


Abstract
Elevated levels of proinflammatory cytokines associated with infection and inflammation can modulate cytochrome P450 enzymes, leading to potential disease-drug interactions and altered small-molecule drug disposition. We established a human-derived hepatocyte-Kupffer cell (Hep:KC) coculture model to assess the indirect cytokine impact on hepatocytes through stimulation of KC-mediated cytokine release and compared this model with hepatocytes alone. Characterization of Hep:KC cocultures showed an inflammation response after treatment with lipopolysaccharide and interleukin (IL)-6 (indicated by secretion of various cytokines). Additionally, IL-6 exposure upregulated acute-phase proteins (C-reactive protein, alpha-1-acid glycoprotein, and serum amyloid A2) and downregulated CYP3A4. Compared with hepatocytes alone, Hep:KC cocultures showed enhanced IL-1β–mediated effects but less impact from both IL-2 and IL-23. Hep:KC cocultures treated with IL-1β exhibited a higher release of proinflammatory cytokines, an increased upregulation of acute-phase proteins, and a larger extent of metabolic enzyme and transporter suppression. IC50 values for IL-1β–mediated CYP3A4 suppression were lower in Hep:KC cocultures (98.0–144 pg/ml) compared with hepatocytes alone (IC50 > 5000 pg/ml). Cytochrome suppression was preventable by blocking IL-1β interaction with IL-1R1 using an antagonist cytokine or an anti-IL-1β antibody. Unlike IL-1β, IL-6–mediated effects were comparable between hepatocyte monocultures and Hep:KC cocultures. IL-2 and IL-23 caused a negligible inflammation response and a minimal inhibition of CYP3A4. In both hepatocyte monocultures and Hep:KC cocultures, IL-2RB and IL-23R were undetectable, whereas IL-6R and IL-1R1 levels were higher in Hep:KC cocultures. In summary, compared with hepatocyte monocultures, the Hep:KC coculture system is a more robust in vitro model for studying the impact of proinflammatory cytokines on metabolic enzymes.
Introduction
Therapeutic proteins (TPs) include monoclonal antibodies (mAbs), cytokines, peptides, growth factors, and enzymes. The increased number of TPs in clinical use has raised the question of whether there is a potential for drug-drug interactions (DDIs) with small-molecule drugs. Although the mechanisms involved in the disposition of TPs and small molecules are fundamentally different (Lu et al., 2013), regulatory agencies, academics, and pharmaceutical companies have developed an interest in this area. Some TPs have been shown to cause a change in the exposure of small molecules (Mahmood and Green, 2007; Huang et al., 2010; Lee et al., 2010). For example, treatment of patients with rheumatoid arthritis with a single 10 mg/kg dose of tocilizumab [an anti-interleukin (IL)-6 receptor mAb] resulted in a 43% reduction in plasma exposure of the CYP3A4 substrate simvastatin (Schmitt et al., 2011), most likely resulting from tocilizumab-mediated normalization of cytochrome P450 (P450) suppression by IL-6. TP-DDI effects are not universal, as observed with denosumab inhibition of cytokine RANKL that resulted in no impact on CYP3A4 in women with osteoporosis (Jang et al., 2014).
In general, the focus for TP-DDIs has been on cytokines [e.g., IL-1, IL-6, and tumor necrosis factor-α (TNF-α)] and cytokine modulators, as these are known to alter the expression levels of various drug-metabolizing enzymes and transporters in hepatocyte in vitro systems (Aitken et al., 2006; Fardel and Le Vée, 2009; Nguyen et al., 2013). Cytokines have differential effects on cytochrome enzymes, suggesting the involvement of diverse mechanisms in P450 downregulation (Gu et al., 2006; Urquhart et al., 2007; Lee et al., 2009; Harvey and Morgan, 2014).
In vitro systems most commonly used for studying effects of TPs on P450 enzymes or drug transporters are fresh or cryopreserved primary human hepatocytes (Dickmann et al., 2011; Dallas et al., 2012). Cultured hepatocytes are useful for mechanistic studies related to the impact of a cytokine on gene expression and activity, but there are inherent limitations (Evers et al., 2013; Kenny et al., 2013): 1) hepatocytes do not reflect the complex interactions between multiple cytokines and cell types that play a role in vivo in inflammatory diseases; 2) high levels of interlaboratory and interdonor variability in cultured hepatocytes make quantitative interpretation of results challenging; and 3) hepatocyte monocultures are relevant only for studying cytokines with receptors directly expressed on hepatocytes. Based on these limitations, applying in vitro data to predict cytokine-mediated DDIs associated with anti-inflammatory TPs is challenging and currently not recommended (Evers et al., 2013).
To address the limitations inherent in hepatocyte monoculture systems, attempts have been made to establish coculture models by incorporating components of the innate immune system such as Kupffer cells (KCs). Sunman et al. (2004) demonstrated in a coculture system in which the presence of KCs caused sustained IL-2 suppression of CYP3A activity in hepatocytes (for 72 hours) compared with 48-hour suppression observed in hepatocyte monocultures. Conversely, KCs did not alter CYP3A activity duration after treatment with IL-1 and IL-6 (positive proinflammatory cytokine controls). Similarly, in cocultures of parenchymal and nonparenchymal liver cells, Chen et al. (2011) demonstrated the downregulation of various P450 isoforms and transporters and upregulation for others. Overall, however, the experience with coculture systems has been limited, and reproducing these observations has been challenging. Attempts at developing standard cocultures with primary human hepatocytes and KCs are limited by the rapid hepatic phenotypic decline. Consequently, the feasibility of standard cocultures for investigating the long-term effects of KC secretions on P450 enzymes and other pathways is restricted. In this study, we have established and characterized a long-term Hep:KC coculture model. The model was achieved by incorporating KCs to a previously established system of hepatocytes cultured on micropatterned collagen domains of empirically optimized dimensions and supported by mouse 3T3-J2 fibroblasts (Khetani and Bhatia, 2008). The Hep:KC coculture system provided more stability and allowed the hepatocytes to retain functionality for several weeks, thus enabling the performance of long-term studies. In the Hep:KC coculture system, we demonstrated the differential effects on metabolic enzymes and drug transporter genes by cytokines for which receptors are (i.e., IL-6, IL-1β) or are not (i.e., IL-2, and IL-23) expressed on hepatocytes. We also propose ways in which data generated in this system can be used in assessing risk for TP-DDIs.
Materials and Methods
Cell Source/Donor Demographics.
The cryopreserved hepatocytes used in creating the cocultures were purchased from Celsis (Baltimore, MD) and came from a 31-year-old man who died of anoxia (secondary to heroin overdose) and who had a history of marijuana and heroin use. The cryopreserved KCs were purchased from Life Technologies (Carlsbad, CA) and came from a 51-year-old white man who died of blunt injury to the head and was known to have smoked marijuana daily for 36 years. The 3T3-J2 fibroblasts were originally obtained from Howard Green at Harvard University (Rheinwald and Green, 1975). These and additional hepatocyte and KC donors were also used in preliminary assessments. A summary of donor demographics can be found in Supplemental Table 1.
Preparation of Human Hep:KC Cocultures.
Cryopreserved primary human hepatocytes and cryopreserved 3T3-J2 murine embryonic fibroblasts were used in the manufacturing of the micropatterned hepatocyte and fibroblast cocultures (HepatoPac; Hepregen Corporation, Medford, MA) in a 96-well format. Briefly, cryopreserved hepatocyte vials were thawed at 37°C for 120 seconds, followed by dilution with 50 ml of prewarmed HepatoPac culture medium (HCM) as described previously (Khetani and Bhatia, 2008; Khetani et al., 2013). The cell suspension was spun at 50g for 5 minutes. The supernatant was discarded, cells were resuspended in HCM, and viability was assessed using trypan blue exclusion (typically 80%–95%). Liver-derived nonparenchymal cells, as judged by their size (∼10 μm in diameter) and morphology (nonpolygonal), were consistently found to be less than 1% in these preparations. To create the micropatterned coculture (MPCC) in 96-well plates, a hepatocyte pattern was first produced by seeding hepatocytes on rat-tail collagen (Corning Life Sciences, Tewksbury, MA) type 1–patterned substrates that mediate selective cell adhesion. The cells were washed with medium 4–6 hours later to remove unattached cells (leaving ∼5000 attached hepatocytes on ∼14 collagen-coated islands within each well of a 96-well plate) and incubated in HCM. The 3T3-J2 murine embryonic fibroblasts (up to passage 12) were seeded 18–24 hours later to create the cocultures. Culture medium was replaced every 2 days (∼64 μl/well of a 96-well plate).
