

Investigation of the Impact of Substrate Selection on In Vitro Organic Anion Transporting Polypeptide 1B1 Inhibition Profiles for the Prediction of Drug-Drug Interactions

Abstract
The risk assessment of organic anion transporting polypeptide (OATP) 1B1–mediated drug-drug interactions (DDIs) is an indispensable part of drug development. We previously reported that in vitro inhibitory potencies of several inhibitors on OATP1B1 depend on the substrates when prototypical substrates, estradiol-17β-glucuronide (E2G), estrone-3-sulfate, and sulfobromophthalein were used as test substrates. The purpose of this study was to comprehensively investigate this substrate-dependent inhibition of OATP1B1 using clinically relevant OATP1B1 inhibitors and substrate drugs. Effects of cyclosporine A (CsA), rifampin, and gemfibrozil on OATP1B1-mediated uptake of 12 substrate drugs were examined in OATP1B1-expressing human embryonic kidney 293 cells. The Ki values (μM) for CsA varied from 0.0771 to 0.486 (6.3-fold), for rifampin from 0.358 to 1.23 (3.4-fold), and for gemfibrozil from 9.65 to 252 (26-fold). Except for the inhibition of torasemide uptake by CsA and that of nateglinide uptake by gemfibrozil, the Ki values were within 2.8-fold of those obtained using E2G as a substrate. Preincubation potentiated the inhibitory effect of CsA on OATP1B1 with similar magnitude regardless of the substrates. R values calculated based on a static model showed some variation depending on the Ki values determined with various substrates, and such variability could have an impact on the DDI predictions particularly for a weak-to-moderate inhibitor (gemfibrozil). OATP1B1 substrate drugs except for torasemide and nateglinide, or E2G as a surrogate, is recommended as an in vitro probe in the inhibition experiments, which will help mitigate the risk of false-negative DDI predictions potentially caused by substrate-dependent Ki variation.
Introduction
Pharmacokinetic drug-drug interaction (DDI) is a concern in clinical practice. Because alteration of pharmacokinetics by a concomitant medication can sometimes significantly affect pharmacologic and/or adverse effects of the victim drug, accurate evaluation of the DDI potentials of drug candidates during drug development is indispensable from the viewpoint of clinical safety. Recent scientific progress has revealed that membrane transporters as well as drug metabolizing enzymes play an important role in the pharmacokinetics of many drugs; thus, they can also serve as the sites of DDIs (Mizuno et al., 2003; Shitara et al., 2005; Yoshida et al., 2013). After publication of the transporter white-paper by International Transporter Consortium (Giacomini et al., 2010), DDI guidelines or draft guidance materials were released by the European Medicines Agency (EMA), the U.S. Food and Drug Administration (FDA), and the Japanese Ministry of Health, Labor and Welfare (MHLW), in which systematic approaches to the evaluation of transporter-mediated DDIs are given (Center for Drug Evaluation and Research, 2012; Committee for Human Medicinal Products, 2012; Ministry of Health, Labor and Welfare, 2014).
Organic anion transporting polypeptide (OATP) 1B1 (encoded by SLCO1B1) is expressed on the sinusoidal membrane of hepatocytes and mediates the uptake of a wide range of anionic drugs (Niemi et al., 2011). The genetic polymorphisms of SLCO1B1 are associated with interindividual variation in the plasma concentrations of 3-hydroxymethylglutaryl-CoA reductase inhibitors (statins) (Shitara and Sugiyama, 2006) and adverse event rates during simvastatin therapy (Link et al., 2008). In clinical situations, DDIs involving OATP substrate drugs and cyclosporine A (CsA) or a single dose of rifampin are considered to be attributable to the inhibition of OATP1B1 and its closely-related isoform, OATP1B3 (Shitara et al., 2003; Maeda et al., 2011; Takashima et al., 2012). Based on the accumulated evidence, OATP1B1 has been widely recognized as a clinically important transporter, and the evaluation of OATP1B1-mediated DDI potential of drug candidates is crucial in the drug development.
For the evaluation of transporter-mediated DDIs, static and dynamic (physiologically based pharmacokinetic) models have been used. The static model that assumes a constant inhibitor concentration is suitable in the early stage of drug development because of its simplicity (Yoshida et al., 2012), while allowing for false-positive prediction. The dynamic model allows more accurate prediction by taking the time-course of the inhibitor concentration into account (Imamura et al., 2011; Kudo et al., 2013). In both models, the inhibition constant (Ki) is an essential parameter to evaluate the clinical relevance of the DDIs, so its accurate estimation for the target transporter is a key step to achieve their purposes.
To determine the Ki values of drug candidates for OATP1B1, in vitro cell-based inhibition assays using representative probe substrates are commonly conducted. However, it was reported that the in vitro inhibitory potencies of gemfibrozil and other inhibitors on OATP1B1 depend on the probe substrates (Noé et al., 2007; Izumi et al., 2013). Therefore, care is needed in probe substrate selection for in vitro OATP1B1 inhibition assays to avoid false-negative DDI predictions. We previously examined the in vitro inhibitory effects of 14 compounds on OATP1B1 using three prototypical probe substrates: estradiol-17β-glucuronide (E2G), estrone-3-sulfate (E1S), and sulfobromophthalein (BSP). Among them, E2G provided the lowest Ki value for all inhibitors examined; thus, we proposed that the use of E2G as an in vitro probe substrate may help mitigate the risk of false-negative DDI prediction (Izumi et al., 2013).
Currently, various clinically used drugs including statins (Sharma et al., 2012), angiotensin II receptor blockers (Yamashiro et al., 2006), endothelin receptor antagonists (Treiber et al., 2007), antidiabetics (Takanohashi et al., 2012), and diuretics (Werner et al., 2010) are known as OATP1B1 substrates. These clinically used OATP1B1 substrate drugs, in addition to prototypical substrates, could also serve as in vitro probe substrates because of their clinical relevance. However, even when using the OATP1B1 substrate drugs, substrate-dependence in the in vitro inhibitory potencies of OATP1B1 inhibitors is a topic of great concern.
Therefore, this study evaluated the inhibitory effects of CsA, rifampin, and gemfibrozil on OATP1B1-mediated uptake of 12 clinically used substrate drugs including statins (pitavastatin, atorvastatin, fluvastatin, rosuvastatin, and pravastatin), antidiabetics (repaglinide, nateglinide, and glibenclamide), and other classes of drugs (bosentan, valsartan, torasemide, and fexofenadine) and compared the Ki values with those we had previously reported with E2G, E1S, and BSP as substrates. In addition, the impact of probe substrate selection on the prediction of OATP1B1-mediated DDIs was evaluated based on a static model.
Materials and Methods
[3H]E2G (48.9 Ci/mmol) and [3H]E1S (45.6 Ci/mmol) were purchased from PerkinElmer Life Sciences (Boston, MA). [3H]BSP (5.5 Ci/mmol) was synthesized by Hartmann Analytic GmbH (Braunschweig, Germany). Unlabeled E2G, rifampin, repaglinide, and gemfibrozil were purchased from Sigma-Aldrich (St. Louis, MO). CsA, pitavastatin calcium, pravastatin sodium, and glibenclamide (glyburide) were obtained from Wako Pure Chemical Industries (Osaka, Japan). Atorvastatin calcium trihydrate, rosuvastatin calcium, and valsartan were purchased from LKT Laboratories (St. Paul, MN), and fluvastatin sodium, fexofenadine hydrochloride, and bosentan were purchased from Toronto Research Chemicals (Toronto, ON, Canada). Torasemide and nateglinide were obtained from Tokyo Chemical Industries (Tokyo, Japan) and Tocris Bioscience (Minneapolis, NM), respectively. All other chemicals were of analytic grade and commercially available.
Uptake and Inhibition Studies Using OATP1B1-Expressing Cells.
