and cryopreserved hepatocytes in the present study is listed in Supplemental Table 1. They display mixed ages and sexes with varied medical history and use of medications. Information on medical history and medications used is cursory, but it is known that two donors of cryopreserved hepatocytes had been taking antihypertensives and two donors of liver tissue and fresh hepatocytes had received cytostatic agents. These drugs are not known to affect expression levels of SLC transporters and are unlikely to explain the differences seen in the present study.
Uptake transporters might be rate-determining for drug clearance, and data from isolated or cryopreserved hepatocytes often underpredict in vivo CL. The modeling of in vivo CL from rat and human hepatocyte uptake data in recent studies needed inclusion of empirical scaling factors ranging from 2 to 67 to achieve accurate CL predictions (Gardiner and Paine, 2011; Menochet et al., 2012). Some researchers have taken a pragmatic approach recommending regression line–based methods to compensate for underpredictions of CL from in vitro systems until the underlying causes can be identified (Sohlenius-Sternbeck et al., 2012). It has been noted that one possible reason for underpredictions could be a change in absorptive capacity of the cryopreserved hepatocytes consistent with a loss of uptake transporters (Foster et al., 2011; Hallifax et al., 2012). In the present study we showed that IVIVE of in vivo hepatic CL was improved using uptake data from freshly isolated hepatocytes over data from cryopreserved cells for several uptake transporter substrates, suggesting that underpredictions of CL are partly due to reduced uptake transporter activity in the cryopreserved hepatocytes.
In our retrospective analysis of in vivo CL predictions, the downregulation of drug uptake transporters in human cryopreserved hepatocytes seemed to limit measurements of drug metabolism for a collection of AstraZeneca compounds. Scaling of in vivo CLint from human cryopreserved hepatocytes in vitro CLint for a large majority of compounds gave lower values than scaling from human microsomal CLint, while scaling from fresh rat hepatocytes gave higher in vivo CLint values than did rat liver microsomes. A series of four AstraZeneca mPGES-1 inhibitors seemed to display uptake rate–limited metabolic clearance that would be consistent with such an uptake limitation present in human cryopreserved hepatocytes that is not present in fresh rat hepatocytes. It is likely that the compounds showing large discrepancies when scaling from microsomes or hepatocytes are substrates of drug uptake transporters. Our data illustrate the need for compensatory scaling methods such as regression line methods that have been proposed by Sohlenius-Sternbeck et al. (2012).
In summary, this study clearly indicates that drug uptake transporters in cryopreserved hepatocytes from both human and rat to a large extent are lost during preparation and may significantly affect the prediction of in vivo clearance when uptake is limiting the metabolism of the studied compounds. Cryopreserved hepatocytes should be used with awareness of their limitations, and it would be prudent to primarily use lots exhibiting high remaining uptake activity. Improved cell isolation and tissue-handling protocols are needed to maintain transporter phenotype in both fresh and cryopreserved hepatocytes.
Acknowledgments
The technical assistance of Annelie Bengtsson, Sveinn Briem, Jessie Dahlström, and Stefan Martinsson is gratefully acknowledged. We thank Dr. Bruno Stieger, Department of Clinical Pharmacology, University of Zürich, Switzerland, for kindly providing us with antibodies.
Authorship Contributions
Participated in research design: Lundquist, Lööf, Sohlenius-Sternbeck, Floby, Johansson, Bylund.
Conducted experiments: Lundquist, Lööf, Floby, Johansson.
Contributed new reagents or analytic tools: Lundquist, Johansson, Bylund, Sohlenius-Sternbeck.
Performed data analysis: Lundquist, Lööf, Sohlenius-Sternbeck, Bylund.
Wrote or contributed to the writing of the manuscript: Lundquist, Sohlenius-Sternbeck, Johansson, Bylund, Hoogstraate, Andersson, Afzelius.
Footnotes
- Received October 2, 2013.
- Accepted January 6, 2014.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ACN
- acetonitrile
- AZ
- AstraZeneca
- Cblood/Cplasma
- blood-to-plasma concentration ratio
- CL
- clearance
- CLint
- intrinsic clearance
- DMSO
- dimethylsulfoxide
- FCS
- fetal calf serum
- fhepatic
- hepatically cleared fraction
- fu
- fraction unbound, free fraction
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- h
- as prefix, human
- HRP
- horseradish peroxidase
- IVIVE
- in vitro–in vivo extrapolation
- KHL
- Krebs-Henseleit buffer
- LOQ
- limit of quantification
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MCT1
- monocarboxylate transporter 1
- mPGES-1
- microsomal prostaglandin E synthase-1
- MS
- mass spectrometry
- NTCP
- Na+-taurocholate cotransporting polypeptide
- OAT
- organic anion transporter
- OATP
- organic anion transporting polypeptide
- OCT
- organic cation transporter
- P450
- cytochrome P450
- PBS
- phosphate-buffered saline
- PEPT1
- peptide transporter 1
- Qh,b
- hepatic blood flow
- r
- as prefix, rat
- SLC
- solute carrier
- TBST
- Tris-buffered saline/0.1% Tween 20
- UGT
- UDP-glucuronosyltransferase
- WE
- Williams’ E medium
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}