


Abstract
Biotransformation pathways and the potential for drug-drug interactions of the orally active antifungal terbinafine were characterized using human liver microsomes and recombinant human cytochrome P-450s (CYPs). The terbinafine metabolites represented four major pathways: 1) N-demethylation, 2) deamination, 3) alkyl side chain oxidation, and 4) dihydrodiol formation. Michaelis-Menten kinetics for the pathways revealed meanKm values ranging from 4.4 to 27.8 μM, andVmax values of 9.8 to 82 nmol/h/mg protein. At least seven CYP enzymes are involved in terbinafine metabolism. Recombinant human CYPs predict that CYP2C9, CYP1A2, and CYP3A4 are the most important for total metabolism. N-demethylation is primarily mediated by CYP2C9, CYP2C8, and CYP1A2; dihydrodiol formation by CYP2C9 and CYP1A2; deamination by CYP3A4; and side chain oxidation equally by CYP1A2, CYP2C8, CYP2C9, and CYP2C19. Additionally, characteristic CYP substrates inhibited pathways of terbinafine metabolite formation, confirming the involvement of multiple enzymes. The deamination pathway was mainly inhibited by CYP3A inhibitors, including troleandomycin and azole antifungals. Dihydrodiol formation was inhibited by the CYP1A2 inhibitor furafylline. Terbinafine had little or no effect on the metabolism of many characteristic CYP substrates. Terbinafine, however, is a competitive inhibitor of the CYP2D6 reaction, dextromethorphan O-demethylation (Ki = 0.03 μM). In summary, terbinafine is metabolized by at least seven CYPs. The potential for terbinafine interaction with other drugs is predicted to be insignificant with the exception that it may inhibit the metabolism of CYP2D6 substrates. Clinical trials are needed to assess the relevance of these findings.
Terbinafine (Lamisil) is an orally active allylamine derivative that has fungicidal activity against dermatophytes and many other pathogenic fungi. The mechanism of terbinafine action is by interference with ergosterol biosynthesis through specific inhibition of lanosterol formation by blocking fungal squalene epoxidase (Ryder, 1989).
The allylamine structure of terbinafine is unrelated to the other leading antifungal agents, which include imidazoles such as ketoconazole and clotrimazole. These compounds also interfere with fungal ergosterol biosynthesis, however, by blocking lanosterol demethylation, which is a cytochrome P-450 (CYP)1-mediated reaction that also occurs in humans (Van den Bossche et al., 1978, 1980). CYP inhibition by azoles has been implicated in a number of drug-drug interactions. For example, ketoconazole is a potent inhibitor of the metabolism of cyclosporine A (CsA), midazolam, and warfarin (Ferguson et al., 1982; Smith, 1984;Shepard et al., 1986; Olkkola et al., 1994). Additionally, clinically important drug-drug interactions of fluconazole with drugs such as warfarin and CsA have been described (Lazar and Wilner, 1990).
Terbinafine is extensively distributed in humans, and its systemic clearance is primarily dependent on biotransformation (Jensen, 1989;Humbert et al., 1995). Terbinafine binds to and is metabolized by human liver microsomal CYP (Schuster, 1987); however, the isoenzymes responsible for its metabolism are not yet defined. The ability of terbinafine to inhibit CYPs seems to be limited. In human liver microsomal preparations, 50 to 100 μM terbinafine inhibited only marginally the metabolism of ethoxycoumarin (CYP1A2), tolbutamide (CYP2C9), or ethynylestradiol, CsA, and cortisol (CYP3A; Back et al., 1989, 1992; Shah et al., 1993). These findings are consistent with minimal or nonexistent interactions of orally administered terbinafine in clinical studies with the CYP1A2 substrates theophylline and caffeine (Wahllander and Paumgartner, 1989; Trepanier et al., 1998a,b), the CYP2C9 substrate warfarin (Guerret et al., 1997), and the CYP3A substrates terfenadine (Sorkin and Heel, 1985; Robbins et al., 1996), nifedipine (Cramer et al., 1996), triazolam (Varhe et al., 1996), and CsA (Long et al., 1994). Additionally, no or only negligible drug-drug interactions have been observed with antipyrine (Seyffer et al., 1989), which is metabolized by at least six human CYP isoenzymes (Engel et al., 1996). However, in a recent case report (Van der Kuy et al., 1998), increased plasma concentrations of the CYP2D6 substrate nortriptyline have been associated with coadministration of terbinafine. Additionally, when terbinafine was given with dextromethorphan, phenotypically extensive metabolizers for dextromethorphan became poor metabolizers (Leeder et al., 1998).