The stably OATP1B1-expressing human embryonic kidney 293 (HEK293) cells and the corresponding control HEK293 cells, as established previously (Izumi et al., 2013), were used in this study. The cells were maintained in Dulbecco’s modified Eagle medium (Invitrogen, Carlsbad, CA) supplemented with 10% (v/v) of fetal bovine serum, penicillin (final concentration, 100 units/ml), streptomycin (100 µg/ml), and hygromycin B (80 μg/ml) in a humidified incubator containing 5% CO2 gas at 37°C. For cell seeding, the cells were trypsinized and uniformly suspended in the designated volume of the culture medium to provide 4 × 105 cells/ml. One ml of the cell suspension was added to each well of a poly-d-lysine–coated 24-well plate (BD Biosciences, San Jose, CA), and the cells were further cultured in the incubator for 48 hours.
The uptake and inhibition studies were performed as described previously elsewhere (Izumi et al., 2013). Briefly, cell culture medium was replaced with prewarmed Krebs Henseleit (KH) buffer (118 mM NaCl, 23.1 mM NaHCO3, 4.83 mM KCl, 0.96 mM KH2PO4, 1.20 mM MgSO4, 12.5 mM HEPES, 5.0 mM glucose, and 1.53 mM CaCl2, pH 7.4), and the cells were preincubated for 5 minutes at 37°C. After aspirating the preincubation buffer, we initiated the uptake reaction by the addition of 250 µl of a prewarmed KH buffer containing a substrate (pitavastatin, atorvastatin, fluvastatin, rosuvastatin, pravastatin, repaglinide, nateglinide, glibenclamide, bosentan, valsartan, torasemide, or fexofenadine) in the presence or absence of an inhibitor (CsA, rifampin, or gemfibrozil). The concentrations of the substrates and inhibitors used are given in the figure legends. At the designated time, the buffer was aspirated, and the cells were washed twice with ice-cold KH buffer.
Effect of Preincubation with CsA on the Inhibition of OATP1B1-Mediated Uptake of [3H]E2G, [3H]E1S, [3H]BSP, Pitavastatin, and Atorvastatin.
The effect of preincubation with CsA on OATP1B1 inhibition was examined according to previous reports with some modifications (Amundsen et al., 2010; Shitara et al., 2013a). After 48-hour cell culture, the cell culture medium was replaced with prewarmed KH buffer. The KH buffer was aspirated, and the OATP1B1-expressing cells and control cells were preincubated for 1 hour at 37°C with KH buffer containing or not containing CsA [or directly coincubated with a substrate ([3H]E2G, [3H]E1S, [3H]BSP, pitavastatin, or atorvastatin) and CsA as the no-preincubation condition]. After the 1-hour preincubation, the preincubation buffer was aspirated, and the cells were coincubated with a substrate and CsA to initiate the uptake reaction. At the designated time (1 minute, [3H]E1S and atorvastatin; 2 minutes, pitavastatin; 5 minutes, [3H]E2G and [3H]BSP), the buffer was aspirated, and the cells were washed twice with ice-cold KH buffer.
Quantification of Test Compounds Taken Up by the Cells.
For the cellular uptake studies with radiolabeled substrates ([3H]E2G, [3H]E1S, and [3H]BSP), the cells were lysed with 0.5 ml of 0.1 N NaOH overnight at room temperature. The resulting cell lysate was neutralized with 50 µl of 1 N HCl. We mixed 400 µl of the aliquot with 4 ml of scintillation fluid (Hionic-Fluor; PerkinElmer Life Sciences), and the radioactivity associated with the cells was measured with a liquid scintillation counter (Tri-Carb 3100TR; PerkinElmer Life Sciences). The remaining neutralized cell lysate samples were used to quantify the protein concentrations (BCA Protein Assay Kit; Thermo Fisher Scientific, Waltham, MA). Radioactivities of the radiolabeled substrates in the incubation buffer were also measured by liquid scintillation counting.
For the cellular uptake studies with unlabeled substrates (pitavastatin, atorvastatin, fluvastatin, rosuvastatin, pravastatin, repaglinide, nateglinide, glibenclamide, bosentan, valsartan, torasemide, and fexofenadine), the cells were vigorously mixed and deproteinized with 250 µl of methanol containing an appropriate internal standard, which was followed by centrifugation. The obtained supernatant was filtrated, and the resulting filtrates were subjected to high-performance liquid chromatography with tandem mass spectrometry analysis. Chromatography was performed using an Atlantis T3 column (3.0 μm, 2.1 mm i.d., 50 mm; Waters, Milford, MA) at a flow rate of 0.3 ml/min. Distilled water containing 0.1% formic acid (solvent A) and acetonitrile containing 0.1% formic acid (solvent B) were used as the mobile phases. The initial condition was 100% solvent A; the percentage of solvent B was linearly increased to 80% over 3 minutes, then to 100% over the next 0.01 minutes, and was maintained at this level for 1 minute. The column was equilibrated with the initial mobile phase before each injection (injection volume, 10 µl). A Quattro Premier mass spectrometer (Waters) was used for the detection.
The analytes were ionized by electrospray ionization in positive or negative (only for pravastatin) ion mode, and the selected ion monitoring transitions were: 422.2 > 274.0 for pitavastatin, 559.3 > 249.9 for atorvastatin, 412.2 > 265.9 for fluvastatin, 482.4 > 258.0 for rosuvastatin, 423.3 > 320.9 for pravastatin, 453.3 > 230.0 for repaglinide, 318.3 > 68.7 for nateglinide, 494.3 > 368.9 for glibenclamide, 552.3 > 201.8 for bosentan, 436.4 > 180.0 for valsartan, 349.3 > 263.9 for torasemide, and 502.7 > 466.3 for fexofenadine.
In the studies with unlabeled substrates, extra cells were prepared to quantify the protein amount per well. The cells were lysed with 0.5 ml of 0.1 N NaOH overnight at room temperature, and the resulting cell lysate was neutralized with 50 µl of 1 N HCl. The neutralized cell lysate samples were used to quantify the protein concentrations by BCA protein assay (Thermo Fisher Scientific).
Determination of Kinetic Parameters.
Uptake of a substrate was expressed as the uptake volume (µl/mg protein), which was given as the amount of the substrate associated with the cells (dpm/well or pmol/well) divided by the concentration in the incubation buffer (dpm/µl or pmol/µl) and the protein amount (mg protein/well). The OATP1B1-mediated net uptake was obtained by subtracting the uptake into control cells from the uptake into OATP1B1-expressing cells. The concentration dependence of the uptake of a substrate mediated by OATP1B1 was analyzed using the following Michaelis-Menten equation: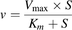 (1)where v is the uptake velocity of the substrate (pmol/min per mg protein), S is the substrate concentration in the incubation buffer (μM), Km is the Michaelis constant (μM), and Vmax is the maximum uptake rate (pmol/min per mg protein). When an additional component was observed at higher substrate concentrations, the following equation was used for the analysis:
(1)where v is the uptake velocity of the substrate (pmol/min per mg protein), S is the substrate concentration in the incubation buffer (μM), Km is the Michaelis constant (μM), and Vmax is the maximum uptake rate (pmol/min per mg protein). When an additional component was observed at higher substrate concentrations, the following equation was used for the analysis: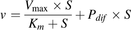 (2)where Pdif represents the nonsaturable uptake clearance (μl/min per mg protein). Fitting was performed by a nonlinear least-squares regression method using the MULTI program (Yamaoka et al., 1981).
(2)where Pdif represents the nonsaturable uptake clearance (μl/min per mg protein). Fitting was performed by a nonlinear least-squares regression method using the MULTI program (Yamaoka et al., 1981).
