


Abstract
The contribution of organic anion transporter OAT2 (SLC22A7) to the renal tubular secretion of creatinine and its exact localization in the kidney are reportedly controversial. In the present investigation, the transport of creatinine was assessed in human embryonic kidney (HEK) cells that stably expressed human OAT2 (OAT2-HEK) and isolated human renal proximal tubule cells (HRPTCs). The tubular localization of OAT2 in human, monkey, and rat kidney was characterized. The overexpression of OAT2 significantly enhanced the uptake of creatinine in OAT2-HEK cells. Under physiologic conditions (creatinine concentrations of 41.2 and 123.5 µM), the initial rate of OAT2-mediated creatinine transport was approximately 11-, 80-, and 80-fold higher than OCT2, multidrug and toxin extrusion protein (MATE)1, and MATE2K, respectively, resulting in approximately 37-, 1850-, and 80-fold increase of the intrinsic transport clearance when normalized to the transporter protein concentrations. Creatinine intracellular uptake and transcellular transport in HRPTCs were decreased in the presence of 50 µM bromosulfophthalein and 100 µM indomethacin, which inhibited OAT2 more potently than other known creatinine transporters, OCT2 and multidrug and toxin extrusion proteins MATE1 and MATE2K (IC50: 1.3 µM vs. > 100 µM and 2.1 µM vs. > 200 µM for bromosulfophthalein and indomethacin, respectively) Immunohistochemistry analysis showed that OAT2 protein was localized to both basolateral and apical membranes of human and cynomolgus monkey renal proximal tubules, but appeared only on the apical membrane of rat proximal tubules. Collectively, the findings revealed the important role of OAT2 in renal secretion and possible reabsorption of creatinine and suggested a molecular basis for potential species difference in the transporter handling of creatinine.
Introduction
Mammalian organic anion transporter (OAT)2 (SLC22A7), along with organic cation transporter (OCT)1 (SLC22A1), OCT2 (SLC22A2), OAT1 (SLC22A6), and OAT3 (SLC22A8), belong to the solute carrier group of membrane transport proteins (i.e., SLC22) that mediate cellular uptake of numerous organic ions, including xenobiotics and endogenous substrates. Oat2 was originally identified as a novel liver-specific transporter because of its predominant mRNA expression in the rat liver (Simonson et al., 1994; Sekine et al., 1998). However, Oat2 mRNA was later found to be expressed at the highest level in the rodent kidney (Buist et al., 2002; Kobayashi et al., 2002). In humans, OAT2 mRNA expression appeared to be comparable in human kidney and liver (Sun et al., 2001; Cropp et al., 2008). Moreover, within the kidney, the abundance of OAT2 mRNA is comparable to that of OAT1, OAT3, and OCT2, the transporters known to play an important role in active uptake of ionized compounds into proximal tubule cells. Although the expression of OAT2 protein in human kidneys has been reported (Cheng et al., 2012), comparatively little is known about its functional role in the renal tubular handling of endogenous substrates, drugs, and toxins.
Glomerular filtration rate (GFR) is an effective indicator of kidney function. Measure of plasma or serum creatinine is the most widely used means for estimating GFR. Creatinine, a low-molecular-weight cation (113 daltons), is not metabolized, is not bound to plasma proteins, and is freely filtered by the renal glomerulus. In a 2009 College of American Pathologist Survey of predominantly North American laboratories, serum (or plasma) creatinine test results are used by the majority participants to estimate GFR (Miller, 2008). A 50% increase of serum creatinine is considered to be a cutoff value for renal functional impairment (Levey et al., 1988). Therefore, understanding creatinine disposition and renal clearance is critical to clinical practice and public health. However, creatinine is also actively secreted by renal tubular cells, accounting for 10–40% of excreted creatinine in normal individuals (Levey et al., 1988). The tubular secretion of creatinine is an active process, and transporters located at basolateral and apical membranes of tubular cells are responsible for the uptake of creatinine from the blood and secretion into the lumen (i.e., urine). Clinical observations indicate that certain drugs such as cimetidine (CMD) (Dubb et al., 1978), pyrimethanmine (PYR) (Opravil et al., 1993), and trimethoprim (Berglund et al., 1975) inhibit tubular creatinine secretion, leading to elevated creatinine levels in the blood, without causing kidney injury. Several renal transporters are reportedly involved in creatinine transport; however, the predominant pathway(s) mediating creatinine tubular secretion, especially at the apical membrane, is unknown. Contradictory data regarding the contribution of OAT2 in tubular creatinine secretion were recently reported. For example, the OAT2-mediated transport of creatinine was found to be either low (Imamura et al., 2011) or high (Lepist et al., 2014). Ciarimboli et al. (2012) studied the transport of creatinine in vivo in transfected human embryonic kidney (HEK) cells and in vivo in wild-type and Oct1/Oct2(−/−) mice. The studies showed that the uptake of creatinine was markedly enhanced in OCT2-HEK cells compared with Mock-HEK cells (5.8-fold), whereas it only increased 2.3-fold in OAT2-HEK cells. In addition, compared with wild-type mice, creatinine clearance was significantly impaired in Oct1/Oct2(−/−) mice. As a result, they concluded that OCT2 plays a decisive role in the renal secretion of creatinine.
The localization of OAT2 in the mammalian kidney has not been convincingly confirmed and perhaps is species-dependent. Oat2 in rat and mouse kidneys was immunolocalized to the brush-border (apical) membrane of the proximal tubule S3 segment in the outer stripe and medullar rays (Kobayashi et al., 2005; Ljubojevic et al., 2007). In addition to the apical membrane of proximal tubule in the rat kidney, Kojima et al. (2002) reported that rat Oat2 protein (∼60 kDa) was also equally distributed in the cortical, outer medullary, and inner medullary tissue and immunolocalized to the apical membrane of thick ascending limb of Henle and cortical and medullary collecting ducts. In contrast, OAT2 was immunolocalized to the basolateral but not apical membrane of proximal tubule in the human kidney (Enomoto et al., 2002). Species differences in the OAT2 expression and its contribution to renal transport of endogenous substrates such as creatinine need to be further elucidated (Enomoto et al., 2002; Cheng et al., 2012). To these aims, the OAT2-mediated creatinine transport was characterized using OAT2-overexpressing HEK cell and human renal proximal tubule cell (HRPTC) models, and its localization in human, monkey, and rat kidney was determined by immunohistochemical analysis with an OAT2-specific antibody.
Materials and Methods
Chemicals and Reagents.