After 7 days of MPCC stabilization, cryopreserved human KCs from unmatched donors (Life Technologies) were thawed and seeded on top of MPCCs at Hep:KC ratios of 1:0 (control cultures without KC addition), 1:0.1, and 1:0.4. These ratios were chosen based on previous coculture studies by Sunman et al. (2004), which approximately represented the ratio of hepatocytes to KCs observed in human liver during the normal physiologic (1:0.1) and inflamed state (1:0.4). Unmatched hepatocyte and KC donors were used based on preliminary assessment showing no obvious deleterious effects to cell cultures when different donors were used for the two cell types (data not shown). Hepatocyte cultures alone and with unmatched KCs showed comparable albumin, urea, and ATP levels (Supplemental Figs. 1–5). Cell cultures used within these studies incorporated human MPCCs (human hepatocytes plus mouse fibroblast supporting cells) with and without KCs. For simplicity, MPCC cultures containing KCs are referred to as Hep:KC cocultures (although technically they are tricultures).
Preliminary Characterization of Hep:KC Cocultures.
Preliminary evaluations of Hep:KC cultures were performed by assessment of hepatocyte metabolic competency in the presence of KCs from unmatched donors. KC functions in the cocultures were also evaluated after 24 hours of lipopolysaccharide (LPS) treatment (50 ng/ml; Sigma Aldrich, St. Louis, MO). At the completion of incubation, culture media samples were collected and analyzed for proinflammatory cytokines. Further characterization was performed by treating Hep:KC cultures with cytokines (IL-1β, IL-2, IL-6, and IL-23 [>97% purity by SDS-PAGE]; 0-200 ng/ml; R&D Systems, Minneapolis, MN) in serum-free dosing media. Treatments were repeated every other day (on initiation of dosing) for 4 days. Concentrations of proinflammatory cytokines were chosen based on preliminary studies considering toxicity (data not shown) and observed physiologic cytokine levels in human plasma. After stabilization of the cocultures, dosing was initiated on day 8 of culture. Supernatants were collected on days 10 and 12 of culture (corresponding to 2 days after the first cytokine addition and 2 days after the second cytokine addition, respectively) for bioanalyses of nondestructive endpoints (albumin secretion, urea synthesis, and CYP3A4 activity) to give an indication of cell health. A 4-day cytokine incubation period was chosen based on preliminary studies showing robust effects on metabolic enzymes out to at least 6 days with peaked effects observed after 4 days of cytokine treatment. Sampling time points were chosen to cover these 4 days of cytokine treatment. After collection of supernatants on day 12 of culture, Hep:KC cultures were washed once and incubated in HCM to recover in the absence of cytokine. Supernatants were collected 6 days after cytokine removal (corresponding to day 18 of culture) to assess hepatocyte metabolic competence (by measurement of albumin secretion, urea synthesis, and CYP3A4 activity), and cell lysates were evaluated for cytotoxicity (by measurement of cellular ATP) relative to untreated controls. Control Hep:KC cultures were treated with vehicle [0.1% dimethylsulfoxide (DMSO)].
A second set of Hep:KC cultures was also dosed with IL-1β, IL-2, IL-6, and IL-23 (0 – 200 ng/ml) in serum-free dosing media, and supernatants were collected 2 days after the first cytokine dose (day 10 of culture) and 2 days after the second cytokine dose (day 12 of culture) and immediately frozen for assessment of secreted proinflammatory cytokines. Control supernatant samples were also collected on day 8 of culture before dosing with cytokines. Cell lysates were obtained at the completion of cytokine treatment on day 12, and mRNA was isolated for gene expression analyses.
Estimation of the IC50 Values for IL-1β and IL-6 inhibition of CYP3A4 Activity in Human Hep:KC Cocultures.
The study contained three groups representing basal control (untreated), cytokine-treated, and recovery group. In the basal control group (n = 3), on day 8 of culture, supernatants were collected from all Hep:KC cultures (1:0, 1:0.1, and 1:0.4) maintained in HCM (without cytokines) for the assessment of basal levels of albumin, urea, and CYP3A4 activities. After supernatant collection, cell lysates were processed for evaluation of basal mRNA expression.
In the treatment group, on day 8 of culture, Hep:KC cultures were dosed with either IL-1β or IL-6 (0.00625, 0.0125, 0.025, 0.050, 0.3125, 0.625, 1.25, 2.5, and 5.0 ng/ml) every other day in serum-free dosing media for a total of two doses over 4 days. At day 10 (2 days after the first cytokine addition) and day 12 (2 days after the second cytokine addition), supernatants were collected (n = 3) for bioanalyses of nondestructive endpoints (albumin secretion, urea synthesis, and CYP3A4 activity). After supernatant collection on each day, cell lysates (n = 3) were processed for evaluation of mRNA expression. Control cells in the treatment group were treated with vehicle (0.1% DMSO) and processed similarly to cytokine-treated cells.
For assessment of recovery from cytokine exposure, Hep:KC cultures were dosed with either IL-1β or IL-6 (0, 50, 5000 pg/ml) on days 8 and 10 of culture. On day 12 (after 4 days of cytokine exposure), dosing solutions were removed, and cells were washed 1× and incubated in HCM without cytokine to initiate recovery. Supernatants were collected (n = 3) on day 18 (6 days postrecovery) and assayed for albumin secretion, urea synthesis, and CYP3A4 activity. After supernatant collection, cell lysates (n = 3) from day 18 cultures were processed for mRNA expression analysis. Control cells in the recovery group were treated with vehicle (0.1% DMSO) and processed similarly to cytokine-treated cells.
Evaluation of the Effects of Anti-IL-1β Antibody and IL-1 Receptor Antagonist on IL-1β-induced Inhibition of CYP3A4 Activity in Human Hep:KC Cocultures.
The study contained three groups representing untreated (basal control), treated (cytokine coincubated with either human IL-1RA [IL-1 receptor antagonist cytokine] or mouse anti-human IL-1β (anti-IL-1β) mAb (R & D Systems) and recovery group. In the basal control group (n = 3), on day 8 of culture, supernatants were collected from all Hep:KC cultures (1:0, 1:0.1, and 1:0.4) maintained in HCM (without cytokines) for assessment of basal levels of albumin, urea, and CYP3A4 activities. After supernatant collection, cell lysates were processed for evaluation of basal mRNA expression.
For the treatment group, on day 8 of culture, Hep:KC cocultures were treated with anti-IL-1β mAb, IL-1RA, or isotype control (mouse IgG1) (R & D Systems) for 2 hours before dosing with proinflammatory cytokines (either IL-1β or IL-6). Varying concentrations of anti-IL-1β mAbs were used to pretreat both Hep:KC 1:0 cultures containing no KCs (10, 50, 100 ng/ml) and Hep:KC 1:0.4 cultures (1, 5, 10 ng/ml). Also, varying concentrations of IL-1RA (100, 500, 1000 ng/ml) were used to pretreat both 1:0 and 1:0.4 Hep:KC cultures. For the isotype control group, all Hep:KC cultures were treated with 100 ng/ml of mouse IgG1. IL-1β study concentrations (2.5 and 0.1 ng/ml) were chosen to be approximately near the IC50 of IL-1β-mediated CYP3A4 inhibition in Hep:KC 1:0 and 1:0.4 cultures, respectively. Study concentrations of IL-1RA and anti-IL-1β mAb were selected based on the manufacturer’s recommendation, and preliminary studies centered on reported ED50 for IL-1RA and ND50 of anti-IL-1β mAb (Symons et al., 1987). Subsequently, the pretreated Hep:KC cultures were codosed with varying IL-1β concentrations [0 and 2.5 ng/ml for Hep:KC 1:0 cultures (containing no KCs]) and 0 and 0.1 ng/ml for Hep:KC 1:0.4 cultures] every other day in serum-free dosing media for a total of two doses over 4 days. Similar pretreatment with anti-IL-1β mAb, IL-1RA, and mouse IgG1 and subsequent codose with IL-6 (0 and 0.5 ng/ml) were also performed in both Hep:KC 1:0 and 1:0.4 cultures. Codosing was also initiated on day 8 and repeated every other day for a total of two doses over 4 days. Concentrations of IL-1β and IL-6 were chosen as approximate of previously determined IC50 values for IL-1β-mediated and IL-6-mediated CYP3A4 inhibition (IL-6 was included in this study as a control against nonspecific effects of IL-1RA and anti-IL-1β mAb). On days 10 and 12 of culture, supernatants were collected (n = 3) for bioanalyses of nondestructive endpoints (albumin secretion, urea synthesis, and CYP3A4 activity). After supernatant collection, cell lysates (n = 3) were collected from day 12 cultures for mRNA evaluation. Control cells (those treated with IL-1RA, anti-IL-1β mAb, or mouse IgG1 but no proinflammatory cytokines) in the treatment groups were processed similarly to cytokine-treated cells.