Half-maximal inhibitory concentration (IC50) of an inhibitor was estimated by examining the inhibitory effect on the OATP1B1-mediated uptake of a substrate (% of control) using the following equation, as reported previously (Izumi et al., 2013): (3)where CL and CLi represent the uptake clearance in the absence and presence of an inhibitor, respectively, and I is the concentration of the inhibitor. P (initial value = 100) was set as a free parameter to achieve the best fit in the nonlinear iterative least-squares regression analysis. The parameters were estimated by a nonlinear least-squares regression method using the MULTI program. With the assumption of competitive inhibition, the inhibition constant, Ki was estimated by eq. 4 (Amundsen et al., 2010):
(3)where CL and CLi represent the uptake clearance in the absence and presence of an inhibitor, respectively, and I is the concentration of the inhibitor. P (initial value = 100) was set as a free parameter to achieve the best fit in the nonlinear iterative least-squares regression analysis. The parameters were estimated by a nonlinear least-squares regression method using the MULTI program. With the assumption of competitive inhibition, the inhibition constant, Ki was estimated by eq. 4 (Amundsen et al., 2010):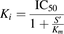 (4)where S′ is the substrate concentration in the incubation buffer used for inhibition studies, and Km is the Michaelis constant of the substrate for OATP1B1.
(4)where S′ is the substrate concentration in the incubation buffer used for inhibition studies, and Km is the Michaelis constant of the substrate for OATP1B1.
Prediction of OATP1B1-Mediated Drug-Drug Interactions with a Static Model.
The degree of inhibition of OATP1B1-mediated hepatic uptake (R value) was calculated for three inhibitors (CsA, rifampin, and gemfibrozil) based on the DDI guidelines and draft guidance materials released by regulatory agencies in the United States (Center for Drug Evaluation and Research, 2012), the European Union (Committee for Human Medicinal Products, 2012), and Japan (Ministry of Health, Labor and Welfare, 2014). The decision tree of the FDA for the inhibition of hepatic uptake transporters including OATP1B1 consists of two steps and a recommendation to evaluate the inhibition potential of an investigational drug on OATP1B1 using the following R value as the first step: (5)where Cmax represents the maximum systemic total (bound + unbound) plasma concentration of inhibitor. The FDA (the second step), EMA, and MHLW recommend evaluating the inhibitory potential of an investigational drug on OATP1B1 using the following R value:
(5)where Cmax represents the maximum systemic total (bound + unbound) plasma concentration of inhibitor. The FDA (the second step), EMA, and MHLW recommend evaluating the inhibitory potential of an investigational drug on OATP1B1 using the following R value: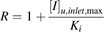 (6)where [I]u,inlet,max represents the estimated maximum unbound inhibitor concentration at the inlet to the liver and is defined as follows (Ito et al., 1998):
(6)where [I]u,inlet,max represents the estimated maximum unbound inhibitor concentration at the inlet to the liver and is defined as follows (Ito et al., 1998):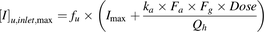 (7)where fu is the unbound fraction of an inhibitor in blood, which was calculated from the unbound fraction in plasma assuming the blood-to-plasma concentration ratio to be unity in this study, Imax is the maximum circulating blood concentration of an inhibitor, ka is the absorption rate constant of the inhibitor, Fa is the fraction of the inhibitor dose absorbed, Fg is the fraction of the absorbed inhibitor dose escaping gut wall extraction, Dose is the inhibitor dose, and Qh is the hepatic blood flow rate (97 l/h) (Committee for Human Medicinal Products, 2012; Ministry of Health, Labor and Welfare, 2014). To minimize the risk of false-negative DDI prediction, ka = 0.1 min−1 and Fa × Fg = 1 were applied to the [I]u,inlet,max calculations in this study. Dose, fu, Cmax, and [I]u,inlet,max of CsA, rifampin, and gemfibrozil (Okerholm et al., 1976; Lemaire and Tillement, 1982; Muck et al., 1999; Burman et al., 2001; Shitara et al., 2004; Maeda et al., 2011; Yoshida et al., 2012) are given in Supplemental Table 1.
(7)where fu is the unbound fraction of an inhibitor in blood, which was calculated from the unbound fraction in plasma assuming the blood-to-plasma concentration ratio to be unity in this study, Imax is the maximum circulating blood concentration of an inhibitor, ka is the absorption rate constant of the inhibitor, Fa is the fraction of the inhibitor dose absorbed, Fg is the fraction of the absorbed inhibitor dose escaping gut wall extraction, Dose is the inhibitor dose, and Qh is the hepatic blood flow rate (97 l/h) (Committee for Human Medicinal Products, 2012; Ministry of Health, Labor and Welfare, 2014). To minimize the risk of false-negative DDI prediction, ka = 0.1 min−1 and Fa × Fg = 1 were applied to the [I]u,inlet,max calculations in this study. Dose, fu, Cmax, and [I]u,inlet,max of CsA, rifampin, and gemfibrozil (Okerholm et al., 1976; Lemaire and Tillement, 1982; Muck et al., 1999; Burman et al., 2001; Shitara et al., 2004; Maeda et al., 2011; Yoshida et al., 2012) are given in Supplemental Table 1.
Results
Uptake of Clinically Used OATP1B1 Substrate Drugs in OATP1B1-Expressing Cells.
The time profiles of the uptake of 12 clinically used OATP1B1 substrate drugs—pitavastatin, atorvastatin, fluvastatin, rosuvastatin, pravastatin, repaglinide, nateglinide, glibenclamide, bosentan, valsartan, torasemide, and fexofenadine—in OATP1B1-expressing cells and control cells are shown in Fig. 1. Single representative substrate concentrations that were equivalent to or lower than the reported Km values for OATP1B1 were selected for respective substrate drugs based on the detection sensitivities in this time-course study. For substrates whose Km values were not available, lower concentrations (0.1 µM, repaglinide and glibenclamide; 1 µM, nateglinide and fexofenadine) were selected based on the detection sensitivities. Our subsequent experiments confirmed that the substrate concentrations selected in this time-course study were lower than the respective Km values we examined (Table 1).
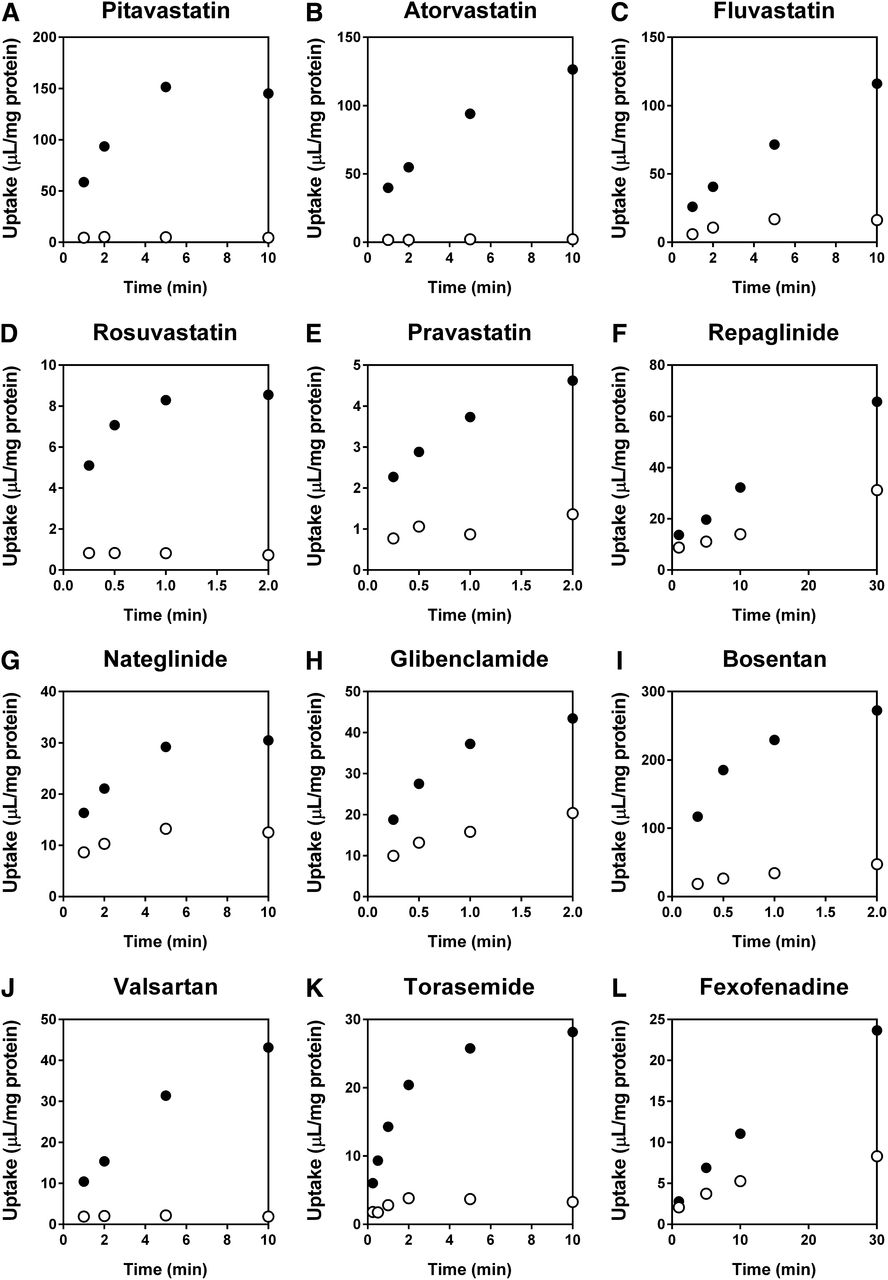
Time profiles of the uptake of clinically used OATP1B1 substrate drugs in OATP1B1-expressing cells and the control cells. Uptake of (A) pitavastatin (0.1 µM), (B) atorvastatin (0.1 µM), (C) fluvastatin (1 µM), (D) rosuvastatin (1 µM), (E) pravastatin (10 µM), (F) repaglinide (0.1 µM), (G) nateglinide (1 µM), (H) glibenclamide (0.1 µM), (I) bosentan (0.1 µM), (J) valsartan (1 µM), (K) torasemide (1 µM), and (L) fexofenadine (1 µM) in OATP1B1-expressing cells (●) and the control cells (○) was examined over a period of 2 (rosuvastatin, pravastatin, glibenclamide, and bosentan), 10 (pitavastatin, atorvastatin, fluvastatin, nateglinide, valsartan, and torasemide), or 30 minutes (repaglinide and fexofenadine) at 37°C. Each point represents the mean ± S.D. (n = 3).