These data indicated that terbinafine is a likely inhibitor of CYP2D6. Nevertheless, this enzyme does not appear to be of importance for terbinafine elimination. Only cimetidine, which inhibits the elimination of many substrates metabolized by different CYPs (Klotz and Reimann, 1983), caused a 30% increase in terbinafine area under the plasma concentration time curve as well as a 30% decrease in plasma terbinafine clearance (Jensen, 1990). In this study, including one poor metabolizer for CYP2D6, there were no differences in the pharmacokinetics of terbinafine. Furthermore, no other coadministered compound has been reported to have a clinically significant and/or enzyme-specific effect on terbinafine metabolism.
The aim of this study was to further investigate the potential of orally administered terbinafine to inhibit the major human CYPs and to characterize the human isoenzymes responsible for terbinafine metabolism.
Materials and Methods
Chemicals.
[14C]Terbinafine (2.18 or 2.4 GBq/mmol) ((E)-N-(6,6-Dimethyl-2-hepten-4-ynyl)-N-methyl-1-naphthalene[14C]methenamine hydrochloride), unlabeled terbinafine, and the synthetic metabolitesN-desmethylterbinafine (D), hydroxyterbinafine (OL), desmethylhydroxyterbinafine (DOL), and carboxyterbinafine (CA) were synthesized by Novartis Pharma AG (Basel, Switzerland). The radiochemical purity of terbinafine was greater than 98% by HPLC. Other compounds obtained from Novartis included CsA, ketotifen, fluconazole, and (S)-mephenytoin. The metabolites 1-naphthoic acid (NA), 1-naphthalenemethanol (NM), and 1-naphtaldehyde (NAL) were obtained from Aldrich (Milwaukee, WI).
[3H]CsA (322 GBq/mmol), [14C]chlorzoxazone (2.18 GBq/mmol), [14C]tolbutamide (2.0 GBq/mmol), and [14C]S-mephenytoin (2.2 GBq/mmol) were obtained from Amersham International plc. (Little Chalfont, UK). [3H]Paclitaxel (618 GBq/mmol) was obtained from Moravec Biochemicals (Brea, CA) and [14C]theophylline (1.93 MBq/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO). [14C]Phenacetin (0.46 GBq/mmol), azidothymidine, chlorzoxazone, clotrimazole, dextromethorphan, disulfiram, ethinyl estradiol, glyburide, miconazole, nifedipine, orphenadrine, phenytoin, quinidine, terfenadine, theophylline and its metabolites (1-methyl uric acid, 1,3-dimethyl uric acid, 3-methyl xanthine, and 1-methyl xanthine), and tolbutamide were acquired from Sigma Chemical Co. (St. Louis, MO). 4-Hydroxymephenytoin, bufuralol, 1-hydroxybufuralol, furafylline, and sulfaphenazole were obtained from Ultrafine Chemicals (Manchester, UK). Itraconazole and ketoconazole were purchased from Janssen Biotech N.V. (Olen, Belgium), phenacetin from Fluka (Buchs, Switzerland), troleandomycin from Pfaltz and Bauer, Inc. (Waterbury, CT), and glibornuride from Roche (Basel, Switzerland). All other reagents used were of the highest grade available and purchased from commercial sources.
Biologicals.