Cell Culture Health and Functionality Assays.
Cellular ATP levels in the cell lysates were measured using the CellTiter-Glo luminescent kits from Promega (Madison, WI) per the manufacturer’s instructions. To calculate hepatocyte-specific responses, cellular ATP levels were also determined in stromal-only cultures (i.e., murine 3T3-J2 fibroblasts). ATP signals in stromal-only control cultures were subtracted from those of Hep:KC cultures to obtain hepatocyte-specific effects. Urea concentration was assayed using a colorimetric endpoint assay kit using diacetylmonoxime with acid and heat (Stanbio Laboratories, Boerne, TX).
Albumin content was measured using an enzyme-linked immunosorbent assay (MP Biomedicals, Santa Ana, CA) with horseradish peroxidase detection and 3,3′,5,5′-tetramethylbenzidine (Fitzgerald Industries, Acton, MA) as a substrate (Khetani and Bhatia 2008). CYP3A4 activity was measured by CYP3A4-Glo assay according to the manufacturer’s protocols (Promega). Proinflammatory cytokines released in cell culture media were measured by electrochemiluminescence detection using Human Pro-inflammatory 7-Plex Kit according to manufacturer’s protocol (Meso Scale Discovery, Gaithersburg, MD).
Gene Expression Analysis.
Cell lysate samples were processed and isolated for total RNA using an RNeasy kit (QIAGEN, Venlo, Limburg, The Netherlands) according to the manufacturer’s protocol. To obtain cDNA, a two-step reverse-transcription polymerase chain reaction (RT-PCR) reaction was conducted by reverse-transcribing an aliquot of total RNA to cDNA using a High-Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA). PCR reactions were then prepared by adding cDNA to a reaction mixture containing TaqMan 2× Universal PCR Master Mix (Applied Biosystems). TaqMan Array microfluidic cards were custom-made by Applied Biosystems and contained probes in triplicate. Samples were applied to low-density microarrays by centrifugation twice for 1 minute at 1200g. Real-time quantitative PCR was performed using an ABI PRISM 7900 Sequence Detector instrument and Sequence Detector 2.1 software (PerkinElmer Instruments, Shelton, CT). Quantitation of the target cDNA in all samples was normalized to 18S ribosomal RNA (Cttarget − Ct18S = ∆Ct), and the difference in expression for each target cDNA in treated cultures relative to the amount in control cultures was calculated (∆Ctcontrol − ∆Cttreated = ∆∆Ct). Fold changes in target gene expression were determined by taking 2 to the power of this number (2-∆∆Ct).
Data Analysis.
All experiments were run in triplicate, and the mean and S.D. were calculated for each of the doses administered. All data were normalized to vehicle controls. IC50 values were determined by nonlinear regression and fitting of the dose response curves to a two-parameter equation with variable slope (eq. 1):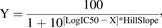 (1)where Y is the normalized response between 0% and 100%, and X is the log (inhibitor concentration). All analyses were performed using GraphPad Prism 5.0 (San Diego, CA).
(1)where Y is the normalized response between 0% and 100%, and X is the log (inhibitor concentration). All analyses were performed using GraphPad Prism 5.0 (San Diego, CA).
Statistical Analysis.
Data are usually presented as mean ± S.D. Statistical analysis was carried out using analysis of variance followed by Tukey’s multiple comparison test. A value of P < 0.05 was considered statistically significant.
Results
Characterization of Hep:KC Cocultures.
Initial characterization of Hep:KC cocultures is summarized in Supplemental Figs. 1–5. Albumin and urea levels, as well as CYP3A4 activity, were sustained from day 7 out to day 15 (Supplemental Fig. 1). Compared with day 3, CYP3A4 activity increased by 10- to 16-fold between day 7 and day 15, the presence of KCs had no impact. After 4 days of cytokine treatment, no changes in ATP levels were observed, indicating the absence of cytotoxicity (Supplemental Figs. 2–5). At the same time, slight decreases in albumin and urea levels were detected after IL-6 and IL-1β treatment, whereas negligible effects were associated with IL-2 and IL-23. Correspondingly, dramatic downregulation of CYP3A4 activity was linked to IL-6 and IL-1β treatment but not to IL-2 or IL-23 treatment (Supplemental Figs. 2–5). After 6 days of recovery in the absence of cytokine treatment, albumin, urea, and CYP3A4 activity levels returned to levels comparable to untreated cultures (data not shown). In summary, Hep:KC cocultures were sustainable for at least 2 weeks as evident by the robust detection of albumin, urea, and CYP3A4 activity. Incorporating unmatched hepatocyte and KC donors and treatment with various cytokines did not compromise cell culture viability.
Coculture of Human Hepatocytes with Kupffer Cells.
Hepatocytes were cultured alone (1:0) and with KC at ratios of Hep:KCs of 1:0.1 and 1:0.4 representing normal and disease states, respectively. Expression of the CD163 cell surface marker is reported to be highly restricted to monocytes and macrophages, and in liver tissues, it has been predominantly observed in KC (Van Gorp et al., 2010). RT-PCR analysis of Hep:KC cultures showed increasing CD163 expression with increasing Hep:KC ratios (1:0.1 and 1:0.4) compared with hepatocytes alone (1:0), and expression was sustained to at least day 15 in culture (Fig. 1). These results confirmed that KC remained present in the coculture model for the entire 2-week study period.

Kupffer cell CD163 cell surface marker mRNA expression in Hep:KC cultures. Hepatocytes were cultured alone (in micropatterned cocultures with 3T3-J2 fibroblasts) ± KCs (at Hep:KC of 1:0, 1:0.1, and 1:0.4). RT-PCR analysis was performed on samples from days 3, 7, 11, and 15 post-KC addition for mRNA expression of CD163. Results were relative to cultures without KCs (Hep:KC 1:0) (n = 3 ± S.D.). Mean values were significantly different from Hep:KC *1:0 and *#1:0.1 cultures (P < 0.05).
Treatment of cell cultures with LPS (50 ng/ml for 24 hours) resulted in substantially higher release of proinflammatory cytokines [IL-1β, IL-6, IL-8, IL-10, TNF-α, and interferon (IFN-γ)] into culture media compared with untreated cells (Fig. 2, A–E). Furthermore, the levels of cytokine measured in culture media appeared to be related to the amount of KCs in the cocultures (highest levels were observed with Hep:KC 1:0.4 ratio followed by 1:0.1 and 1:0, respectively). In the absence of LPS stimulation, very low or even undetectable levels of cytokines were observed in culture media. These observations were consistent with previously generated results in our laboratories (data not shown) from separate studies involving cocultures using various combinations of two different hepatocyte lots and six different KC lots. These studies produced similar effects whereby cytokine secretion (such as IL-6 and TNF-α) was detected after treatment with LPS, and higher KC ratio equated to higher cytokine levels measured in cell culture media. Together, the presence of the KC-specific CD163 cell surface marker expression and cytokine release data confirmed that KCs were both present and functional in the cocultures during the study period.

Cytokine secretion in Hep:KC cultures after LPS treatment. Hepatocytes were cultured alone (in micropatterned cocultures with 3T3-J2 fibroblasts) ± Kupffer cells (at Hep:KC of 1:0, 1:0.1, and 1:0.4). Secreted proinflammatory cytokines (IL-1β, IL-6, IL-8, IL-10, TNF-α, and IFN-γ) were measured in cell culture media 24 hours after exposure to LPS (50 ng/ml) (n = 3 ± S.D.). Mean values were significantly different from *LPS-untreated Hep:KC cultures of matched cell ratios and *#LPS-treated 1:0 cultures (P < 0.05). aLevels were undetectable.