Saturation kinetics of OATP1B1-mediated uptake of clinically used OATP1B1 substrate drugs
Kinetic parameters were estimated by nonlinear least-squares regression analysis based on eqs. 1 or 2 as described under Materials and Methods, and are shown as mean of two independent experiments or mean ± S.D. (n = 3 or 4).
The OATP1B1-expressing cells showed higher transport activities for all compounds examined compared with the control cells, indicating that they are substrates of OATP1B1. On the basis of the time profiles and detection sensitivities, the following incubation times were selected for subsequent in vitro studies: 0.5 minutes for rosuvastatin, pravastatin, glibenclamide, bosentan, and torasemide; 1 minute for atorvastatin; 2 minutes for pitavastatin and nateglinide; 5 minutes for fluvastatin, repaglinide, and valsartan; and 10 minutes for fexofenadine.
Saturation Kinetics of OATP1B1-Mediated Uptake of Clinically Used Substrate Drugs.
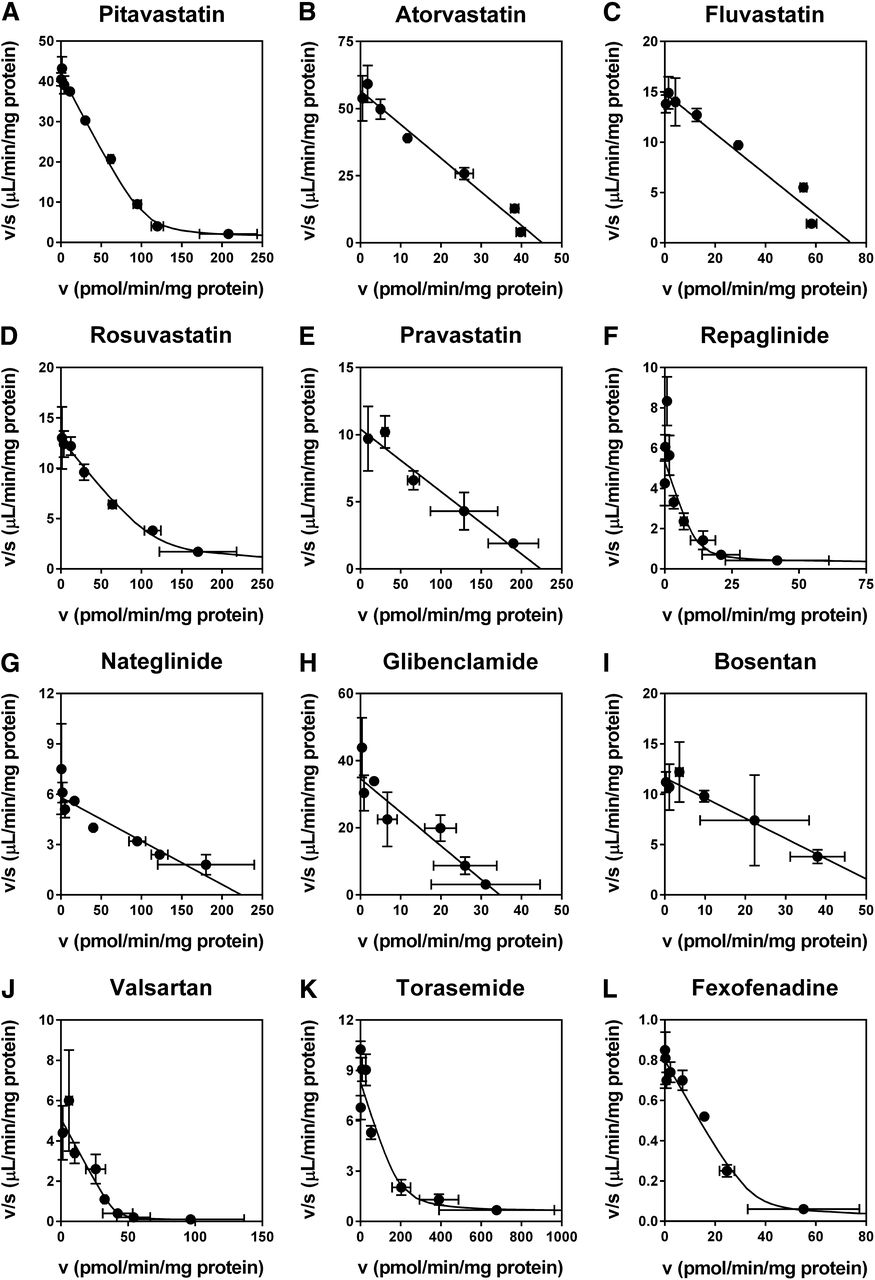
The concentration dependence of the uptake of the 12 substrate drugs via OATP1B1 was examined to determine the saturation kinetic parameters. Representative Eadie-Hofstee plots of the uptake are shown in Fig. 2, and the obtained kinetic parameters (Km, Vmax, and Pdif) are summarized in Table 1. The OATP1B1-mediated uptake of the compounds examined consisted of one saturable component with or without a minor nonsaturable component, and the Km values ranged from 0.761 (atorvastatin) to 61.6 μM (fexofenadine). The Km values of pitavastatin (2.48 µM), atorvastatin (0.761 µM), fluvastatin (4.80 µM), rosuvastatin (9.31 µM), pravastatin (27.0 µM), valsartan (7.48 µM), and torasemide (20.9 µM) determined in this study were within the range of those previously reported elsewhere using OATP1B1-expressing mammalian cells (Table 1). To our knowledge, this is the first report to examine the Km values of repaglinide (1.36 µM), nateglinide (36.4 µM), glibenclamide (1.24 µM), and fexofenadine (61.6 µM) for OATP1B1 using OATP1B1-expressing mammalian cells.