Human liver tissue, not suitable for transplantation, was obtained from the International Institute for the Advancement of Medicine (Exton, PA) GGM002; from Vitron Inc. (Tucson, AZ) HL-4, HL-5, HL-44, HL-45, and HL-46; from In Vitro Technologies, Inc. (Baltimore, MD), HHM0011; and from the Novartis liver bank, M8 (Basel, Switzerland; Ball et al., 1992). Microsomes from livers GGM002 and M8 were prepared in house by differential centrifugation as described previously (Ball et al., 1992). Microsomal protein was determined by the Bradford method (Bradford, 1976) using bovine γ globulin as the standard (Bio-Rad, Glattbrug, Switzerland). CYP content was determined according to the method of Omura and Sato (1964). Total CYP content, expressed as nanomoles CYP per milligram microsomal protein, was 0.44 (GGM002), 0.20 (HL-4), 0.29 (HL-5), 0.23 (HL-44), 0.26 (HL-45), 0.37 (HL-46), 0.29 (M8), and 0.90 (HHM0011).
Baculovirus insect cell-expressed human CYP enzymes (BTI-TN-5B1–4), were available as microsomal preparations from Gentest Corp. (Woburn, MA). Enzyme content and relative activity factors (RAFs; Crespi, 1995) compared with human liver microsomes were as follows: CYP1A2 (185.18 pmol CYP/mg protein; RAF = 9 for phenacetinO-deethylase), CYP2A6 (175.43 pmol CYP/mg protein), CYP2B6 (128.20 pmol CYP/mg protein; RAF not available), CYP2C8 (500 pmol CYP/mg protein; RAF = 13 for paclitaxel 6α-hydroxylase), CYP2C9*1 (555.55 pmol CYP/mg protein; RAF = 4 for diclofenac 4′-hydroxylase), CYP2C19*1 (357.14 pmol CYP/mg protein; RAF = 230 for (S)-mephenytoin 4′-hydroxylase), CYP2D6 (172.41 pmol CYP/mg protein), CYP2E1 (588.23 pmol CYP/mg protein), CYP3A4 (99 pmol CYP/mg protein; RAF = 3.8 for testosterone 6β-hydroxylase). Human lymphoblast-expressed human CYP1A2 (140 pmol CYP/mg protein), CYP2A6 (140 pmol CYP/mg protein), CYP2B6 (86 pmol CYP/mg protein), CYP2C8 (28 pmol CYP/mg protein), CYP2C9R144 (47 pmol CYP/mg protein), CYP2C19*1 (14 pmol CYP/mg protein), CYP2D6 (42 pmol CYP/mg protein), CYP2E1 (150 pmol CYP/mg protein), and CYP3A4 (65 pmol CYP/mg protein) were also obtained from Gentest. Chinese hamster ovary cell-expressed human CYP3A5 was prepared as described previously (Fischer et al., 1998).
Human Liver Metabolism.
Microsomal incubations were carried out in 0.1 M phosphate buffer at pH 7.4 and 37°C in a shaking incubator. Terbinafine (in water) was added to 100 μg microsomal protein in a final volume of 200 (inhibition studies) or 600 μl (kinetic studies). Metabolism was initiated by the addition of 0.2 mM β-NADPH and a NADPH-regenerating system (final concentrations of 1 mM NADP+, 5 mM isocitrate, 5 mM MgCl2, and 1 U of isocitrate dehydrogenase). The reaction was stopped after 20 min by the addition of an equal volume of cold methanol on ice. The samples were stored at −20°C until analysis.
Chemical inhibitors were added in dimethyl sulfoxide, ethanol, acetonitrile and in 60 mM potassium hydroxide (chlorzoxazone), not to exceed 0.5% v/v. Incubations containing disulfiram, furafylline, or troleandomycin were preincubated in the presence of β-NADPH and regenerating system for 15 min at 37°C before the addition of terbinafine.
Recombinant Human CYP Microsomal Incubations.