IL-6– and IL-1β–Mediated Immune Response in Hep:KC Cocultures.
After incubation with either IL-6 or IL-1β (0.05–200 ng/ml), Hep:KC (1:0, 1:0.1, and 1:0.4) culture media were sampled and measured for cytokine release. In general, after 4 days of treatment, the presence of IL-6 stimulated a release of proinflammatory cytokines (IL-8, TNF-α, and INF-γ) in a concentration-dependent manner (i.e., higher cytokine measurements corresponded with higher IL-6 exposure) (Fig. 3, A–C). For IL-8, higher levels measured in culture media also corresponded with a higher KC ratio. Similarly, exposure to IL-1β also caused a strong release of proinflammatory cytokines (Fig. 3, D–F). The response appeared to be related to both IL-1β concentrations (i.e., higher IL-1β exposure resulted in higher IFN-γ secretion) and KC ratios (i.e., higher Hep:KC ratios produced higher levels of IL-8, and TNF-α in culture media). These outcomes showed that the Hep:KC coculture system was responsive to both IL-6 and IL-1β stimulation and that the presence of KC further enhanced this response.

Cytokine secretion in Hep:KC cultures after IL-6 and IL-1β treatment. Hepatocytes were cultured alone (in micropatterned cocultures with 3T3-J2 fibroblasts) ± KCs (at Hep:KC of 1:0, 1:0.1, and 1:0.4). Secreted proinflammatory cytokines such as IL-8, TNF-α, and IFN-γ were measured in cell culture media 4 days after exposure to IL-6 (A–C) and IL-1β (D–F) (n = 3 ± S.D.). aLevels were undetectable.
IC50 of IL-1β-Mediated Inhibition of CYP3A4 Activity in Hep:KC Cocultures.
IC50 characterization of the IL-1β–mediated CYP3A4 suppression in Hep:KC cultures (1:0 vs. 1:0.4) was performed at cytokine concentrations (6.25–5000 pg/ml) lower than those previously studied (50–200,000 pg/ml) as a result of observed >50% CYP3A4 suppression at 50 pg/ml (Supplemental Fig. 3). After IL-1β treatment, a greater extent of CYP3A4 suppression was detected in Hep:KC (1:0.4) cocultures (at most studied concentrations) compared with hepatocytes alone (Hep:KC of 1:0) (Fig. 4A). Correspondingly, IC50 was reduced to 98.0 ± 11.9 pg/ml in Hep:KC 1:0.4 cocultures after 4 days of treatment (compared with IC50 > 5000 pg/ml observed in Hep:KC 1:0 cultures (assuming IL-1β is capable of fully suppressing CYP3A4 activity). In contrast, incubation with IL-6 did not show similar IC50 shifts (Fig. 4C) as values observed for IL-6-mediated CYP3A4 inhibition were not dependent on the presence of KCs. After 6 days of recovery in the absence of either IL-1β or IL-6 treatment, CYP3A4 activity returned to levels similar to those of untreated controls (Fig. 4, B and D, respectively).
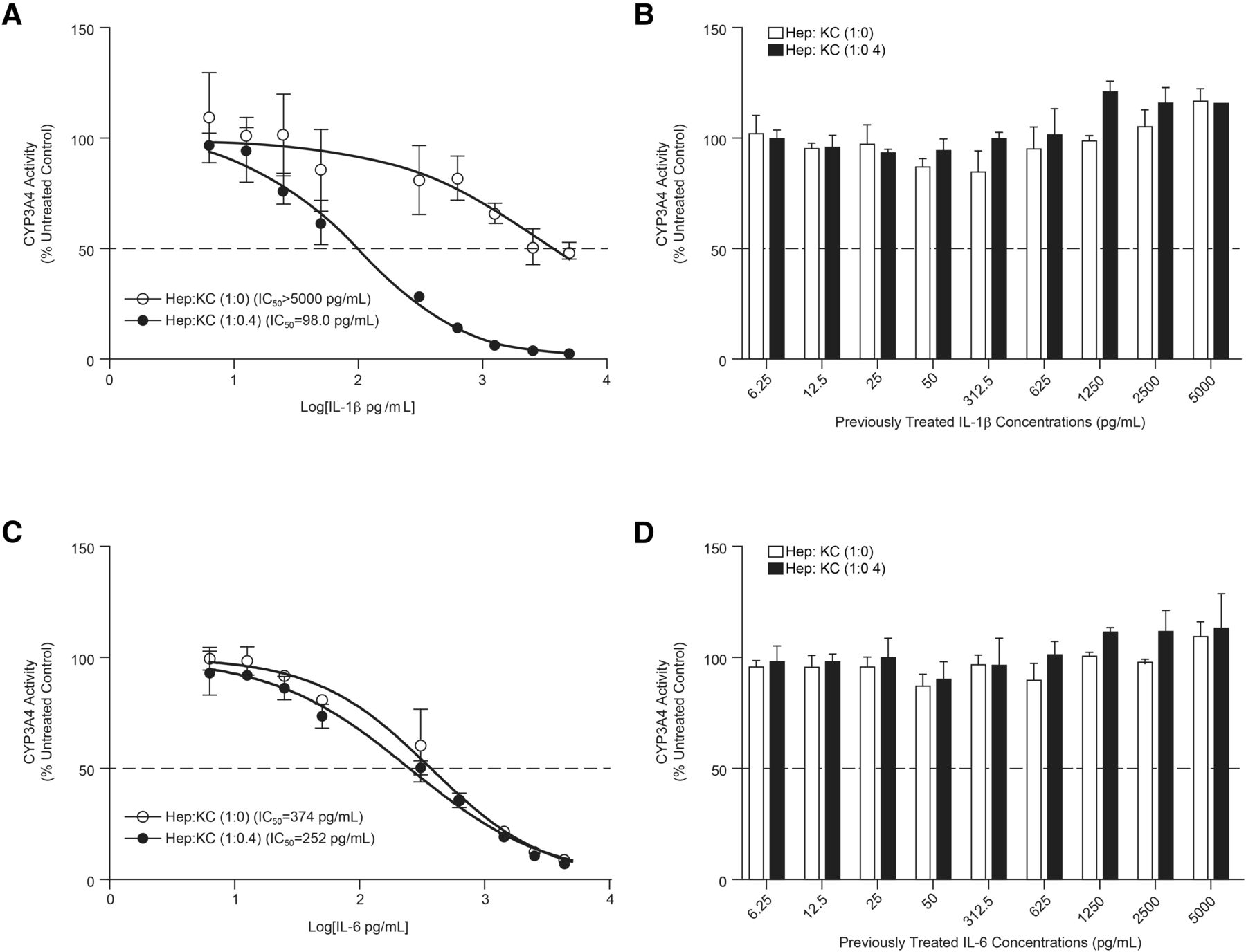
IC50 of CYP3A4 suppression in Hep:KC cultures after IL-1β and IL-6 incubation. Hepatocytes were cultured alone (in micropatterned cocultures with 3T3-J2 fibroblasts) ± KCs (at Hep:KC of 1:0 and 1:0.4). After IL-1β and IL-6 (6.25–5000 pg/ml) incubation, CYP3A4 activity levels were measured using the CYP3A4-Glo assay at 4 days after cytokine exposure (A and C) and 6 days after recovery in cytokine-free media (B and D). IC50 values were measured using GraphPad Prism (n = 3 ± S.D.).
The differences in observed IL-1β– and IL-6–mediated CYP3A4 downregulation after cytokine treatment may be attributed to variation in levels of receptor expression on hepatocytes and KCs (Fig. 5, A and B). For the IL-6 receptor (IL-6R), results from RT-PCR analysis indicated that the addition of KCs to hepatocyte cultures only modestly increased mRNA levels (IL-6R expression in Hep:KC 1:0.1 and 1:0.4 cocultures were 78%–135% and 88%–151% of 1:0 control cultures, respectively). In contrast, the presence of KCs corresponded to a more significant increase in IL-1β receptor, type 1 (IL-1R1) measurements compared with cultures without KC (Hep:KC 1:0) (IL-1R1 mRNA levels in Hep:KC 1:0.1 and 1:0.4 cocultures were 186%–263% and 284%–376% relative to 1:0 control cultures, respectively).