Concentration dependence of the OATP1B1-mediated uptake of (A) pitavastatin (0.01–100 µM), (B) atorvastatin (0.01–10 µM), (C) fluvastatin (0.03–30 µM), (D) rosuvastatin (0.1–100 µM), (E) pravastatin (1–100 µM), (F) repaglinide (0.01–100 µM), (G) nateglinide (0.1–100 µM), (H) glibenclamide (0.01–10 µM), (I) bosentan (0.03–10 µM), (J) valsartan (0.3–1000 µM), (K) torasemide (0.1–100 µM), and (L) fexofenadine (0.1–1000 µM). The uptake was determined for 0.5 (rosuvastatin, pravastatin, glibenclamide, bosentan, and torasemide), 1 (atorvastatin), 2 (pitavastatin and nateglinide), 5 (fluvastatin, repaglinide, and valsartan), or 10 minutes (fexofenadine) at 37°C. Representative data from two to four independent experiments are shown as Eadie-Hofstee plots, and each point represents the mean ± S.D. (n = 3). Fitted lines that were obtained by a nonlinear least-squares regression analysis based on eqs. 1 or 2 are also presented as solid lines.
Inhibitory Effects of CsA, Rifampin, and Gemfibrozil on OATP1B1-Mediated Uptake of Substrate Drugs.
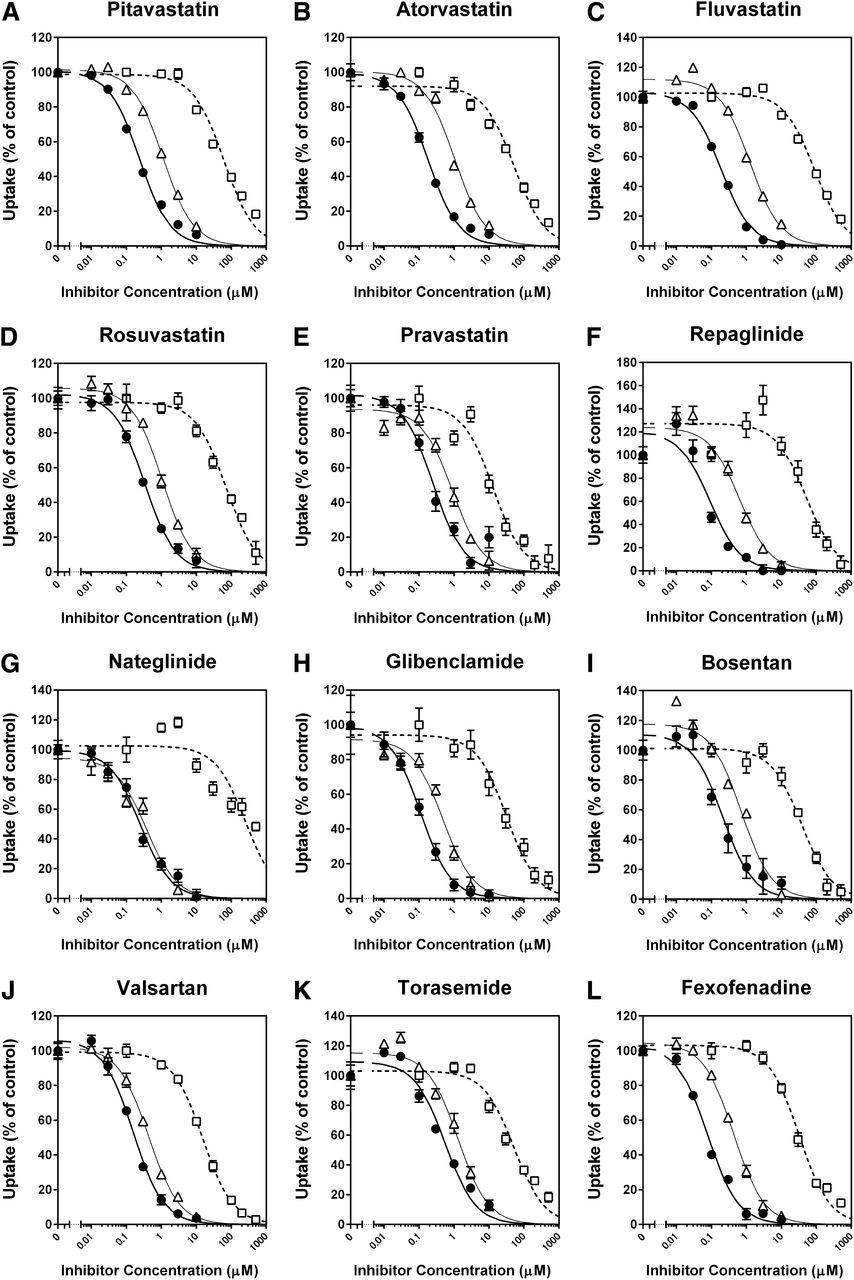
The inhibitory effects of CsA, rifampin, and gemfibrozil on the OATP1B1-mediated uptake of 12 substrate drugs were investigated without any preincubation of the cells in the presence of inhibitors (Fig. 3). CsA, rifampin, and gemfibrozil inhibited the uptake of all substrates examined in a concentration-dependent manner, and all the inhibition curves were well described by the equation for monophasic inhibition (eq. 3). Because the majority of OATP1B1-mediated uptake of clinically used substrate drugs consisted of one saturable component at the selected substrate concentrations in this inhibition assay, the Ki values of CsA, rifampin, and gemfibrozil for the OATP1B1-mediated uptake of the substrate drugs were estimated by applying the respective IC50 values (Supplemental Table 2), substrate concentrations, and Km values (Table 1) into eq. 4, as summarized in Table 2. For comparison, the Ki values of CsA, rifampin, and gemfibrozil for OATP1B1-mediated uptake of prototypical in vitro probe substrates (E2G, E1S, and BSP) that we examined under the same experimental conditions (as reported previously by Izumi et al., 2013) are also listed in Table 2. In addition, the reported Ki or IC50 values of CsA, rifampin, and gemfibrozil for the OATP1B1-mediated uptake of the prototypical substrates and substrate drugs are summarized in Supplemental Table 3.