[14C]Terbinafine (1, 5, 10, 20, 30, 50, 70, 90, and 100 μM) was incubated in 0.1 M phosphate buffer (pH 7.4, 200 μl) for 20 min at 37°C with microsomes from the expressed cells; CYP1A2 or CYP3A4 at 12.5 pmol CYP/incubation, CYP2A6, CYP2B6, CYP2D6, or CYP2E1 at 25 pmol CYP/incubation, CYP2C19 at 6.25 pmol CYP/incubation, or CYP2C8 or CYP2C9 at 16.6 pmol CYP/incubation. All reactions were stopped by the addition of 200 μl of cold methanol.
HPLC Analysis.
The proteins of all incubations were sedimented by centrifugation at 40,000g for 15 min at room temperature, and supernatant aliquots (200–350 μl) were used for analysis. The HPLC separations were performed on an Alliance (Waters, Milford, MA) or a Kontron system controlled by a 450 data system (Kontron Instruments, Zurich, Switzerland) with on-line detection using either a fluorescence detector F1000 (Merck, Darmstadt, Germany), or an LB 507A radioactivity monitor (Berthold AG, Wildbad, Germany). In addition, an Alliance (Waters) system with a Beta Ram detector (IN/US Inc., Tampa, FL) was used.
Terbinafine and its metabolites were separated on two columns connected in series (20 × 4.6 mm and 150 × 4.6 mm, 5 μm Supelcosil LC-18DB; Supelco Inc., Bellefonte, PA). The mobile phases consisted of 10 mM NH4HCO3 in water, pH 7.8, (solvent A) and acetonitrile (solvent B). The proportion of solvent B was increased linearly from 0% to reach 20% at 10 min, 50% at 70 min, and 100% at 90 min. The total flow rate was 0.8 ml/min and the column temperature was 35°C. Metabolite structures were assigned by cochromatography of synthetic reference compounds and UV detection at 210 and 280 nm, as well as by liquid chromatography-mass spectrometry (LC-MS) analysis. The quantitation of parent compound and metabolites of radiolabeled samples was calculated from the integration of the relative peak areas.
CYP marker substrates were incubated in the absence or presence of terbinafine (0–150 μM) with human liver microsomes and analyzed as described previously (Kronbach et al., 1988; Fischer et al., 1994,1998). Interference of terbinafine metabolites with dextrorphan determinations by fluorescence detection was corrected for using identical experiments in the absence of dextromethorphan.
Mass Spectrometric Analysis.
LC-MS of terbinafine metabolites was performed on a TSQ 7000 triple stage quadrupole mass spectrometer (Finnigan MAT, San Jose, CA) equipped with an electrospray ionization (ESI) LC-MS interface. The HPLC conditions were as described above; however, after the column the total flow was split such that 70% of the volume went to the liquid scintillation detector and 30% to the ESI interface. The latter was operated with methanol as sheath liquid (0.2 ml/min) and nitrogen as sheath (40 psi) and auxiliary gas (5 flowmeter units). The ESI spray voltage was 4.5 kV and the transfer capillary was heated to 210°C. Single-stage mass spectra were taken at unit mass resolution by using the first quadrupole as mass analyzer. For declustering and/or induction of fragmentation in the ion source region, an offset voltage of 29 V (positive ions) or 10 V (negative ions) between the skimmer and the transfer-octopole was applied.
Data Analysis.
Metabolic rates were calculated from mean substrate concentrations over the incubation period. IC50 values were determined graphically by plotting the percentage of the control activity against the inhibitor concentration. Michaelis-Menten parameters Km andVmax were determined by nonlinear curve fitting using Fig.P (Biosoft, Cambridge, UK) with the following equation: V =Vmax · [S]/Km+ [S].
Human liver microsomal velocities and the intrinsic clearance were predicted from terbinafine metabolite formation by individually expressed human CYPs using RAF values as defined by Crespi (1995).