[3H]Penciclovir (PCV; 1.1 Ci/mmol), [14C]creatinine (58.2 mCi/mmol), and [14C]metformin (98.0 mCi/mmol) were purchased from Moravek Biochemicals (Brea, CA). [3H]cGMP (25.0 Ci/mmol) was purchased from American Radiolabeled Chemicals (St. Louis, MO). [3H]Para-aminohippuric acid (4.3 Ci/mmol), [3H]1-methyl-4-phenylpyridinium (83.9 Ci/mmol), and [14C]mannitol (57.1 mCi/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). Creatinine-d3 (methyl-d3) was obtained from Medical Isotopes (Pelham, NH). Nonradiolabeled PCV, creatinine, CMD, IMC, and PYR were obtained from Toronto Research Chemicals (North York, Ontario, Canada). Bromosulfophthalein (BSP) was from Sigma-Aldrich (St. Louis, MO). Hygromycin, Flp recombinase expression plasmid (pOG44), lipofectamine 2000, phosphate-buffered saline (PBS), Hanks’ balanced salt solution (HBSS), Dulbecco’s modified Eagle’s growth medium, and HEK Flp-In cells were from Invitrogen (Carlsbad, CA). The primary antibody against human OAT2 was obtained from Sigma-Aldrich (St. Louis, MO). Cryopreserved and freshly isolated HRPTCs and InVitroGRO proximal tubule (PT) medium were from BioreclamationIVT (Baltimore, MD). The freshly isolated HRPTCs were seeded directly onto 0.4-μm pore-size 24-well polycarbonate Corning Costar Transwell permeable supports (Tewksbury, MA). All other chemicals used were of the highest purity available from standard sources.
Generation of Stable Human OAT2-Transfected HEK Cell Line (OAT2-HEK) and Cell Cultivation.
The open reading frame of human OAT2 cDNA variant 1 (OAT2-546aa, GenBank accession number NM_006672) was subcloned into pcDNA5/Flp recombination target (pcDNA5/FRT) expression plasmid, according to the manufacturer’s instructions (Invitrogen). Plasmid DNA was prepared using standard methods (Qiagen, Valencia, CA), and sequences were confirmed to be identical to the one reported in the National Center for Biotechnology Information database. The stable transfection of HEK cells with OAT2, using Flp-In expression system, was conducted according to a procedure previously described for the preparation of OCT2-, multidrug and toxin extrusion protein (MATE)1-, and MATE2K-transfected cells (Han et al., 2010; Shen et al., 2013). In brief, HEK Flp-In cells were seeded into a six-well plate at a density of 0.1 million cells/cm2 in Dulbecco’s modified Eagle’s growth medium supplemented with 10% fetal calf serum, 0.1 mM nonessential amino acids, and 2 mM L-glutamate, and incubated overnight. Lipofectamine 2000 reagent (10 µL) was diluted in 250 µL serum-free Opti-MEM I reduced serum medium and incubated at room temperature for 5 minutes. Plasmid DNA (0.4 µg pCDNA5/FRT/OAT2 plasmid and 3.6 µg pOG44 plasmid) was added to the Lipofectamine mixture and incubated for an additional 20 minutes at room temperature. The DNA-Lipofectamine mixture was then added dropwise to the cells, and the culture plate was incubated for 6 hours before the medium was changed. Transfected cells were then trypsinized and plated to 10% confluence in fresh medium containing 1% penicillin-streptomycin. After 24 hours, the medium was replaced with fresh medium containing 200 µg/ml hygromycin. Media were changed every 3–4 days until hygromycin-resistant colonies formed. Twenty clones of cells were picked and plated in 24-well plate and amplified. Then the cells were screened by uptake experiment using a probe substrate (i.e., [3H]cGMP), leading to the identification of high expression clone of the OAT2 gene.
The established OAT2-HEK cells, and OCT2-, MATE1-, and MATE2K-HEK cells and mock cells were cultured at 37°C in an atmosphere of 95% air/5% CO2 and subcultured once per week (Han et al., 2010; Shen et al., 2013). OAT2-HEK cells and other HEK cells used in the current study were passaged less than 10 and 30 times, respectively, to retain consistent transporter expression and functional activity.
Uptake and Inhibition Studies Using Transporter-Expressing HEK Cells.
Transporter-expressing cells were grown to confluence in 24-well poly-D-lysine–coated plates (BD Biosciences, San Jose, CA). All experiments were conducted at 37°C using a transport solution containing HBSS supplemented with 10 mM HEPES (pH 7.4 for OAT2 and OCT2, and pH 8.4 for MATE1 and MATE2K, respectively), along with a radiolabeled probe substrate or creatinine-d3. The probe substrates used for OAT2 were PCV and cGMP, and that used for OCT2, MATE1, and MATE2K was metformin. Creatinine-d3 was used as a test substrate, rather than unlabeled creatinine, because a low-level interference was observed for the selective reaction monitoring transition of unlabeled creatinine in the liquid chromatography–tandem mass spectrometry (LC-MS/MS) assay; there was no interference for the quantification of deuterium-labeled creatinine. The concentration of labeled substrate(s) and the incubation time(s) used for transport studies are indicated in the figure legends. The transport solution was aspirated after the incubation period, and the cells were rinsed with three changes of ice-cold HBSS. The cells were then solubilized with 0.3 mL lysis buffer (0.1% Triton X-100 or methanol). Compound concentration in the cell lysates was measured by either liquid scintillation counting (Tri-Carb 2910 TR Liquid Scintillation Analyzer; PerkinElmer Life and Analytical Sciences, Waltham, MA) or LC-MS/MS (Triple Quad 5500, AB Sciex, Foster City, CA; Shimadzu Nexera uHPLC system, Shimadzu, Kyoto, Japan). Protein concentration was determined with a BCA Protein Assay kit (Pierce Chemical, Rockfold, IL). Transport experiments were conducted under linear uptake conditions and at probe substrate concentrations well below the Km values of test compounds. For kinetic analysis of creatinine transport, experiments were carried out under linear conditions with increasing concentrations of creatinine-d3. A wide range of creatinine-d3 concentrations was used (from 4.6 to 10,000 µM) because of the high Km values associated with the transport.
Uptake and Transcellular Transport Studies Using HRPTCs.