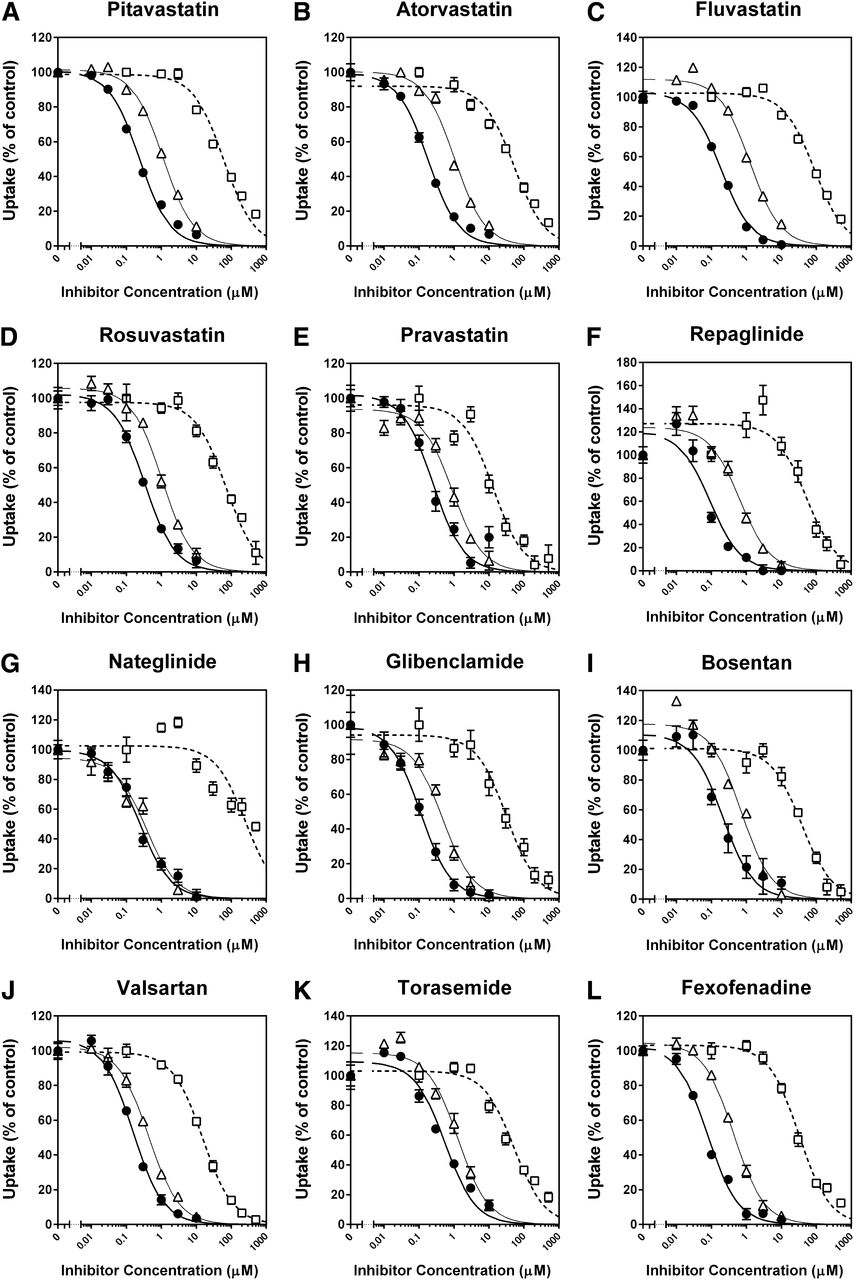
Inhibitory effects of CsA, rifampin, and gemfibrozil on OATP1B1-mediated uptake of clinically used substrate drugs. Uptake of (A) pitavastatin (0.1 µM), (B) atorvastatin (0.1 µM), (C) fluvastatin (1 µM), (D) rosuvastatin (1 µM), (E) pravastatin (10 µM), (F) repaglinide (0.1 µM), (G) nateglinide (1 µM), (H) glibenclamide (0.1 µM), (I) bosentan (0.1 µM), (J) valsartan (1 µM), (K) torasemide (1 µM), and (L) fexofenadine (1 µM) were investigated for 0.5 (rosuvastatin, pravastatin, glibenclamide, bosentan, and torasemide), 1 (atorvastatin), 2 (pitavastatin and nateglinide), 5 (fluvastatin, repaglinide, and valsartan), or 10 minutes (fexofenadine) at 37°C in the presence and absence of CsA (●), rifampin (▵), or gemfibrozil (□). The data are shown as the percentage of control. Thick, thin, and dashed lines represent fitted lines for the inhibition by CsA, rifampin, and gemfibrozil, respectively, which were obtained by a nonlinear least-squares regression analysis based on eq. 3, and the estimated IC50 values were summarized in Supplemental Table 2. Each point represents the mean ± S.D. (n = 6 or 9).
Ki values of CsA, rifampin, and gemfibrozil for OATP1B1-mediated uptake of clinically used OATP1B1 substrate drugs
OATP1B1-mediated uptake of 12 clinically used OATP1B1 substrate drugs was examined in the presence and absence of CsA, rifampin, or gemfibrozil as shown in Fig. 3. The IC50 values were estimated by a nonlinear least-squares regression analysis. The Ki values (parameter estimate ± parameter S.D.) were estimated using substrate concentrations, Km, and IC50 values by eq. 4 (n = 6 or 9). The Ki values of CsA, rifampin, and gemfibrozil for the OATP1B1-mediated uptake of prototypical substrates (E2G, E1S, and BSP) are cited from our previous study (Izumi et al., 2013).
The Ki values of CsA showed a 6.3-fold difference depending on the substrates selected, ranging from 0.0771 (fexofenadine as a substrate) to 0.486 μM (torasemide). Except for the Ki value for torasemide uptake (0.486 µM), the Ki values (range: 0.0771–0.301 µM) were within 2.6-fold of that obtained using E2G as a substrate (0.118 µM), but were overestimated by those given with E1S and BSP as substrates (0.732 and 0.694 µM, respectively).
The Ki values of rifampin showed a 3.4-fold difference depending on the substrate selected, ranging from 0.358 (nateglinide) to 1.23 μM (torasemide). These Ki values were within 2.1-fold of that obtained using E2G as a substrate (0.585 µM), but were overestimated by those given with E1S and BSP as substrates (6.96 and 2.75 µM, respectively).
The Ki values of gemfibrozil showed a 26-fold difference depending on the substrates selected, ranging from 9.65 (pravastatin) to 252 μM (nateglinide). Except for the Ki value for nateglinide uptake (252 µM), the Ki values (range: 9.65–72.7 µM) were within 2.8-fold of that obtained using E2G as a substrate (26.4 µM), but were overestimated by those given with E1S and BSP as substrates (381 and 173 µM, respectively).
Impact of Substrate Selection on Prediction of Clinical OATP1B1-Mediated DDIs Using a Static Model.
To evaluate the impact of substrate selection on the prediction of OATP1B1-mediated DDIs by a static model, the R values of CsA, rifampin, and gemfibrozil were calculated for the 12 clinically used OATP1B1 substrate drugs and three typical prototypical substrates (E2G, E1S, and BSP) using the Ki values estimated for each substrate (Table 2). Based on the DDI guidelines or draft guidance materials released by the FDA, EMA, and MHLW, the maximum systemic total plasma concentration of the inhibitor (Cmax) or the estimated maximum unbound inhibitor concentration at the inlet to the liver ([I]u,inlet,max) was used as [I], the inhibitor concentration. The Cmax and [I]u,inlet,max of CsA, rifampin, and gemfibrozil that were used for the R value calculations are given in Supplemental Table 1. The calculated R values and observed AUCR, which is the ratio of the area under the plasma concentration-time curve (AUC) of a substrate drug with an inhibitor to that without the inhibitor in clinical DDI studies, are summarized in Table 3.
Prediction of OATP1B1-mediated DDIs with a static model
The R values of CsA, rifampin, and gemfibrozil were determined using Ki values obtained from each probe substrate and [I] (Cmax or [I]u,inlet,max) based on eqs. 5 and 6. The Ki values were taken from Table 2. Cmax and [I]u,inlet,max of the inhibitors are given in Supplemental Table 1.
The R values of CsA calculated with [I]u,inlet,max were slightly higher than those using Cmax as the [I]. When Cmax was used, the R values of CsA for the 12 clinically used substrate drugs ranged from 2.95 (torasemide as substrate) to 13.3 (fexofenadine), and those for E2G, E1S, and BSP were 9.05, 2.30, and 2.37, respectively. When [I]u,inlet,max was used as [I], the R values of CsA for the clinically used substrate drugs ranged from 3.47 (torasemide) to 16.6 (fexofenadine), and those for E2G, E1S, and BSP were 11.2, 2.64, and 2.73, respectively. Therefore, the R values of CsA for clinically used substrate drugs were within 2.2-fold of that for E2G under the both [I] conditions except for torasemide, which yielded 3.1- to 3.2-fold greater Ki values than E2G, but the R values for E1S and BSP uptake were equal to or less than the lower limit of the R values obtained for the clinically used substrate drugs. Although the R values of CsA showed some variation depending on the Ki values determined with various substrates, all clinically used substrate drugs and prototypical substrates provided R values greater than the corresponding cutoff values recommended by the regulatory agencies in both Cmax (R ≥ 1.1, FDA) and [I]u,inlet,max (R ≥ 1.25, FDA and MHLW; R ≥ 1.04, EMA) conditions.
The R values of rifampin calculated with [I]u,inlet,max were lower than those using Cmax as the [I]. When Cmax was used, the R values of rifampin for the clinically used substrate drugs ranged from 19.7 (torasemide) to 65.2 (nateglinide), and those for E2G, E1S, and BSP were 40.3, 4.30, and 9.36, respectively. When [I]u,inlet,max was used as [I], the R values of rifampin for the clinically used substrate drugs ranged from 9.13 (torasemide) to 28.9 (nateglinide), and those for E2G, E1S, and BSP were 18.1, 2.44, and 4.64, respectively. Therefore, the R values of rifampin for clinically used substrate drugs were within 2.0-fold of that for E2G under both [I] conditions, but the R values for E1S and BSP uptake were less than the lower limit of the R values obtained for clinically relevant substrate drugs. Although the R values of rifampin showed some variation depending on the Ki values determined with various substrates, all clinically used substrate drugs and prototypical substrates provided R values greater than the corresponding cutoff values in both [I] conditions.