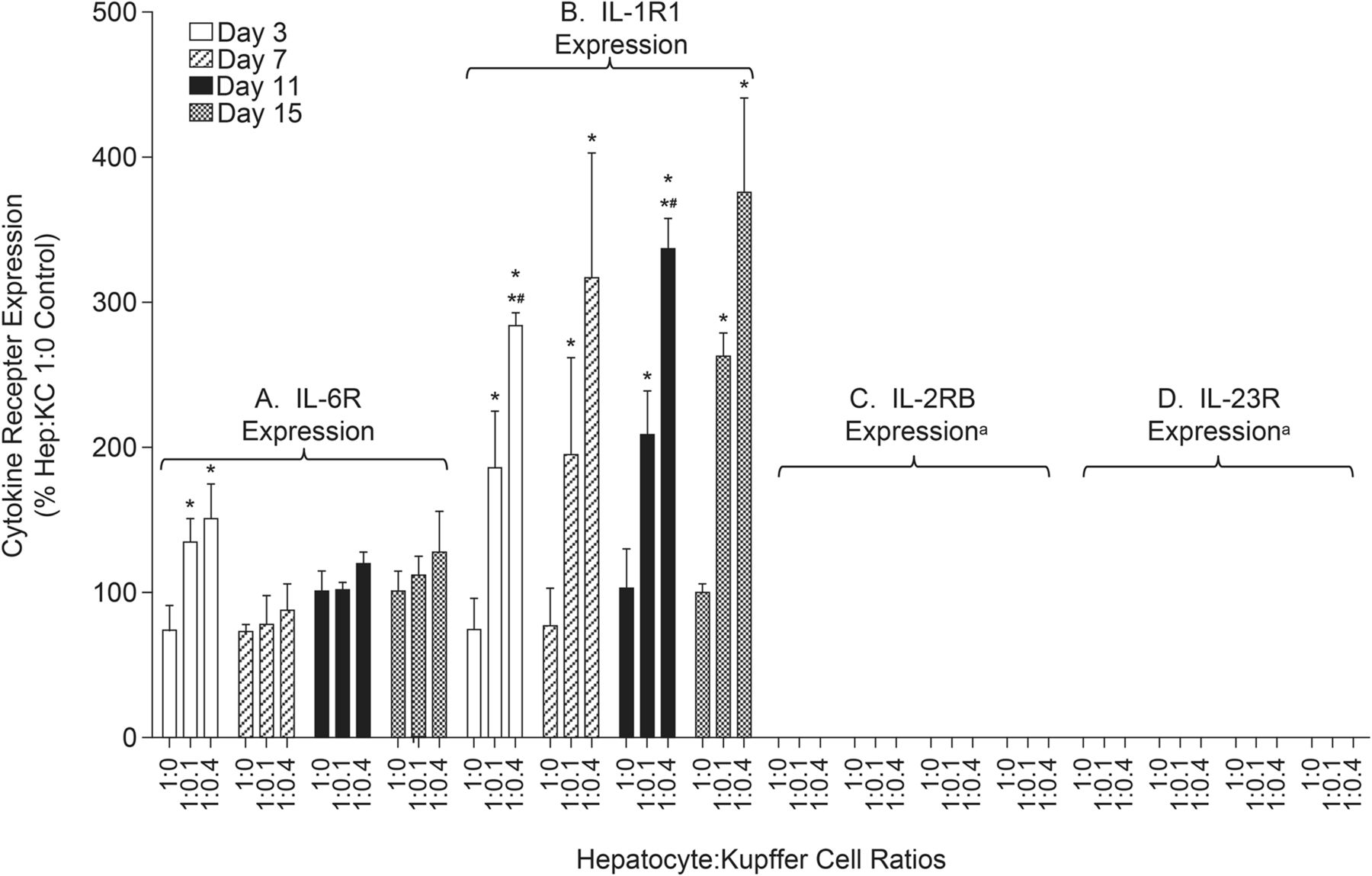
Expression of cytokine receptors in Hep:KC cultures. Hepatocytes were cultured alone (in micropatterned cocultures with 3T3-J2 fibroblasts) ± KCs (at Hep:KC of 1:0, 1:0.1, and 1:0.4). RT-PCR analysis was performed on samples from days 3, 7, 11, and 15 for gene expression of (A) IL-6R, (B) IL-1R1, (C) IL-2RB, and (D) IL-23R. Results were relative to cultures without KC (Hep:KC 1:0) (n = 3 ± S.D.). Mean values were significantly different from Hep:KC *1:0 and *#1:0.1 (P < 0.05). aLevels were undetectable (Ct values for 18s housekeeping gene were consistent across all cell samples, but Ct values for IL-2RB were >35 and undetectable for IL-23R).
Specificity of IL-1β Effects in Hep:KC Cocultures.
The observed suppression of CYP3A4 after cytokine treatment discussed herein is not believed to be a general effect seen with all cytokines. Rather, these observations are to be specific to a limited number of cytokines (e.g., IL-1β). The proposed mechanism mediating cytochrome suppression is most likely a result of direct interaction with cell surface receptors, such as IL-1R1. Blocking this interaction with either IL-1RA or anti-IL-1β mAb prevented IL-1β–mediated CYP3A4 suppression (Fig. 6). IL-1RA is a naturally occurring cytokine that binds to IL-1R type 1 and 2 and competitively inhibits its interaction with IL-1α and IL-1β (Gabay et al., 1997). Conversely, anti-IL-1β mAb binds to the cytokine and prevents it from interacting with surface receptor IL-1R. In the current studies, hepatocyte cultures without KC (Hep:KC 1:0) were coadministered with 2.5 ng/ml of IL-1β and either IL-1RA (100-1000 ng/ml) or anti-IL-1β mAb (10–100 ng/ml). Likewise, Hep:KC (1:0.4) cocultures were coadministered with 0.1 ng/ml of IL-1β (∼IC50 for CYP3A4 suppression in the coculture) and either IL-1RA (100–1000 ng/ml) or anti-IL-1β mAb (1–10 ng/ml). Results of cotreatment of cell cultures with IL-1β and either IL-1RA or anti-IL-1β mAb are summarized in Fig. 6.
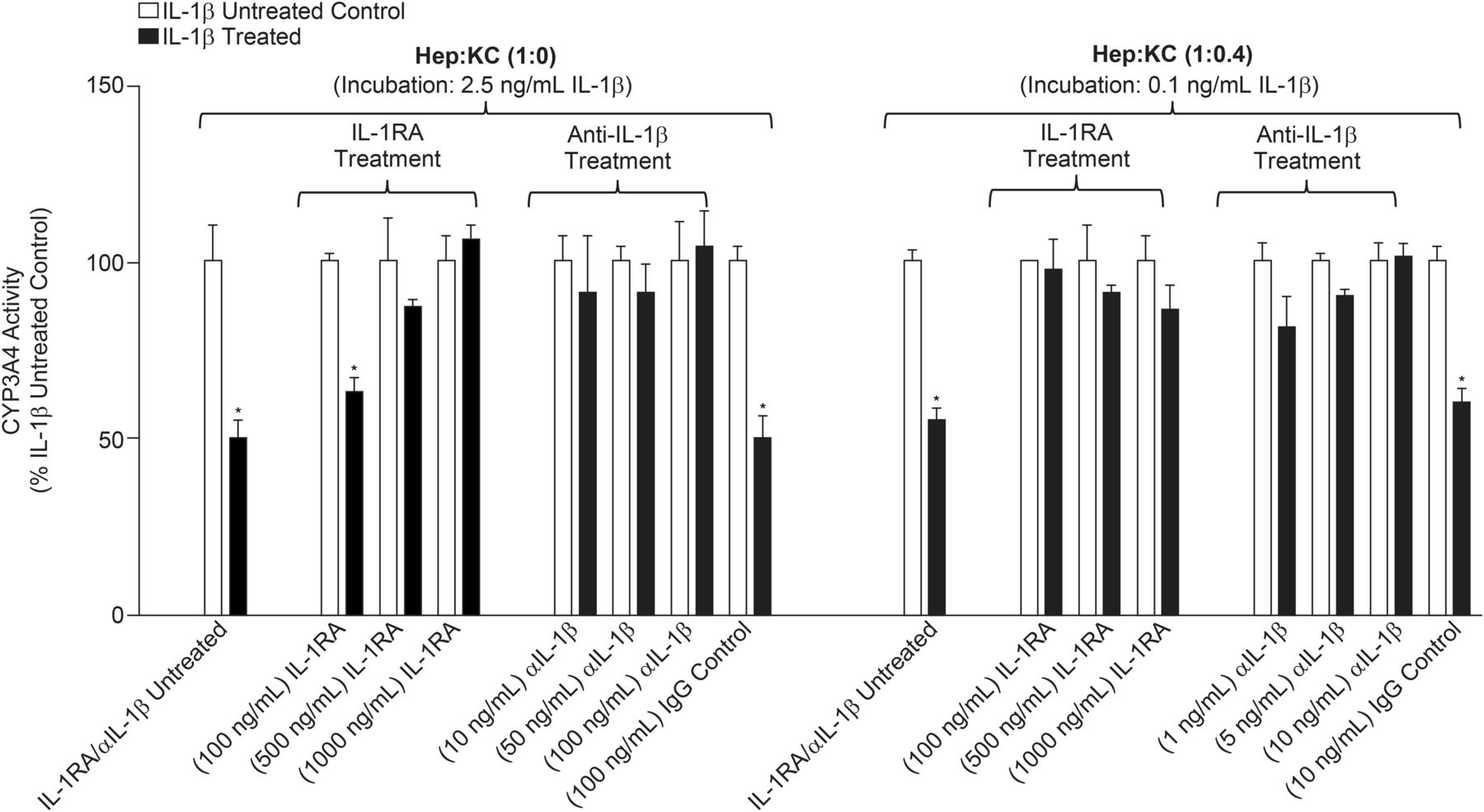
Effects of IL-1 receptor antagonist (IL-1RA) cytokine and anti-IL-1β mAb (αIL-1β) on IL-1β–mediated CYP3A4 suppression. Hepatocytes were cultured alone (in micropatterned cocultures with 3T3-J2 fibroblasts) ± KCs (at Hep:KC of 1:0 and 1:0.4) and codosed with IL-1β (2.5 ng/ml or 0.1 ng/ml for Hep:KC of 1:0 or 1:0.4, respectively) and either IL-1RA (100–1000 ng/ml) or αIL-1β (10–100 ng/ml). CYP3A4 activity levels were measured using CYP3A4-Glo assay at 4 days after treatment (n = 3 ± S.D.). *Mean values were significantly different from IL-1β-untreated matched Hep:KC cultures (P < 0.05).