Ki values were calculated using a model for mixed-type inhibition by the following equation (Segel, 1993):Ki = [I]/[(Km,i/Km,u)(Vmax,u/Vmax,i) − 1], where Km,i andKm,u are the Michaelis-Menten constants andVmax,i andVmax,u are the maximal velocities in the presence and absence of inhibitor, respectively. The in vitro intrinsic clearance was calculated by dividing the maximal rate of metabolism by the Michaelis-Menten constant, according to the equation: CLint =Vmax/Km.
Human liver in vivo intrinsic metabolic clearance was predicted from in vitro microsomal clearance assuming an adult body weight of 70 kg, a liver weight of 1.69 kg, and a yield of 52.5 mg of microsomal protein from 1 g of human liver (Iwatsubo et al., 1997).
Results
Biotransformation Pathways.
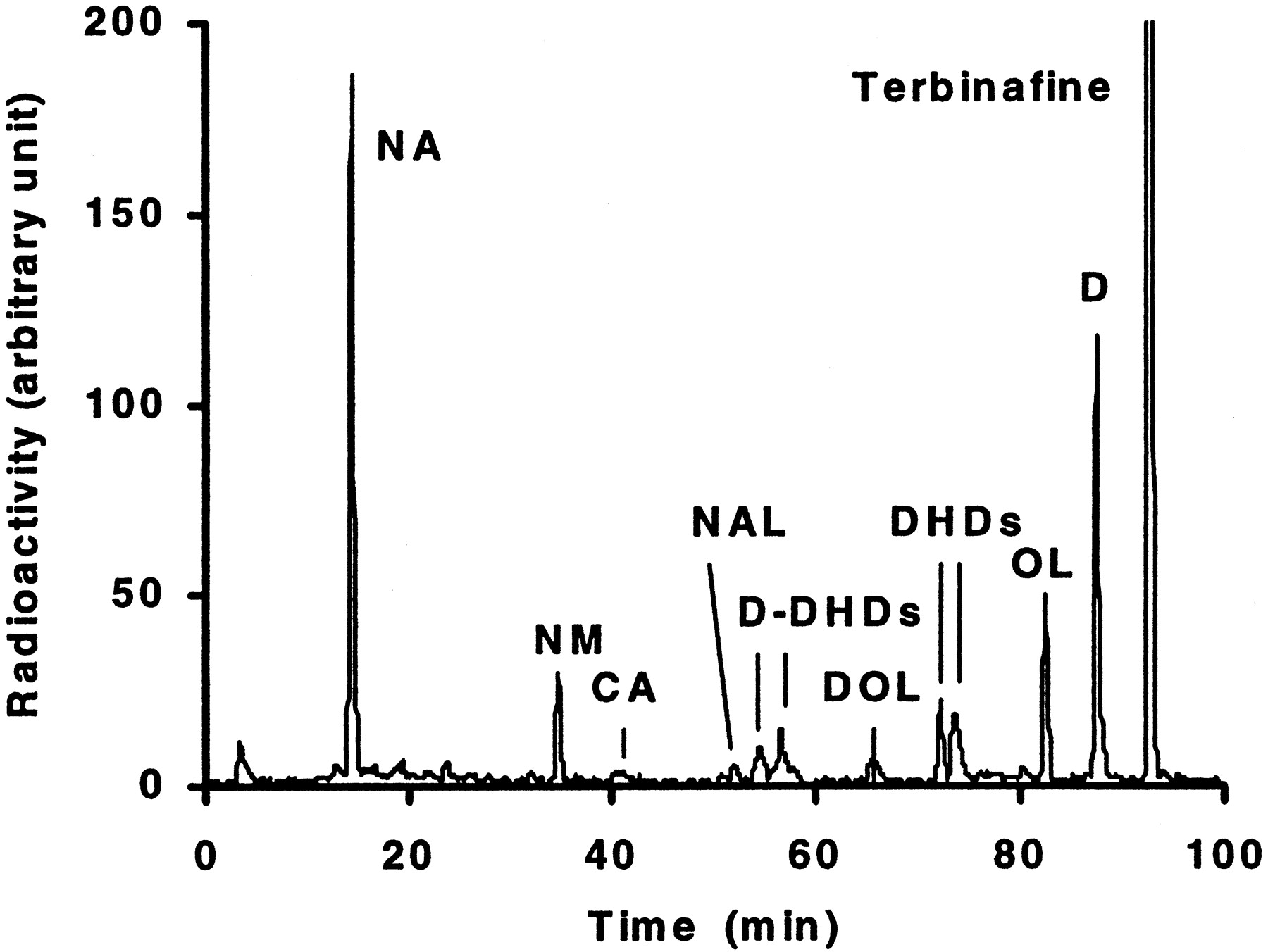
Metabolism of 20 μM [14C]terbinafine by human liver microsomal preparations resulted in several chromatographically well separated peaks (Fig. 1). The metabolite structures were assigned by cochromatography with synthetic reference compounds and/or by LC-MS (Table1). The main pathways of terbinafine metabolism by human liver microsomes were: 1)N-demethylation (metabolites D, DOL,d-DHDs); 2) deamination forming NAL followed by oxidation to NA or reduction to NM; 3) alkyl side chain oxidation, initially by hydroxylation at the tert-butyl group (metabolites OL, DOL) followed by further oxidation to the carboxylic acid; and 4) formation of arene oxides (not observed) followed by rapid hydrolysis to form dihydrodiols (DHDs, d-DHDs; Fig. 2).

Terbinafine metabolite separation by HPLC with radioactivity monitoring.
Typical HPLC radiochromatogram of 20 μM [14C]terbinafine and its metabolites formed by human liver microsomes incubated for 20 min at 37°C.
Mass spectrometric data from LC-ESI/MS analyses of pooled human liver microsomal incubates (20 μM, 20 min)