Cryopreserved HRPTCs were thawed according to the vendor’s instructions (BioreclamationIVT, Baltimore, MD). Briefly, HRPTCs were thawed at 37°C for 1.5 minutes and transferred to a conical tube containing 5 mL prewarmed InVitroGRO PT medium. The viability was assessed by trypan blue exclusion method, and the average post-thawing viability of HRPTCs was greater than 85%. One lot of cryopreserved human renal proximal tubule cells (Lot AFA; male organ donor; African American) was used, based on availability. The cell suspension was then diluted to the appropriate density of viable cells in PT medium prewarmed to 37°C. For inhibition analysis, a 10-minute preincubation with PT medium containing an inhibitor was conducted. Creatinine-d3 (10 µM) was added into HRPTC suspensions and incubated at 37°C in the presence and absence of cimetidine (1 mM), indomethacin (100 µM), or BSP (50 µM) for a period of 30 seconds. This time was demonstrated to fall in the linear region of the uptake velocity (unpublished data). The 100-µL reaction mixtures were removed and overlaid onto preprepared 0.4-mL microcentrifuge tubes containing 50 µL 2 M ammonium acetate (bottom layer) and 100 µL filtration oil (top layer; 84.5:15.5 silicon oil-mineral oil mix, final density of 1.015). Samples were centrifuged immediately at 10,000g for 15 seconds using a benchtop centrifuge to pellet the cells. The tubes were placed on dry ice and then cut, and the cell pellet was digested in 2:1 (volume/volume) ratio of acetonitrile and water at room temperature. The contents were filtered through a 96-well filter plate (0.45 µm low-binding hydrophilic polytetrafluoroethylene), and the filtrate was dried under nitrogen gas. The dried sample was further reconstituted with acetonitrile, and the concentrations of creatinine were analyzed by LC-MS/MS.
In transcellular transport experiments, freshly isolated HRPTCs were seeded on 24-well polycarbonate Transwell plates at a density of 0.1 million cells/cm2. One lot of fresh human renal proximal tubule cells (Lot THU-K-063014; female organ donor; Caucasian) was used based on availability. Media in the cultures were replaced every other day, and transepithelial electrical resistance was measured daily using the Millicell electrical resistance meter (Millipore, Billerica, MA). Cell monolayers were considered fully differentiated when transepithelial electrical resistance values exceeded 150Ω × cm2. The incubation solution for transport experiments was HBSS containing 5.6 mM glucose and 10 mM HEPES (pH 7.4 or pH 6.5). After the culture medium was removed from both sides of the monolayers, cells were washed three times with prewarmed incubation solution. Experiments were then initiated by adding creatinine-d3 (10 µM) to either the apical or basolateral compartment. In all experiments, the pH of the solution on the basolateral side was 7.4, and that on the apical side was 6.5, unless stated otherwise. Cell monolyers were incubated at 37°C in 95% air/5% CO2 for 60 minutes, and 50 µL aliquots from both compartments were taken for LC-MS/MS measurement. The effect of varying apical pH values on creatinine permeability was tested. For inhibition analysis, a 15-minute preincubation in both compartments with transport solution containing inhibitor was performed. [14C]Mannitol (1.8 µM) was added to the donor compartment to assess integrity of the cell monolayer. The radioactivity of the collected sample was determined by liquid scintillation counting.
Immunohistochemistry.
The kidneys from human, cynomolgus monkey, and Sprague-Dawley rat were removed and fixed in 10% neutral buffered formalin solution. After fixation, the specimens were processed for embedding into paraffin blocks. Five-micron tissue sections were cut from blocks and mounted on positively charged microscope slides (Superfrost/Plus slides; Erie Scientific, Portsmouth, NH). The tissue slides were deparaffinized by immersing in xylene and rehydrated through a series of ethanol solutions in descending series from 100% ethanol up to water. After rehydration, the slides were washed twice with PBS at room temperature. Endogenous peroxidase was blocked by incubating with PBS containing 3% hydrogen peroxide (Biocare Medical, Concord, CA) at room temperature for 10 minutes. Antigen retrieval in pH 6.0 citrate buffer (Dako Target Retrieval Solution) was performed in Biocare Medical decloaking device for 3 minutes at 110°C. After rinsing with water and washing with PBS containing 0.05% Tween 20, the specimens were incubated with Biocare Medical blocking solution at room temperature for 10 minutes. They were then incubated with polyclonal anti-human OAT2 antibody (Sigma-Aldrich) in blocking solution at a dilution of 1:200 (5 µg/mL) or rabbit IgG matched to the protein concentration of the primary antibody for 60 minutes, respectively. After washing with PBS containing 0.05% Tween 20, the specimens were treated with an anti-rabbit probe and then horseradish peroxidase-polymer, using Biocare Medical’s Mach 3 Rabbit kit. Positive staining was visualized using a dark brown color developing 3,3′-diaminobenzidine horseradish peroxidase substrate (Biocare Medical). The slides were counterstained with hematoxylin.
LC-MS/MS Analysis of Creatinine-d3.
The LC-MS/MS analysis was performed on a Triple Quad 5500 (AB Sciex) coupled with a Shimadzu Nexera uHPLC system (Shimadzu, Kyoto, Japan). The chromatographic separation was performed on a Luna Silica column (4.6 × 150 mm, 5 µm) from Phenomenex (Torrence, CA) using mobile phases of 2 mM ammonium acetate in water and acetonitrile (45:55, volume/volume). The flow rate was 1.5 ml/min, and total run time was 2.5 minutes. The LC column was maintained at 40°C. The analyte was monitored using selective reaction monitoring in positive ion electrospray mode with the optimized nebulizing, turbo, and curtain gases set at 55, 60, and 35. The turbospray voltage was set at 2000 V, and turbo probe temperature was set at 650°C; declustering potential and collision energy were optimized to be 60 V and 30 eV. Quantitation was performed using the transition of m/z 117→47. The system control and data processing were performed on the Analyst v1.5.1 software.
Dried cell samples were extracted using organic precipitation. Specifically, 300 µL methanol was added, followed by gentle mixing on a mixer for 1 hour at room temperature. The mixture was transferred to a clean 96-well plate and evaporated under nitrogen. The dried sample was further reconstituted with 300 µL acetonitrile. After mixing and centrifugation, 100 µL supernatant was aliquoted to a clean 96-well plate, followed by the addition of 100 µL acetonitrile. For transcellular transport experiments, samples from apical or basolateral compartment were extracted directly with acetonitrile. The final extracts were mixed and centrifuged and stored at 15°C in the autosampler before injection. The injection volume was 10 µL. The assay was qualified over the analytical range of 10.0–10,000 pg/mL using a linear 1/x2 weighed regression.
LC-MS/MS Quantification of OAT2, OCT2, MATE1, and MATE2K Proteins in Transporter-Overexpressing Cells.
The membrane protein fraction was extracted from transporter-overexpressing cells using the native membrane protein extraction kit, as previously described (Li et al., 2008). Membrane protein samples were prepared and digested, as described previously (Qiu et al., 2013). Briefly, samples containing 200 µg membrane proteins were reduced with 10 mM dithiothreitol at 95°C for 5 minutes in 25 mM ammonium bicarbonate buffer with 1% deoxycholate. The protein was then alkylated with 15 mM iodoacetamide in the dark for 30 minutes, followed by trypsin (1:50 enzyme:protein) digestion at 37°C overnight with shaking. The digestion was stopped by addition of an equal volume of water with 0.2% formic acid containing the stable isotope- labeled internal standards to each sample. Synthetic unlabeled peptides were used as standards for quantification.