In Hep:KC 1:0 cultures, 4 days of treatment with IL-1β (2.5 ng/ml) with increasing IL-1RA concentrations resulted in normalized CYP 3A4 activity (63%, 87%, and 106% of controls corresponding to 100, 500, and 1000 ng/ml IL-1RA, respectively). The same was observed in Hep:KC (1:0.4) cocultures after coadministration with both IL-1β (0.1 ng/ml) and IL-1RA (100-1000 ng/ml). Enzyme activity returned to levels (86%–97%) comparable to untreated controls (control cultures were those treated with IL-1RA but not IL-1β). Similarly, when IL-1β was coincubated with anti-IL-1β mAb (10–100 ng/ml in Hep:KC 1:0 and 1–10 ng/ml in Hep:KC 1:0.4), CYP3A4 activity returned to levels comparable to untreated controls (91%–104% and 81%–101% of untreated controls in Hep:KC 1:0 and 1:0.4 cultures, respectively). Additionally, cell cultures were cotreated with mouse IgG1 (100 ng/ml) and IL-1β to control for nonspecific isotype effects related to mouse anti-human IL-1β mAb. Results showed no effects on IL-1β–mediated CYP3A4 suppression (Fig. 6). Control cultures were those treated with mouse IgG1 but not IL-1β. Similar studies with IL-6 (0.5 ng/ml) coincubation with either IL-1RA or anti-IL-1β mAb did not show any effect on IL-6-associated CYP3A4 downregulation (data not shown). In summary, results from cotreatment with IL-1RA, anti-IL-1β mAb, and IgG1 isotype suggest that CYP3A4 suppression was mediated through specific interactions of IL-1β with IL-1R1.
IL-2 and IL-23 Effects on Immune Response and CYP3A4 Activity in Hep:KC Cocultures.
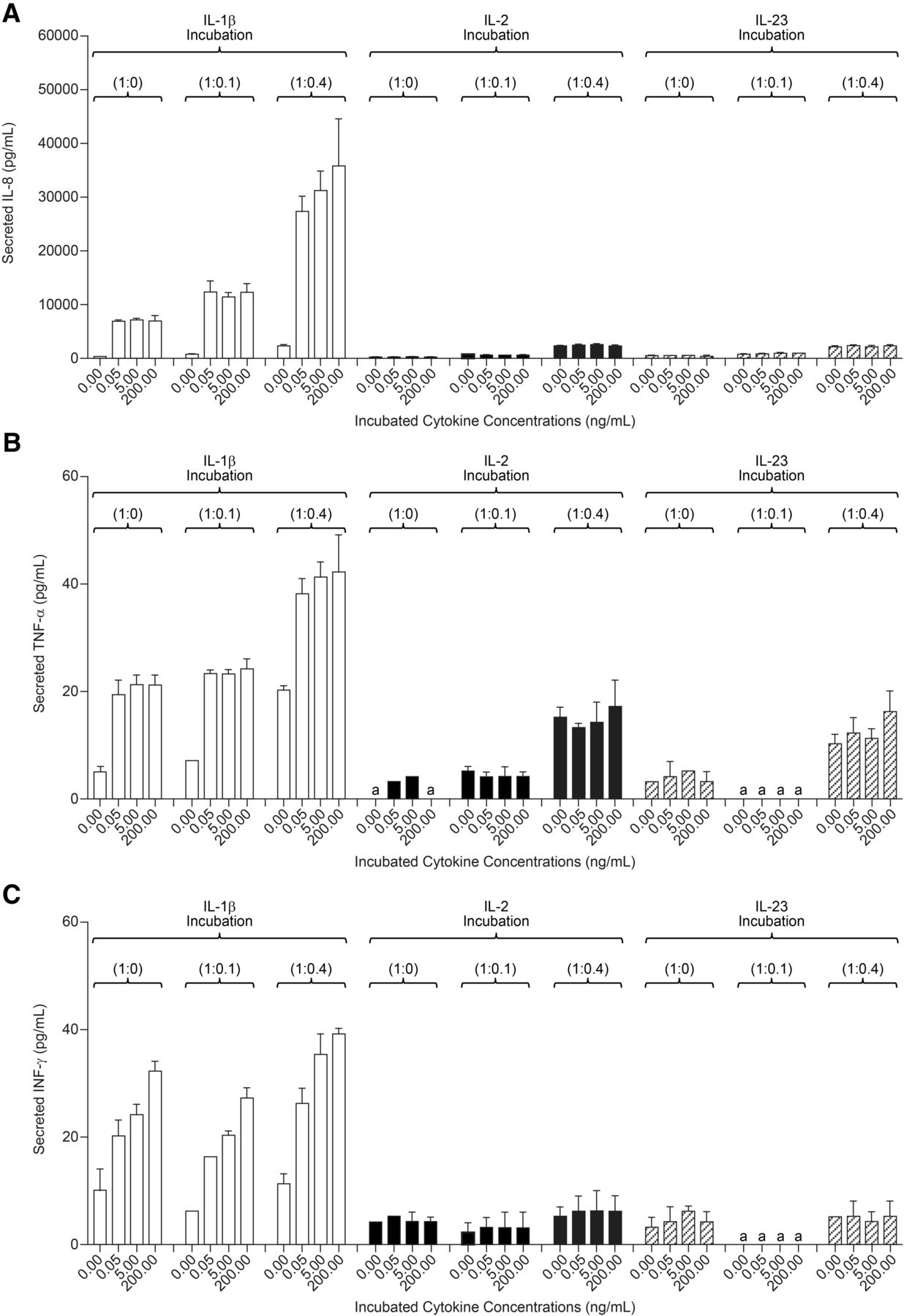
After 2 and 4 days of treatment with either IL-2 or IL-23 (0.05–200 ng/ml) Hep:KC (1:0, 1:0.1, and 1:0.4), culture media samples were measured for cytokine release. Levels of IL-8, TNF-α, and INF-γ secreted in cell culture media after 4 days of cytokine treatment are summarized in Fig. 7. Exposure of Hep:KC cultures to either IL-2 or IL-23 resulted in cytokine release of IL-8, TNF-α, and INF-γ at levels similar to untreated controls (Fig. 7, A–C). Thus, the release of these cytokines appeared to be less related to IL-2 and IL-23 treatment and more with the presence of KC. These results suggested that the presence of IL-2 and IL-23 was less relevant than IL-1β on stimulating proinflammatory cytokine release in Hep:KC cultures. Not surprisingly, CYP3A4 activity was also less affected by IL-2 and IL-23 treatment (all activity levels remained nearly above 50% relative to untreated controls) (Fig. 8). It should be noted that although a modest downregulation of cytochrome activity was seen in 1:0 and 1:0.1 Hep:KC cultures, this result was not reproducible in 1:0.4 cultures. The minimal interaction of both IL-2 and IL-23 was likely attributed to a lack of cell surface receptor expression [IL-2RB (IL-2 receptor, β chain) and IL-23R (IL-23 receptor)] on either hepatocytes or KC (Fig. 5, C and D).