Proposed metabolic pathways of terbinafine in human liver microsomes and human CYPs.
The mass spectra of the metabolites revealed molecular weights (M + H+/[M − H]−) as well as structural details (Table 1) and were in agreement with those of reference compounds (data not shown). The prominent cleavage of the C-N bond next to the ring system (fragment A) allowed localizing metabolic changes in one or the other half of the molecule. This fragmentation was less pronounced in the case of dihydrodiols, likely due to the loss of aromaticity in one of the two rings of the naphthalene system, but was followed by a typical dehydration. The14C labeling (88%) resulted in characteristic doublets in the mass spectra, confirming that the observed components were indeed terbinafine-related. Metabolites NAL and NM did not produce signals in ESI-MS but eluted at identical retention times as the corresponding synthetic references.
Enzyme Kinetics.
The rate of [14C]terbinafine metabolite formation was determined over the concentration range of 2 to 220 μM in three human liver microsomal preparations (GGM002, HL-44, and HL-46). The metabolite formation exhibited monophasic Michaelis-Menten kinetics (Fig. 3) in all three human livers. The apparent Michaelis constant (Km) for total metabolism was about 20 μM. The maximal velocity of total metabolite formation (Vmax) varied 2-fold with a mean of approximately 180 nmol/h/mg protein (Table2). The resulting mean in vitro intrinsic metabolic liver clearance calculated from these parameters was 8.9 ml/h/mg microsomal protein, or scaled to in vivo, 13 liters/min.

Kinetics of terbinafine metabolism in human liver microsomes.
[14C]Terbinafine (2–220 μM) was incubated with human liver microsomes (GGM002). Terbinafine metabolism via the major routes was analyzed by nonlinear regression analysis as described inMaterials and Methods.