Peptide quantification was conducted using a Shimadzu Nexera UHPLC (LC-30A) system (Columbia, MD) coupled to an API-6500 triple quadrupole mass spectrometer (AB Sciex). The LC column used for peptide separation and elution was a Waters (Milford, MA) BEH300 C18 2.1 × 100 mm Peptide Separation Technology column with a 1.7 µm particle size and 300 Å pore size. Mobile phase A was water with 0.1% formic acid (v/v), and mobile phase B was acetonitrile with 0.1% formic acid (v/v). A linear gradient was used to achieve chromatographic separation with the following linear gradients: 1 minute, 5% B; 17 minutes, 27.2% B; 17.5 minutes, 90% B; 20.5 minutes, 90% B; 21 minutes, 5% B; 33 minutes, 5% B. A sample volume of 10 µL was injected onto the LC column at a flow rate of 0.2 ml/min. The precursor-to-product transition for the native/unlabeled peptide monitored represented the double-charged precursor ions to the single-charged product y ions, as shown in Table 1.
Summary of peptide sequences and multiple reaction monitoring (MRM) transitions used to quantitate the four renal transporter proteins with LC-MS/MS
Data Analysis.
Concentration-dependent uptake of creatinine by OCT2, OAT2, MATE1, or MATE2K was determined after subtracting the uptake in mock-transfected cells at each concentration tested from uptake into the cells expressing the individual transporters; this was done to account for potential endogenous transport activity in the control cells. The kinetics was best fit to a single Michaelis-Menten term (Phoenix WinNonlin; Certara, St. Louis, MO): where V and C are the uptake rate and concentration of substrate, respectively, and Km and Vmax represent the half saturation concentration (Michaelis constant) and the maximum transport rate, respectively.
where V and C are the uptake rate and concentration of substrate, respectively, and Km and Vmax represent the half saturation concentration (Michaelis constant) and the maximum transport rate, respectively.
IC50, the concentration required to inhibit the transport of creatinine by 50%, was calculated by fitting the data to the following equation using Phoenix WinNonlin (Certara). where γ is the Hill coefficient that describes the steepness of inhibition curve, and I is the inhibitor concentration.
where γ is the Hill coefficient that describes the steepness of inhibition curve, and I is the inhibitor concentration.
Data were reported as mean ± S.D., unless otherwise noted. Statistical differences between two groups were determined by an unpaired one-tailed Student t test. Multiple group comparisons were performed using one-way analysis of variance, followed by Dunnett’s test, in which uptake in the absence of inhibitor served as the control group (GraphPad Prism v5.0; GraphPad Software, San Diego, CA).
Results
Characterization of OAT2-HEK Cell Lines.
Stable transfection of the HEK cell line with SLC22A7 cDNA encoding human OAT2 variant 1 yielded three clones with substantial OAT2-546aa expression. All three clones had a comparable expression level, as verified by real-time reverse-transcription polymerase chain reaction. Expression of OAT2 mRNA in the stably transfected cell lines was approximately 5,000,000-fold higher relative to the mock cell line. The OAT2-HEK cell line selected for further study was subjected to LC-MS/MS analysis to determine OAT2 protein content, and the results also revealed significant overexpression of OAT2 at the protein level over the mock-HEK cell line (54.1 pmol/mg membrane protein in OAT2-HEK cells versus undetectable level in Mock-HEK cells). Functional activities of OAT2-HEK cells were confirmed by substrate uptake assays. As shown in Fig. 1, the OAT2-HEK cell line was capable of transporting [3H]PCV and [3H]cGMP, two known substrates of OAT2, at concentrations below the reported Km values (284 and 88 µM, respectively) (Cropp et al., 2008; Cheng et al., 2012), with uptake ratio of 10.8- and 7.7-fold, respectively, compared with Mock-HEK cell lines (15.1 ± 1.5 versus 1.4 ± 0.13 pmol/mg/min for [3H]PCV and 5.7 ± 0.83 versus 0.74 ± 0.14 pmol/mg/min for [3H]cGMP) (Fig. 1). Furthermore, the OAT2-mediated uptake of [3H]PCV and [3H]cGMP was almost abolished in the presence of 50 µM BSP or 100 µM IMC, and significantly reduced by 200 µM CMD (P < 0.001).

Characterization of OAT2-mediated transport of PCV and cGMP. Uptake in HEK cells stably transfected with the control vector (Mock-HEK) and OAT2-546aa (OAT2-HEK) was measured after a 2-minute incubation at 37°C with [3H]PCV (0.14 μM) (A) and [3H]cGMP (0.005 μM) (B). Incubations were conducted in the absence and presence of CMD (200 μM), BSP (50 μM), or IMC (100 μM) to evaluate the effects of these inhibitors on creatinine uptake. Each value represents the mean ± S.D. (n = 3). ***P < 0.005, significantly different from uptake in the absence of an inhibitor.
Transport of Creatinine by OAT2 Expressed in HEK Cells.
To determine whether creatinine is a substrate for human OAT2, the cellular uptake of creatinine-d3 was measured at two concentrations in the physiologic range (41.2 and 123.5 µM or 0.48 and 1.43 mg/dL) in OAT2-HEK cells after a 2-minute incubation. The uptake in OAT2-expressing cells was approximately 200-fold higher than that in the control cells (13.9 ± 1.1 versus 0.07 ± 0.01, and 13.1 ± 1.1 versus 0.06 ± 0.01 µL/min/mg at 41.2 and 123.5 µM, respectively) (Fig. 2A). There was also significant OCT2-, MATE1-, and MATE2K-mediated uptake of creatinine-d3 compared with the control (approximately 20-, 3-, and 3-fold, respectively). In addition, the uptake was sensitive to treatment with BSP and IMC, two potent OAT2 inhibitors (Fig. 3, B and C). We observed that OAT2-HEK cells lose OAT2-specific activity quickly as the cells grow beyond 10 passages (data not shown). Therefore, to avoid a substantial reduction in OAT2 functional activity, OAT2-HEK cells used in the current experiments had undergone less than 10 passages after establishing the cell line. The cell lines were analyzed by LC-MS/MS methods, and the results showed that the mean transporter membrane protein content was 54.1, 58.7, 329, and 18.6 pmol/mg for OAT2, OCT2, MATE1, and MATE2K, respectively.