Cytokine secretion in Hep:KC cultures resulting from IL-1β, IL-2, and IL-23 exposure. Hepatocytes were cultured alone (in micropatterned cocultures with 3T3-J2 fibroblasts) ± KCs (at Hep:KC of 1:0, 1:0.1, and 1:0.4). Secreted proinflammatory cytokines such as IL-8 (A), TNF-α (B), and IFN-γ (C) were measured in cell culture media 4 days after cytokine treatment (n = 3 ± S.D.). aLevels were undetectable.

CYP3A4 activity in Hep:KC cultures after IL-2 and IL-23 treatment. Hepatocytes were cultured alone (in micropatterned cocultures with 3T3-J2 fibroblasts) ± KCs (at Hep:KC of 1:0, 1:0.1, and 1:0.4). CYP3A4 activity levels were measured after 4 days of IL-2 (A) and IL-23 (B) treatment (n = 3 ± S.D.). *Mean values were significantly different from IL-2-untreated Hep:KC cultures of matched cell ratios (P < 0.05).
Effects of Cytokines on Gene Expression of Metabolism Enzymes, Drug Transporters, and Acute-Phase Proteins.
RT-PCR analysis was performed to evaluate effects of IL-1β on gene expression of various metabolic enzymes, transporters, and acute phase proteins. Results were considered upregulated or downregulated when mRNA levels were ≥150% or ≤50% of untreated controls, respectively, and observations were concentration-dependent. Although translating changes in gene expression to meaningful changes in enzyme activity is difficult, the chosen arbitrary cutoff levels were deemed sufficient to indicate the presence of a cytokine effect that might affect enzyme and transporter activity. Representative dose-response profiles of CYP3A4 and acute-phase protein (C-reactive protein [CRP]) after IL-1β and IL-6 treatment are illustrated in Fig. 9. A summary of gene expression data for other proteins is summarized in Tables 1, 2, and 3. Gene expression of various phase 1 enzymes (CYP1A2, CYP2C8, CYP2C9, CYP3A4) and phase 2 enzymes (GSTA1, GSTA2, UGT1A1, and UGT2B7) was generally downregulated in the presence of IL-1β and IL-6 (Table 1). As illustrated in Fig. 9A, a concentration-dependent downregulation of CYP3A4 mRNA was observed that corresponded to increasing IL-1β exposure (6.25–5000 pg/ml). Additionally, Hep:KC (1:0.4) cocultures produced a greater extent of mRNA suppression compared with cultures without KC (Hep:KC 1:0). Studies with IL-6 (6.25–5000 pg/ml) also produced a concentration-dependent downregulation of CYP3A4 expression (Fig. 9C) but showed no KC-dependent effects. Consistent with observed enzyme activity, cultures containing KC (Hep:KC 1:0.4) enhanced IL-1β-mediated gene suppression, but had no effect on IL-6.

Gene expression of CYP3A4 and CRP in Hep:KC cultures after cytokine treatments. Hepatocytes were cultured (in micropatterned cocultures with 3T3-J2 fibroblasts) ± Kupffer cells (at Hep:KC of 1:0 and 1:0.4). After IL-1β (A and B) and IL-6 (C and D) incubation (6.25–5000 pg/ml), CYP3A4 and CRP mRNA levels were assessed by RT-PCR (n = 3 ± S.D.).
Effects of IL-1β and IL-6 on metabolism enzymes mRNA expression in Hep:KC (1:0 and 1:0.4) cell cultures
Effects of IL-1β and IL-6 on uptake and efflux transporters mRNA expression in Hep:KC (1:0 and 1:0.4) cell cultures
Effects of IL-1β and IL-6 on acute-phase proteins mRNA expression in Hep:KC (1:0 and 1:0.4) cell cultures
Analogous to metabolic enzymes, gene expression of drug transporters was also downregulated by the presence of both IL-1β and IL-6. Gene expression results for various influx and efflux transporters are summarized in Table 2. After cytokine treatments (6.25–5000 pg/ml), similar IL-1β- and IL-6-mediated downregulation of expression was observed for NTCP, OATP1B3, and OCT1 (mRNA levels were ≤50% of untreated control, and effects were concentration-dependent). The extent of IL-1β-mediated suppression of these transporters was generally greater in the presence of KC (Hep:KC 1:0.4 cocultures) than in hepatocytes alone (Hep:KC 1:0 cultures). No KC-dependent effects were observed after IL-6 treatment. Additionally, OATP1B1 and OATP2B1 expression were also affected by IL-1β treatment and the presence of KCs further enhanced the impact on OATP1B1.
Treatment with cytokines IL-1β and IL-6 (6.25–5000 pg/ml) resulted in an upregulation of CRP mRNA expression, indicative of an inflammation response. At the highest concentration studied (5000 pg/ml IL-1β), CRP mRNA levels reached 1133 ± 260% and 53,132 ± 15,568% of untreated controls in Hep:KC 1:0 and 1:0.4 cultures, respectively (Fig. 9B; Table 3). Similar to the effects on metabolic enzymes and drug transporters, the presence of KC also enhanced IL-1β–mediated CRP upregulation. At the highest IL-6 concentration studied (5000 pg/ml), CRP mRNA levels reached 95,220% ± 12,671% and 80,426% ± 25,395% of untreated controls in Hep:KC 1:0 and 1:0.4 cultures, respectively (Fig. 9D). With IL-6, no obvious differences in effects were seen in the presence and absence of KC.
In addition to CRP, the gene expression of acute phase proteins α-1-acid glycoprotein (AAG) and serum amyloid A2 (SAA2) protein were also evaluated. Incubation of Hep:KC 1:0 and 1:0.4 cultures with IL-1β (6.25 –5000 pg/ml) also resulted in upregulation of AAG (effects were also enhanced in the presence of KC) and SAA2 (Table 3). Similar treatment with IL-6 (6.25–5000 pg/ml) also led to upregulation of AAG and SAA2 mRNA levels. No differences in effects were detected in cultures with and without KC, however (Table 3).
Discussion
The aim of this work was to establish a viable, functional, and long-term human Hep:KC coculture model that is more physiologic relevant than hepatocyte monocultures for studying the effects of cytokines and cytokine modulators on drug metabolism enzymes and transporters. The utility of standard cocultures with human hepatocytes and KCs are limited by a short working window (∼3 days) and a rapid decline of hepatic functions. The micropatterned hepatocyte system previously established by Khetani and Bhatia (2008) offers flexibility in the timing of KC introduction and experimental design. In this study, KCs were incorporated into the micropatterned hepatocyte platform to produce a Hep:KC coculture model that provided a longer experimental window (∼2 weeks) compared with conventional in vitro models. The contribution of 3T3 fibroblasts helped to stabilize hepatocyte functions within the system (although their secretory products cannot be formally excluded from having any direct influence on cytokine-mediated responses reported here). Analysis of appropriate hepatocyte-fibroblast control cultures suggested that excretion of different cytokines in the absence of KCs was consistent with what would be expected based on literature data. Based on findings that Hep:KC cocultures were more responsive to LPS and IL-1β compared with hepatocyte monocultures, we feel that this system is a first step toward a more physiologically relevant in vitro model to study the effects of cytokines and cytokine modulators on drug metabolism enzymes and transporters.