The R values of gemfibrozil calculated with [I]u,inlet,max were lower than those using Cmax as the [I]. When Cmax was used, the R values of gemfibrozil for the clinically used substrate drugs ranged from 1.40 (nateglinide) to 11.4 (pravastatin), and those for E2G, E1S, and BSP were 4.79, 1.26, and 1.58, respectively. Therefore, the R values for clinically used substrate drugs were within 2.4-fold of that for E2G except for nateglinide, which yielded 3.4-fold greater Ki values than E2G, but the R values for E1S and BSP uptake (1.26 and 1.58, respectively) were equal to or less than the lower limit of the R values obtained for clinically used substrate drugs. Although the R values of gemfibrozil showed some variation depending on the Ki values determined with various substrates, all clinically used substrate drugs and prototypical substrates provided R values greater than the cutoff value (R ≥ 1.1).
When [I]u,inlet,max was used as [I], the R values of gemfibrozil for the clinically used substrate drugs ranged from 1.01 (nateglinide) to 1.26 (pravastatin), and those for E2G, E1S, and BSP were 1.09, 1.01, and 1.01, respectively. Based on the cutoff value recommended by the FDA and MHLW (R ≥ 1.25), only pravastatin (R = 1.26) showed an R value greater than the threshold. Based on the cutoff value recommended by the EMA (R ≥ 1.04), the R values of 11 out of 15 substrates examined were greater than the threshold.
Effect of Preincubation with CsA on the Inhibition of OATP1B1-Mediated Uptake of [3H]E2G, [3H]E1S, [3H]BSP, Pitavastatin, and Atorvastatin.
Amundsen et al. (2010) and Shitara et al. (2013a) reported that the inhibitory effect of CsA on OATP1B1 was potentiated by preincubating OATP1B1-expressing cells with CsA before coincubation with a substrate and CsA, but whether the effect depends on the substrate selected remained to be clarified. To address this, the OATP1B1-mediated uptake of [3H]E2G (0.1 μM), [3H]E1S (0.01 μM), [3H]BSP (0.01 μM), pitavastatin (0.1 μM), and atorvastatin (0.1 μM) was examined in the presence of CsA after 1-hour preincubation with or without CsA. For comparison, coincubation of CsA and each of the substrates without the 1-hour preincubation process was also conducted as a no-preincubation condition.
The IC50 values of CsA for the five substrates after 1-hour preincubation without CsA were comparable to or slightly lower than those under a reference, no-preincubation condition (Fig. 4; Table 4). One-hour preincubation with CsA enhanced its inhibitory effect on OATP1B1 compared with 1-hour preincubation without CsA (Fig. 4), and the IC50 values of CsA for E2G, E1S, BSP, pitavastatin, and atorvastatin uptake decreased by 3.29-, 5.08-, 3.15-, 3.88-, and 4.32-fold, respectively (Table 4). The OATP1B1 inhibitory effect potentiated by preincubation with CsA resulted in higher R values for the substrates examined (Table 4).

Effect of preincubation with CsA on the inhibition of OATP1B1-mediated uptake of (A) [3H]E2G (0.1 µM), (B) [3H]E1S (0.01 µM), (C) [3H]BSP (0.01 µM), (D) pitavastatin (0.1 µM), and (E) atorvastatin (0.1 µM). Cells were coincubated with a substrate and CsA for 1 ([3H]E1S and atorvastatin), 2 (pitavastatin), or 5 minutes ([3H]E2G and [3H]BSP) after 1-hour preincubation with (□) or without (▵) CsA. As a reference, cells were coincubated with a substrate and CsA without the 1-hour preincubation step (●). Representative data from three independent experiments are shown as a percentage of control. Thick, thin, and dashed lines represent fitted lines for coincubation of a substrate and CsA without preincubation, coincubation after 1-hour preincubation without CsA, and coincubation after 1-hour preincubation with CsA, respectively that were obtained by a nonlinear least-squares regression analysis based on eq. 3. Each point represents the mean ± S.D. (n = 3).
Effect of preincubation with CsA on the inhibition of OATP1B1-mediated uptake of [3H]E2G, [3H]E1S, [3H]BSP, pitavastatin, and atorvastatin
The inhibitory effect of CsA on OATP1B1-mediated uptake of [3H]E2G (0.1 µM), [3H]E1S (0.01 µM), [3H]BSP (0.01 µM), pitavastatin (0.1 µM), and atorvastatin (0.1 µM) was examined in the absence and presence of 1-hour preincubation with or without CsA as shown in Fig. 4. The IC50 values were estimated by a nonlinear least-squares regression analysis (mean ± S.D., n = 3). The R values of CsA were calculated by eqs. 5 and 6 with the exception that IC50 values instead of Ki values were used for the calculations because the applicability of eq. 4 to preincubation-dependent inhibition by CsA remains to be established.
Discussion
We previously reported that the in vitro inhibitory effects of several OATP1B1 inhibitors showed remarkable substrate-dependence using prototypical substrates, E2G, E1S, and BSP (Izumi et al., 2013). In addition to the prototypical substrates, clinically used OATP1B1 substrate drugs could also serve as in vitro OATP1B1 probe substrates, for which the potential substrate-dependent inhibition has not been comprehensively evaluated. To identify representative in vitro OATP1B1 probe substrates that could mitigate the risk of false-negative DDI prediction, this study investigated the impact of in vitro substrate selection on OATP1B1 inhibition and the subsequent DDI prediction for 12 clinically used OATP1B1 substrate drugs compared with the prototypical probe substrates.
Twelve OATP1B1 substrate drugs—including statins (pitavastatin, atorvastatin, fluvastatin, rosuvastatin, and pravastatin), antidiabetics (repaglinide, nateglinide, and glibenclamide), a dual endothelin receptor antagonist (bosentan), an angiotensin II receptor blocker (valsartan), a loop diuretic (torasemide), and a histamine H1 receptor antagonist (fexofenadine)—were selected in this study because accumulated evidence suggests the involvement of OATP1B1 in their hepatic uptake in vivo. As expected, all drugs were confirmed to be substrates of OATP1B1 (Fig. 1), and their OATP1B1-mediated uptake showed saturation kinetics (Fig. 2). Although the majority of the obtained Km values were within the range of reported values (Table 1), the Km value of bosentan (4.27 µM) was, for unknown reasons, approximately 10-fold lower than the reported value (44 µM) (Treiber et al., 2007).
To investigate substrate-dependence in the inhibitory potency against OATP1B1, we examined the in vitro inhibitory effects of CsA, rifampin, and gemfibrozil on the OATP1B1-mediated uptake of the clinically used substrate drugs (Fig. 3) and compared them with those on E2G, E1S, and BSP uptake (Table 2). CsA, rifampin, and gemfibrozil were used as the strong (CsA and rifampin) or weak (gemfibrozil) OATP1B1 inhibitors in this study because they showed substrate-dependent OATP1B1 inhibition with 6- to 14-fold difference in the Ki values when E2G, E1S, and BSP were used as in vitro test probes (Izumi et al., 2013), and administration of the inhibitors has caused clinical DDIs with OATP1B1 substrate drugs (Shitara et al., 2013b). In fact, inhibition of hepatic uptake by rifampin was demonstrated in healthy subjects by positron emission tomography using 15R-[11C]TIC as an OATP substrate (Takashima et al., 2012).