Creatinine-d3 uptake mediated by OAT2. (A) Uptake of creatinine-d3 (41.2 and 123.5 µM) was determined in either OAT2-, OCT2-, MATE1-, MATE2K-, or Mock-HEK cells after 2 minutes of incubation. Significant uptake of creatinine-d3 was found in HEK cells expressing OAT2, OCT2, MATE1, or MATE2K (relative to mock cells). Each value represents the mean ± S.D. (n = 3). ***P < 0.005, uptake was significantly different from Mock-HEK cells. (B) Concentration-dependent creatinine uptake was determined over a range of creatinine-d3 concentrations in OAT2-, OCT2-, MATE1-, MATE2K-, and Mock-HEK cells. Uptake in Mock-HEK cells was subtracted to give individual transporter-specific uptake. Nonlinear least-squares regression analysis of the data was used to generate the Km and Vmax values (Table 2). Inset in (B) depicts same data on creatinine transport scale. Each value represents the mean ± S.D. (n = 3).
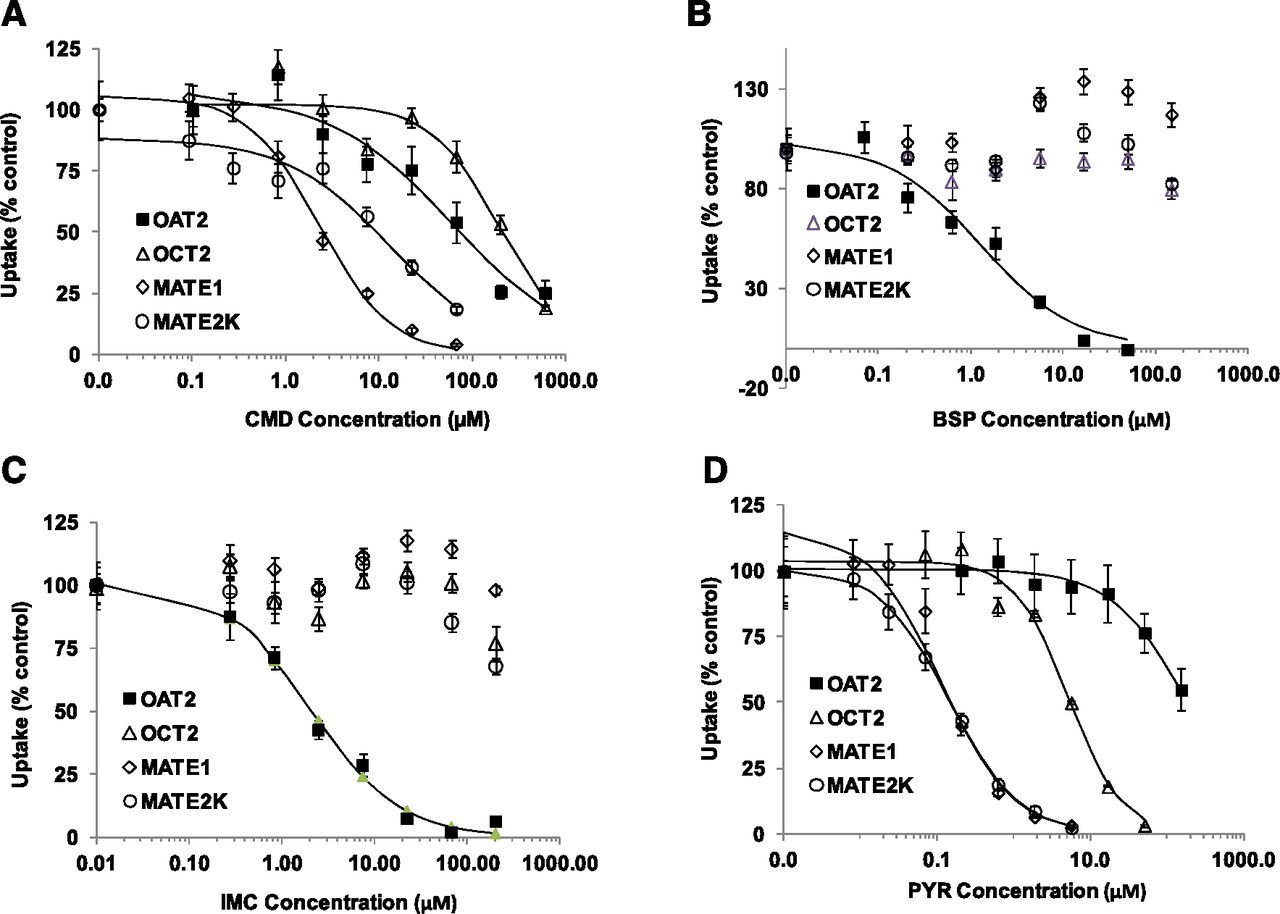
Kinetics of inhibition of transporter-mediated uptake by CMD (A), BSP (B), IMC (C), and PYR (D). A range of CMD, BSP, IMC, and PYR concentrations was tested for their effect on the uptake of 2 µM creatinine-d3 into OAT2-HEK cells, and 2 µM [14C]metformin into OCT2-, MATE1-, or MATE2K-HEK cells (IC50 reported in Table 3). The extent of inhibition of transporter-mediated uptake is expressed as a percentage of the uptake in the absence of inhibitor. Nonlinear regression analysis of the data was used to determine apparent IC50 values. Each value represents the mean ± S.D. (n = 3).
To compare the efficiency for creatinine renal transport, the uptake of creatinine-d3 was tested over a range of creatinine-d3 concentrations (5–10,000 µM) in OAT2-, OCT2-, MATE1-, and MATE2K-HEK cells after a 2-minute incubation, which was under linear-uptake conditions (data not shown). As shown in Fig. 2B and Table 2, analysis of the resultant concentration-dependent uptake curves gave apparent Km values of 795 ± 138, 18,771 ± 1,139, 10,234 ± 339, and 21,597 ± 27,066 µM for OAT2, OCT2, MATE1, and MATE2K, respectively. The corresponding Vmax values were 24,987 ± 1,204, 17,546 ± 739, 1,058 ± 20, and 2,948 ± 3,477 pmol/min/mg total protein. The creatinine transport efficiency, as measured by Vmax/Km, was approximately 37- to 1850-fold higher for OAT2 than the other three transporters, when normalized for transporter protein levels (Table 2).
Parameters describing the kinetics of creatinine transport by OAT2, OCT2, MATE1, and MATE2K
Data are expressed as mean ± SD (n = 3). The kinetic parameters were determined as indicated in Materials and Methods.
Characterization of BSP and IMC as Inhibitors for OAT2.