Characterization of the Hep:KC coculture model showed sustained KC presence for at least 2 weeks. During this period, the cocultures maintained: 1) their metabolic capacity as indicated by continued production of albumin and urea; 2) functionality as demonstrated by sustained CYP3A4 activity; 3) capacity to secrete proinflammatory cytokines in response to LPS exposure; and 4) reversibility of the effects on CYP3A4 downregulation after withdrawal of IL-6 or IL-1β. Furthermore, we demonstrated that IL-1β–mediated cytokine release was KC-dependent, whereas IL-6 stimulated cytokine release was not. Additionally, weak cytokine response was observed for IL-2 and IL-23, most likely the result of minimal or undetectable IL-2RB or IL-23R on either hepatocytes or KC.
The extent of CYP3A4 activity downregulation observed in Hep:KC cultures by IL-6 (61%–97%) was comparable to those reported in numerous studies using human hepatocyte monocultures (20%–80%) (Dickmann et al., 2011; Dallas et al., 2012; Nguyen et al., 2013). This finding indicates that the coculture model is responding as expected to proinflammatory cytokine stimulation. In this system, IL-1β produced predictable results such as stimulating an immune modulatory response (Fig. 3) and downregulating both CYP3A4 activity and gene expression (Fig. 4). To our knowledge, this is the first time in which the incorporation of KCs was demonstrated to enhance IL-1β effects (compared with hepatocytes alone). In essence, the addition of another component of the liver to the conventional hepatocyte monoculture model allowed the coculture system to more fully capture the impact of IL-1β. As evident, the IC50 value for CYP3A4 inhibition in Hep:KC 1:0.4 decreased by greater than 51-fold compared with 1:0 cultures (Fig. 4) after IL-1β treatment.
Another advantage of the Hep:KC coculture model over conventional hepatocyte monocultures is the increased sensitivity to cytokine stimulation, thus allowing in vitro studies to be performed at physiologically relevant cytokine concentrations. Reported IC50 values of IL-1β– and IL-6–mediated cytochrome inhibition (Fig. 4) were within the physiologic range of serum IL-1β and IL-6 in patients experiencing inflammation-related diseases. In humans, many proinflammatory cytokines (i.e., IL-1β and IL-6) circulate under normal conditions in the picomolar range and may increase by as much as 1000-fold during infection or trauma (Cannon, 2000). Earlier in vitro studies that used primary hepatocyte monocultures to assess cytokine-CYP450 enzyme interactions often required much higher supraphysiological concentrations of IL-1 and IL-6 (in the ng/ml range) to demonstrate an effect (Sunman et al., 2004; Aitken and Morgan, 2007). In contrast, the IC50 values of CYP3A4 inhibition in the Hep:KC system can be observed in the pg/ml range (Fig. 4). It is conceivable that the sensitivity of the Hep:KC coculture model is likely attributed to the KC-mediated release of additional proinflammatory cytokines that can interact with the hepatocytes.
In addition to CYP3A4, effects were also note for other metabolism enzymes and transporters. In general, downregulation of gene expression associated with various phase 1 and 2 metabolism enzymes and drug transporters was observed after IL-1β and IL-6 treatment, consistent with observations reported in the literature (Nguyen et al., 2013). Gene-suppression trends appeared more pronounced in the presence of KCs after IL-1β treatment, but not IL-6 (Tables 1–3), suggesting that the addition of KCs increased the capacity of Hep:KC co-cultures to capture better the differential in cytokine impact. For CYP3A4, gene expression downregulation (Fig. 9) corresponded with a decrease in enzyme activity (Fig. 4), consistent with the previous observations of a simultaneous decrease in activity and expression in the same study using IL-1β (Abdel-Razzak et al., 1993) and IL-6 (Dickmann et al., 2011). It is currently difficult to predict whether these results will lead to important clinical implications because of the lack of clinical TP-DDI data for marketed biologics related to IL-1β (e.g., canakinumab and anakinra). As a result, additional clinical TP-DDI data and more standardized in vitro cell culture models across laboratories are needed before IVIVC can be explored.
Application of the Hep:KC coculture model to study IL-2 produced conflicting results to observations made by Sunman et al. (2004). Using conventional hepatocytes and KCs at similar Hep:KC 1:01 and 1:0.4 ratios, they reported sustained IL-2–mediated CYP3A suppression. This discrepancy may be attributed to interlaboratory variability associated with differences in experimental conditions, donors used, and/or purity of the hepatocyte-KC cell populations. The negligible impact of IL-2 on CYP3A4 in our coculture system is consistent with the observed minimal IL-2RB expression, which would limit IL-2 interactions leading to diminished cytokine release following treatment. Others have also reported difficulty in measuring IL-2R expression in normal liver compared with those infected with hepatitis C (Morshed et al., 1993).
Elevated IL-23 levels are often associated with psoriasis and rheumatoid arthritis (El-Hadid et al., 2008; Melis et al., 2010). Dallas et al. (2012) previously showed that IL-23 had little impact on CYP3A4 in hepatocyte monocultures. Similar results were confirmed in the current Hep:KC coculture model whereby IL-23 treatment generated only low levels of CYP3A4 suppression. Together with negligible cell surface receptor expression of IL-23R and minimal stimulation of proinflammatory cytokine release, it can be argued that cytokines with such characteristics may pose less DDI risk with small-molecule drugs; however, in vitro data should not be interpreted in isolation, but rather in conjunction with clinical evidence to provide a better assessment of TP-DDI risks (i.e., are elevated proinflammatory cytokine and CRP levels associated with the disease state) (Evers et al., 2013).
In summary, the current opinion from regulatory agencies is that conventional in vitro systems are not yet fully established to provide meaningful prediction of clinical TP-DDI (Evers et al., 2013; Kenny et al., 2013). However, to advance this area of research, the U.S. Food and Drug Administration encourages continued research effort and data gathering. Toward this objective, the Hep:KC coculture system discussed here offers a potentially improved in vitro system compared with conventional hepatocyte monocultures. The Hep:KC system is arguably more physiologically relevant as it incorporates an additional innate immune component (a closer resemblance of the liver than hepatocyte monocultures). This immune component plays an important role in mediating the effects of therapeutic proteins (i.e., cytokines and mAbs) on metabolism enzymes and drug transporters. As a result, the more holistic Hep:KC system has the potential to generate a complex network of cross-talk between cytokines and inflammatory mediators, thus making it more sensitive to stimulation by exogenous cytokines. Consequently, in vitro studies in Hep:KC cocultures can be performed at lower, more clinically relevant cytokine concentrations, and its effects can be more fully captured (such as the case with IL-1β). Furthermore, the Hep:KC cocultures can potentially mediate interactions from a broader spectrum of cytokines (i.e., those that lack receptors on hepatocytes but can still indirectly interact through stimulation of KCs). Although more research is needed to explore the full potential and limitations of this coculture model, the work presented here demonstrated its value as a tool for providing further understanding into the mechanisms by which cytokines (i.e., IL-1β) interact with hepatocytes and KCs to impact metabolic enzymes and drug transporters.
Acknowledgments
The authors thank Dr. Diana Montgomery for the valuable scientific discussions, input, and help with editing.
Authorship Contributions
Participated in research design: Nguyen, Ukairo, Khetani, McVay, Evers.
Conducted experiments: McVay, Kanchagar.
Contributed new reagents or analytic tools: McVay, Kanchagar, Seghezzi, Ayanoglu.
Performed data analysis: Nguyen, Ukairo, Khetani, McVay, Kanchagar, Irrechukwu
Wrote or contributed to the writing of the manuscript: Nguyen, Ukairo, Khetani, Irrechukwu, Evers.
Footnotes
- Received September 23, 2014.
- Accepted March 4, 2014.
This work was funded in part by the Merck New Technology Review and Licensing Committee.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AAG
- α-1-acid glycoprotein
- CRP
- C-reactive protein
- DDI
- drug-drug interactions
- DMSO
- dimethylsulfoxide
- HCM
- HepatoPac culture medium
- Hep:KC
- hepatocyte-Kupffer cell
- IFN
- interferon
- IL
- interleukin
- IL-1RA
- interleukin-1 receptor antagonist cytokine
- IL-1R1
- IL-1 receptor, type 1
- IL-2RB
- IL-2 receptor, β-chain
- IL-6R
- IL-6 receptor, IL-23R, IL-23 receptor
- KC
- Kupffer cells
- LPS
- lipopolysaccharide
- mAb
- monoclonal antibody
- MPCC
- micropatterned coculture
- P450
- cytochrome P450
- RT-PCR
- reverse-transcription polymerase chain reaction
- SAA2
- serum amyloid A2
- TNF-α
- tumor necrosis factor-α
- TP
- therapeutic protein
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}