The Ki values of CsA, rifampin, and gemfibrozil for the clinically used substrate drug uptake showed 6.3-, 3.4-, and 26-fold variations depending on the substrates, respectively (Table 2). This large variation was mainly attributable to the lower inhibitory effect of CsA on torasemide uptake and that of gemfibrozil on nateglinide uptake. Except for these two cases, the variation decreased to 3.9- and 7.5-fold for CsA and gemfibrozil, respectively. Notably, the Ki values for the clinically used substrate drugs, except for the two cases, were within 2.8-fold of those given by E2G as a substrate. In contrast, E1S and BSP yielded the higher Ki values of all three inhibitors compared with the clinically used substrate drugs. Therefore, E2G behaved similarly to the clinically used substrate drugs in terms of the susceptibility to OATP1B1 inhibition in general.
To evaluate the impact of substrate selection on OATP1B1-mediated DDI prediction, we calculated the R values of CsA, rifampin, and gemfibrozil based on a static model using the Ki values determined for each substrate (Table 3). According to the regulatory DDI guidelines and draft guidance materials, the R values were calculated using Cmax (FDA) or [I]u,inlet,max (FDA, EMA, and MHLW) as the [I] and compared with the corresponding cutoff values. CsA, a clinically relevant inhibitor of OATPs as well as CYP3A and other transporters in the liver and intestine (e.g., breast-cancer resistance protein and P-glycoprotein) (Yoshida et al., 2012), showed the observed AUCRs for OATP1B1 substrate drugs, ranging from 2.0 (bosentan) to 23 (pravastatin). Although the R values of CsA showed some variation depending on the Ki values determined with various substrates, the variation had no impact on the DDI prediction for CsA, and CsA was classified as a potential in vivo OATP1B1 inhibitor regardless of substrates because of its strong inhibitory potency against OATP1B1 (Table 3).
A single dose of rifampin is also used as a clinical OATP inhibitor (Maeda et al., 2011) with the observed AUCRs for OATP1B1 substrate drugs ranging from 2.2 (glibenclamide) to 12 (atorvastatin). As in the case of CsA, rifampin was classified as a potential in vivo inhibitor regardless of substrates because of its strong inhibitory potency against OATP1B1 (Table 3).
Gemfibrozil is a weak OATP1B1 inhibitor, and the observed AUCRs of statins and nateglinide ranged from 1.1 (fluvastatin) to 2.0 (pravastatin). Although the observed AUCRs of repaglinide were 7.0 to 8.2, this is thought to be mainly attributable to the mechanism-based inhibition of CYP2C8-mediated metabolism of repaglinide by gemfibrozil-1-O-glucuronide (Tornio et al., 2008). The R values of gemfibrozil calculated with Cmax showed some variation but were greater than the cutoff value (1.1), regardless of substrates. However, the R values calculated with [I]u,inlet,max suggested that substrate selection as well as regulatory standards applied could affect the decision as to a need for clinical DDI studies. Indeed, gemfibrozil was considered as a potential in vivo OATP1B1 inhibitor when E2G and several clinically used substrate drugs (e.g., pravastatin, valsartan) were used as in vitro probes under the regulations in the European Union (R ≥ 1.04), or pravastatin was used under the regulations in the United States and Japan (R ≥ 1.25) (Table 3).
Therefore, care should be exercised with in vitro OATP1B1 probe substrate selection, particularly for weak-to-moderate inhibitors, because the decision regarding the need for clinical DDI studies is more susceptible to substrate-dependent Ki variation. Based on the susceptibility to OATP1B1 inhibition in vitro (Table 2), clinically used substrate drugs except for torasemide and nateglinide, or E2G as a surrogate could be used as an in vitro probe to mitigate the risk of false-negative predictions.
Although the static model is widely used for DDI prediction because of its simplicity, we need to understand its limitations. In vivo DDIs can involve multiple elimination pathways of a victim drug, but the R value calculation based on the regulatory guidelines or draft guidance materials considers only a single pathway (e.g., 100% contribution of OATP1B1 to the overall hepatic uptake of drugs in this study). In addition, not only an unchanged drug but also its metabolite(s) could serve as an inhibitor in vivo, but our R value calculation considers only the inhibitory effects of unchanged drugs on OATP1B1.
Regarding clinical DDIs involving gemfibrozil, it is suggested that gemfibrozil-1-O-glucuronide as well as unchanged gemfibrozil has an inhibitory effect on OATP1B1 and partly contributes to clinical DDIs (Shitara et al., 2004). Indeed, Yoshida et al. (2012) demonstrated that a static model with [I]u,inlet,max provided R values greater than the cutoff values (R ≥ 1.25) for clinical DDIs between gemfibrozil and statins (e.g., atorvastatin, fluvastatin, pitavastatin, pravastatin, and rosuvastatin) by considering both gemfibrozil and its glucuronide as OATP1B1/1B3 inhibitors. R values considering the [I]u,inlet,max of only unchanged gemfibrozil resulted in less than 1.25 for all substrates except for pravastatin in this study (Table 3). Because the quantitative contribution of gemfibrozil and its glucuronide to clinical DDIs with OATP1B1 substrate drugs remains to be fully elucidated, we cannot judge whether the R values of unchanged gemfibrozil at less than the cutoff value in this study should be considered as false negatives or true negatives. However, this may highlight the importance of considering not only an unchanged drug but also its metabolite(s) as an inhibitor in DDI prediction with a static model when the metabolite(s) has a potential to inhibit OATP1B1.
Amundsen et al. (2010) and Shitara et al. (2013a) reported that the inhibitory effect of CsA on OATP1B1 was potentiated by preincubating OATP1B1-expressing cells with CsA before coincubation of a probe substrate and CsA. As reported previously, the inhibitory effect of CsA was 3- to 5-fold potentiated by the 1-hour preincubation in our study, resulting in higher R values (Table 4). However, remarkable substrate-dependence was not observed in the degree of the potentiation. Therefore, even if the inhibitory effect of an inhibitor on OATP1B1 is potentiated after preincubation, E2G or clinically used substrate drugs such as pitavastatin and atorvastatin could sensitively detect the inhibition potential as in the case of the no-preincubation condition. However, this effect has been observed only for a limited number of inhibitors (CsA, saquinavir, and ritonavir) (Shitara et al., 2013a), and it remains to be elucidated whether lower Ki values obtained by inhibitor preincubation are clinically relevant. Further evaluation is needed of OATP1B1 inhibitors in the light of the preincubation effect to see whether this type of experiment would be useful for further mitigating the risk of false-negative DDI predictions.
Our study has demonstrated that E2G and clinically used OATP1B1 substrate drugs except for torasemide and nateglinide behave similarly in terms of susceptibility to OATP1B1 inhibition by CsA, rifampin, and gemfibrozil. Therefore, we recommend that clinically used OATP1B1 substrate drugs or E2G as a surrogate should be used as in vitro probes for OATP1B1 inhibition assays to help mitigate the potential risk of false-negative DDI prediction caused by substrate-dependent Ki variation. These findings will contribute to the establishment of optimal in vitro evaluation systems for clinically relevant OATP1B1-mediated DDIs.
Acknowledgments
The authors thank Hisae Noguchi (Eisai Co., Ltd.) for technical support.
Authorship Contributions
Participated in research design: Izumi, Nozaki, Komori, Maeda, Takenaka, Sugiyama.
Conducted experiments: Izumi.
Performed data analysis: Izumi, Nozaki, Komori, Maeda.
Wrote or contributed to the writing of the manuscript: Izumi, Nozaki, Komori, Maeda, Takenaka, Kusuhara, Sugiyama.
Footnotes
- Received May 13, 2014.
- Accepted November 20, 2014.
This study was partly supported by Grants-in-Aid for Research on Publicly Essential Drugs and Medical Devices from the Ministry of Health, Labour and Welfare of Japan.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the plasma concentration-time curve
- BSP
- sulfobromophthalein
- CL
- clearance
- CsA
- cyclosporine A
- DDI
- drug-drug interaction
- DMSO
- dimethylsulfoxide
- E2G
- estradiol-17β-glucuronide
- E1S
- estrone-3-sulfate
- EMA
- European Medicines Agency
- FDA
- U.S. Food and Drug Administration
- OATP
- organic anion transporting polypeptide
- MHLW
- Ministry of Health, Labour and Welfare (Japan)
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}