To search for a potent inhibitor of OAT2, the inhibitory effect was evaluated on the uptake of 2 µM creatinine-d3 (OAT2) or [14C]metformin (OCT2, MATE1, and MATE2K) in the presence of increasing concentrations of CMD, BSP, IMC, PYR, cyclosporine A, metformin, and rosuvastatin in transporter-expressing cells. Metformin was used as a probe substrate for the organic cation transporter assessments because it provides more robust uptake activity and dynamics than creatinine. As shown in Fig. 3, B and C, BSP and IMC substantially inhibited OAT2-mediated uptake of creatinine in a concentration-dependent manner with IC50 values of 1.3 ± 0.4 and 2.1 ± 0.4 μM, respectively (Table 3). In contrast, BSP and IMC showed very little inhibition of the uptake of [14C]metformin in OCT2-, MATE1-, and MATE2-K-HEK cells (IC50 values were >100 µM; Table 3). The other five compounds, CMD, PYR, cyclosporine A, metformin, and rosuvastatin, demonstrated weaker inhibition toward OAT2 with IC50 values of 62.3 ± 25.1, >50, >11.1, and 33.6 ± 5.1 µM, respectively. PYR was the most potent inhibitor of OCT2, MATE1, and MATE2K, with IC50 values ranging from 0.11 to 5.1 µM, but showed little inhibition of OAT2 (the IC50 value was >50 µM).
The effect of different inhibitors on creatinine transport by OAT2, or metformin transport by OCT2, MATE1, and MATE2K
Data are expressed as mean ± SD (n = 3).
Creatinine Transport in HRPTCs.
To explore the role of OAT2 in creatinine transport in human proximal tubular cells, the impact of modulators of OAT2 upon intracellular accumulation of creatinine-d3 in HRPTCs was assessed. As shown in Fig. 4A, in the presence of the OAT2 inhibitor IMC (100 µM), the cellular uptake of creatinine-d3 decreased by approximately 50–60% over control (n = 3, P < 0.05). The control inhibitor CMD, at concentration of 1 mM, also showed the inhibition at a similar extent (∼50%) of creatinine-d3 uptake in HRPTCs (n = 3, P < 0.05).

Intracellular uptake and transepithelial transport of creatinine in HRPTCs. (A) The uptake of creatinine-d3 (10 µM) was determined in HRPTCs in suspension after 30 seconds of incubation by oil filtration method. The uptake of creatinine was significantly decreased in the presence of 1 mM CMD and 100 µM IMC compared to the control (*p < 0.05). The uptake of creatinine was reduced in the presence of 50 µM BSP although the difference was not statistically significant. Each value represents the mean ± S.D. (n = 3). (B) Unidirectional fluxes of [3H]1-methyl-4-phenylpyridinium+ (1 µM), PAH (1 µM), and creatinine (3.4 µM) from both basolateral-to-apical side and apical-to-basolateral side were measured across HRPTC monolayer. The efflux ratios, calculated by dividing the B-to-A flux by A-to-B flux, were greater than the unity for the probe substrates. The pH values of the solutions on the basolateral and apical sides for each probe are listed in parentheses. Each value represents the mean ± S.D. (n = 3). (C) Unidirectional permeability of creatinine-d3 (10 µM) from basolateral-to-apical side was measured across HRPTC monolayer. The B-to-A permeability was significantly decreased in the presence of 500 µM CMD, 50 µM BSP, and 100 µM IMC compared with the control (*P < 0.05, **P < 0.01, and ***P < 0.005). Each value represents the mean ± S.D. (n = 4).
To further demonstrate the importance of OAT2 in renal excretion of creatinine, the transepithelial transport of creatinine-d3 across human renal proximal tubule cell monolayer was assessed. The integrity of the HRPTC cell monolayers was determined using transepithelial electrical resistance and mannitol permeability. Cell monolayers were considered fully differentiated when showing electrical resistances of >150 Ω × cm2. The mannitol permeability values assessed in the experiments were consistent with the values calculated using literature data (i.e., ∼170 nm/s; Supplemental Fig. 1). In addition, there was asymmetry in the unidirectional fluxes of organic cations (i.e., [3H]1-methyl-4-phenylpyridinium+ and creatinine) and anion (i.e., para-aminohippuric acid) across the monolayers in that the secretory fluxes were greater than the ones in the absorptive direction, resulting in the efflux ratios [basolateral to apical (B-to-A) flux/apical to basolateral (A-to-B) flux] of 1.33 ± 0.11, 1.22 ± 0.08, and 1.10 ± 0.24, respectively (Fig. 4B). We also examined the effect of varying apical pH values on creatinine flux, and the apical pH of 6.5 showed better handling of creatinine unidirectional flux that that of 7.4 with higher efflux ratio (1.43 ± 0.27 versus 1.22 ± 0.08).
Consistent with significant inhibition of creatinine uptake in HRPTC suspension by BSP and IMC, the modulation of OAT2 activity by BSP (50 µM) or IMC (100 µM) resulted in an approximately 40–50% decrease in creatinine-d3 transport across the HRPTC monolayer in the B-to-A direction. In the presence of CMD (500 µM), the control inhibitor, the transcellular transport of creatinine-d3 decreased by 25% compared with control (n = 4, P < 0.05). There was also inhibition of creatinine-d3 transport in the A-to-B direction in the presence of BSP and IMC (Supplemental Fig. 2).
Localization of OAT2 in Human, Cynomolgus Monkey, and Rat Renal Cortex by Immunohistochemistry.
Immunohistochemical analysis was performed using affinity-purified rabbit anti-human OAT2 antibody that reacts with human, monkey, and rat OAT2 protein (Fig. 5). Appropriate controls and separate staining experiments confirmed that there was no cross-staining (see above). The immunostaining against OAT2 protein was detected in tubules of the renal cortex from all species. In both human and monkey kidneys, OAT2 was located along the basolateral infoldings of the cell membrane, but an apical localization was also visible (Fig. 5, A and B). In contrast, rat Oat2 was restricted to the brush-border (apical) membrane. There was no staining for rat Oat2 protein at the basolateral membrane (Fig. 5C).

Immunohistochemistry analysis of OAT2 protein in human (A), monkey (B), and rat (C) proximal tubule epithelial cells. Paraffin-embedded sections (5 µm thickness) of kidneys were stained with OAT2 antibody. Representative immunostaining images show the localization of OAT2 in both basolateral and apical membranes of proximal tubule cells in sections from human and monkey kidneys, and in the apical membrane only in sections from rat kidney. GL, glomerulus; PT, proximal tubule.
Discussion
OCT2 and MATEs, localized to the basolateral and apical membrane of the proximal tubule, respectively, have been shown to act concertedly in the excretion of creatinine (Urakami et al., 2004; Tanihara et al., 2007, 2009; Imamura et al., 2011). More recently, Lepist et al. (2014) reported that OAT2 most likely played a relevant role in creatinine transport. The present study was undertaken to confirm and further explore the possible role of OAT2 in the renal excretion of creatinine.
To evaluate the role of OAT2 in the transport of creatinine and determine its relative contribution to creatinine renal excretion, OAT2 inhibition was assessed using transporter-expressing HEK cell lines. Of seven selected compounds tested, only BSP and IMC acted as potent inhibitors for OAT2-mediated creatinine-d3 uptake with apparent IC50 values of 1.3 ± 0.4 and 2.1 ± 0.4 μM, respectively (Fig. 3; Table 3). In contrast, [14C]metformin uptake in HEK cells expressing OCT2, MATE1, and MATE2K was not significantly inhibited by either BSP or IMC at concentrations up to 100 and 200 µM, respectively (Fig. 3, B and C). These results are consistent with the observations that IMC is a potent inhibitor of organic anion transporters OAT1 and OAT3, but not organic cation transporters OCT1 and OCT2 (Mulato et al., 2000; Khamdang et al., 2002; Takeda et al., 2002; Nozaki et al., 2007; Ingraham et al., 2014). IMC was previously reported to be a weak inhibitor for OAT2 with IC50 of 64.1 µM in mouse second segment (S2) proximal tubule cells stably expressing OAT2 (Khamdang et al., 2002). The difference in IC50 is probably due to the use of different systems (HEK cells versus S2 cells) and the probe substrate used (creatinine-d3 versus [3H]prostaglandin F2α). Furthermore, the presence of endogenous mouse Oat2, Oct2, and Mate1 in the S2 cells may complicate human OAT2 inhibition assessment. The propensity of BSP to inhibit OCT2, MATE1, and MATE2K has not been determined previously. To our knowledge, this is the first time potent inhibitors of OAT2 transporter have been reported. The results from the current study suggested that BSP and IMC were relatively selective inhibitors of OAT2 over other known creatinine transporters. These inhibitors can be used in experiments to provide new insight for the study of OAT2. Interestingly, it has been reported that IMC caused significant reduction in creatinine urine output with concomitant elevations of blood creatinine in various clinical studies (Berg and Talseth, 1985; Al-Waili, 2002). These effects in most neonates are transient, disappearing with cessation of therapy with IMC (Kang et al., 1999; Walker et al., 2011). However, IMC is a potent prostaglandin synthesis inhibitor, and the mechanisms other than transporter inhibition have been implicated in its inhibitory effects. In addition, although there are reports that IMC had no effect on creatinine renal clearance in humans (Levey et al., 1988; Prescott et al., 1990), the unchanged clearance could be confounded by consumption of a high-protein meal (Levey et al., 1988). Information about the inhibition of creatinine clearance by BSP in humans is lacking, because BSP is a contrast agent used as a diagnostic aid in liver function test.
In the present study, the uptake of creatinine-d3 in the OAT2-HEK cells and Mock-HEK cells was quantitated using LC-MS/MS. There have been few studies of the OAT2-specific handling of creatinine at a molecular level. Of those, Imamura et al. (2011) showed that creatinine was not a substrate for OAT2 in transfected S2 mouse kidney cells. In contrast, Lepist et al. (2014) reported that creatinine is a substrate for OAT2 expressed in Madin-Darby canine kidney II cells. They also reported that CMD inhibited OAT2-mediated cGMP uptake with an inhibitory constant of 72.6 ± 17.0 μM. Similarly, Ciarimboli et al. (2012) also reported OAT2-specific transport of creatinine using transporter-transfected HEK cells, although less intracellular accumulation was observed. In the present study, we confirmed the observation of Lepist et al. (2014) that creatinine was a substrate of OAT2 and this transporter was found to cause markedly greater accumulation of creatinine compared with OCT2, MATE1, or MATE2K (Fig. 2A). However, different experimental conditions between the transporter assays such as expression level and transport solution pH may complicate the comparison. A splice variant of OAT2 has also been reported (Cropp et al., 2008). The 6-bp insertion (TCCCAG) in cDNA between exons 1 and 2 of the registered National Center for Biotechnology Information reference sequence for OAT2 (NM_06672) resulted in an in-frame addition of two amino acids (Ser and Gln) in the OAT2 slice variant (i.e., OAT2-548aa). Both OAT2-546aa and OAT2-548aa isoforms are expressed at equal levels in the kidney, liver, and other tissues (Cropp et al., 2008). But, the small difference between the two isoforms produced a clear distinction in the activity of human OAT2, with the splice variant OAT2-548 resulting in complete loss of transport function. The mechanism for the loss of function of OAT2-548aa was related to a reduced expression level of the protein on the plasma membrane. When expressed in HEK cells, although the OAT2-546aa isoform trafficked to the plasma membrane and was functional, OAT2-548aa variant was retained in an intracellular compartment (Cropp et al., 2008). The OAT2-546aa isoform was capable of transporting various compounds, including cGMP, acyclovir, and penciclovir (Cheng et al., 2012). Taken together, these findings warrant the careful selection of OAT2 isoform for OAT2 investigation. Interestingly, it has been reported that human and animal OAT2 expression is highly regulated by cytokines, hormones, and the drugs that are activators of xenosensors, such as aryl hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, and nuclear factor E2-related factor 2 (Jigorel et al., 2006; Ljubojevic et al., 2007; Aoki et al., 2008; Chen et al., 2011; Noel et al., 2013). Additional experiments in our laboratory reveal that OAT2-HEK cells lose transport activity quickly when grown in culture (data not shown). This may be the result of protein regulation and possible trafficking from membrane to intracellular compartment. Therefore, cell culture conditions must be optimized for OAT2 assessment.
To compare the transport efficiency of creatinine by OAT2 with other renal creatinine transporters, we first determined kinetic parameters in OAT2-, OCT2-, MATE1-, and MATE2K-expressing HEK cells in relation to the absolute transporter protein expression levels. To our surprise, OAT2 demonstrated both the highest affinity and highest velocity for creatinine transport, and therefore the most efficient transport of creatinine among the transporters evaluated. The transporter efficiency as calculated by Vmax/Km was 31.4 µL/min/mg total protein for OAT2, 0.93 µL/min/mg total protein for OCT2, 0.10 µL/min/mg total protein for MATE1, and 0.14 µL/min/mg total protein for MATE2K (Fig. 2B; Table 2). In addition, when normalized for individual transporter protein levels, the creatinine transport efficiency was approximately 37- to 1850-fold higher for OAT2 than OCT2, MATE1, and MATE2K (Table 2).
To confirm the findings, we investigated creatinine transport in HRPTCs. The HRPTCs are a valuable tool for developing a system-level understanding of creatinine disposition, as the cells retain functionality and polarization for a number of transport systems, and are considered as a more physiologically relevant model. Some limitations are also anticipated, for example, limited availability of the cells, laborious preparation, low productivity, higher interdonor variability, and greater cost (Brown et al., 2008). If the isolated HRPTs are handled properly, the vast majority of renal transporters will be present in native environments at endogenous levels and natural localizations. Therefore, the HRPTC model offers a distinct advantage toward a system-level understanding of renal creatinine transporters, especially when the predominant clearance pathway(s) of creatinine are unknown. It has been suggested that the native environment provided by the isolated HRPTCs allows for more accurate evaluation of renal transporter inhibition (Brown et al., 2008). In HRPTC suspension, creatinine-d3 uptake was inhibited by OAT2 inhibitors BSP (50 µM) and IMC (100 µM). In the transporter-overexpressing cells, BSP and IMC almost completely abolished the uptake of creatinine-d3 into OAT2-HEK cells at concentrations of 50 µM and 100 µM, respectively (Fig. 3, B and C). To further explore the potential role of OAT2 in renal excretion of creatinine, the transcellular transport creatinine-d3 was assessed using primary cultures of freshly isolated HRPTCs in a Transwell plate. Consistent with HRPTC suspension results, the potent OAT2 inhibitors (50 µM BSP and 100 µM IMC) showed significant inhibition (∼40–50%, P < 0.05) of creatinine transport in the B-to-A or excretion direction. Taken together these data suggest that OAT2 plays a substantial role in creatinine renal excretion. In addition, we observed that the protein expression level of OAT2 is comparable to that of OCT2 and MATE1 in frozen human kidney cortex tissue and cryopreserved human renal proximal tubular cells using the LC-MS/MS method (unpublished observation). However, given the overlapping creatinine substrate and inhibition specificities and varying potencies of the inhibitors, we cannot rule out further contribution from other unknown transporters to creatinine transport across the basolateral and apical membranes of renal tubules.
Differing localization of OAT2 in human, monkey, and rat kidneys could suggest a potential for species difference in the renal handling of creatinine. Rat Oat2 protein was exclusively localized to the brush-border (apical) membranes of renal tubules (Fig. 5C). In contrast, the OAT2-specific staining is diffusively distributed to both basolateral and apical membranes of renal proximal tubules in human and cynomolgous monkey (Fig. 5, A and B). Fork et al. (2011) reported bidirectional transport of glutamate by OAT2. In addition, because glutamate is the most abundant intracellular amino acid (range from 2 to 20 mM), OAT2 effectively operates as a glutamate efflux carrier. It is notable that, even in the absence of extracellular substrates to be exchanged, the transporter functions to release glutamate from the cells (Fork et al., 2011); this may be one of the primary physiologic functions of OAT2. As OAT2-mediated transport is found to be bidirectional and OAT2 is the most efficient creatinine transporter among the transporters tested, the localization of OAT2 in both basolateral and apical membranes suggests that renal secretion of creatinine is most likely handled efficiently by OAT2 through initial basal influx of creatinine into proximal tubule cells, followed by apical efflux of creatinine in humans and monkeys. Imamura et al. (2011) reported that the renal clearance of creatinine on day 4 was less than the GFR in 8 of 11 healthy subjects after i.v. administration of 800 mg DX-619 once daily for 4 days, suggesting that creatinine has undergone reabsorption from the urine in these subjects; they further speculated that OAT4 may mediate the reabsorption of creatinine (Imamura et al., 2011). The current study indicated that creatinine is a high-affinity substrate for OAT2, and the transporter is expressed on both apical membrane of HRPTC and the basolateral membrane in humans and cynolmogus monkeys. These results imply that OAT2 may play a role in the reabsorption of creatinine from the lumen as well as basolateral uptake in both species. In contrast, in rats, creatinine renal secretion may be mediated by the basolateral organic cation transporter Oct1 and Oct2 and apical transporters Mate 1 and Oat2. Due to similiar OAT2 transporter expression, cynomolgus monkey is likely to be an appropriate preclinical model for studying OAT2 function in human kidney. It is worthwhile to note that species differences in substrate specificity and interactions of compounds with OAT2 have been reported. For example, mouse Oat2 did not mediate the transport of salicylate, despite the fact that salicylate is a good substrate of rat and human OAT2 (Sekine et al., 1998; Khamdang et al., 2002; Kobayashi et al., 2002). Rat Oat2-mediated uptake of salicylate was inhibited by ketoprofen, diclofenac, and ibuprofen with Ki values of 1.84, 49.3, and 155 µM, respectively, whereas the IC50 values for human OAT2 were 400, 14.3, and 692 µM, respectively (Morita et al., 2001; Khamdang et al., 2002). Further study to investigate the affinity of creatinine with rat and monkey Oat2 is warranted to understand the impact of species-dependent tubular localization of OAT2 in creatinine renal disposition. In addition, different subcellular localization of OAT2 protein in epithelial cells between species may be related to the driving force(s) for OAT2-mediated transport, which has not been clearly resolved. The OAT2 antibody used in this study is affinity isolated. There are two bands detected by the antibody in Western blotting analysis with human fetal liver tissue lysate, and molecular weight of the bands are approximately 50 and 20 kDa. As a result, it is unlikely that this OAT2 antibody cross-reacts with human OAT4 transporter. However, experiments with a different OAT2 antibody should be conducted to confirm the apical expression of OAT2 in human renal proximal tubules.
In summary, our investigations showed that creatinine is a good substrate for OAT2. The OAT2-mediated creatinine transport was far more efficient than other creatinine transporters, including OCT2, MATE1, and MATE2K, suggesting that OAT2 plays an important role in active renal-handling creatinine. In addition, the remarkable species differences in OAT2 localization on renal tubular cells most likely affects renal elimination of xenobiotics and endogenous compounds, including creatinine, between species. With the involvement of OAT2 in the elimination of toxic compounds and drugs, the present results may improve the interpretation of renal toxicity studies and aid in understanding drug disposition.
Authorship Contributions
Participated in research design: Shen, Morse, Lai.
Conducted experiments: Shen, T. Liu, Morse, Zhao, Zhang, Qiu, Chen, Lewin.
Contributed new reagents or analytic tools: Shen, T. Liu, Morse, Zhao.
Performed data analysis: Shen, T. Liu, Morse, Wang, G. Liu, Lai.
Wrote or contributed to the writing of the manuscript: Shen, Zhao, Christopher, Marathe, Lai.
Footnotes
- Received November 22, 2014.
- Accepted April 22, 2015.
This work was supported by Bristol-Myers Squibb Company.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- A-to-B
- apical to basolateral
- B-to-A
- basolateral to apical
- BSP
- bromosulfophthalein
- CMD
- cimetidine
- GFR
- glomerular filtration rate
- HBSS
- Hanks’ balanced salt solution
- HEK
- human embryonic kidney
- HRPTC
- human renal proximal tubule cell
- IMC
- indomethacin
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MATE
- multidrug and toxin extrusion protein
- OAT
- organic anion transporter
- OCT
- organic cation transporter
- PBS
- phosphate-buffered saline
- PCV
- penciclovir
- PT
- proximal tubule
- PYR
- pyrimethanmine
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}