Enzyme kinetic parameters for terbinafine metabolism
The contribution of the four metabolic pathways of terbinafine metabolism i.e., N-demethylation (D + DOL +d − DHDs), deamination (NAL + NM + NA), alkyl side chain oxidation (OL + CA + DOL), and dihydrodiol (DHDs + d − DHDs) formation was also assessed by calculating the respective in vitro intrinsic clearance values. Mean Km values ranged from 4.4 to 27.8 μM with dihydrodiol formation, exhibiting the lowest value, followed by side chain oxidation, N-demethylation, and deamination. Maximal pathway velocities followed the same rank order, resulting in similar clearance values for all four pathways.N-demethylation and deamination were thus estimated to contribute ∼30% each to the total in vitro intrinsic clearance and contribute to ∼20% each side chain oxidation and dihydrodiol formation.
Similar contributions of each pathway were found when [14C]terbinafine (6 μM) metabolism was screened in a series of human liver microsomal preparations to estimate the variation in human metabolism (Table3). Total terbinafine metabolite formation varied ∼2.5-fold, whereas side chain oxidation varied ∼6-fold, N-demethylation 2.5-fold, and deamination and dihydrodiol formation ∼4-fold.
Terbinafine (6 μM) metabolism in seven human liver microsomal preparations
Terbinafine Metabolism by Recombinant Human CYPs.
Seven of the 10 human baculovirus insect cell-expressed CYP isoenzymes investigated clearly metabolized [14C]terbinafine (Fig.4, CYP3A5 data not shown). Only CYP2E1, CYP2D6, and CYP2A6 (0.125 nmol CYP/ml) did not result in any detectable [14C]terbinafine (50 μM) metabolism. TheKm values for total terbinafine metabolite formation by the recombinant CYPs were 3.9 to 18.0 μM (Table4), which was similar to values from human liver microsomes. The most efficient enzyme (Vmax/Km) for total terbinafine metabolite formation was CYP2C19, followed equally by CYP1A2 and CYP3A4 and then by CYP2C8 and CYP2C9. The least effective CYP in metabolizing terbinafine was CYP2B6. The most efficient in forming the dihydrodiols was CYP1A2 and to a lesser extent CYP2C9. The terbinafine pathway of side chain oxidation was formed by CYP2C19 followed by CYP2C8 and CYP1A2. The N-demethylation pathway was produced by CYP2C19 and CYP1A2 followed by CYP2C9, CYP2C8, CYP3A4, and CYP2B6. Terbinafine deamination was mainly detected in CYP2C19 and CYP3A4. No difference in metabolite formation was observed when lymphoblast-expressed enzymes were used, except for dihydrodiol formation, which required epoxide hydrolase (data not shown). CYP3A5 produced identical metabolites as 3A4 (data not shown); however, a rate calculation was not possible because the CYP in these cells was spectrally not detectable.

Terbinafine metabolite profile in recombinant human CYPs.
HPLC separation with radioactivity monitoring of terbinafine and its metabolites formed in incubations of 20 μM [14C]terbinafine during 20 min with baculovirus insect cell-expressed CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4.
Individual enzyme kinetic parameters for terbinafine metabolism
A prediction of CYP isoenzyme contribution toward terbinafine metabolite formation was based on the RAF for each isoenzyme and revealed that CYP2C9 would contribute the most to human liver terbinafine metabolism followed by CYP1A2, CYP3A4, and CYP2C8. The least important isoenzyme for terbinafine metabolite formation is predicted to be CYP2C19. For the individual metabolic pathways of terbinafine, dihydrodiol formation is predicted to be catalyzed primarily by CYP2C9 and CYP1A2, N-demethylation mainly by CYP2C9, CYP2C8, and CYP1A2, deamination mainly by CYP3A4, and side chain oxidation about equally by CYP2C8, CYP1A2, CYP2C19, and CYP2C9. The contribution of CYP2B6 was not possible to assess because of the unavailability of a specific reference activity.
Inhibition of Terbinafine Metabolism.
The effect of 25 characteristic CYP isoenzyme inhibitors and potentially coadministered compounds toward 20 μM [14C]terbinafine metabolism was investigated (Table 5). At low concentrations no compound could inhibit all pathways of terbinafine metabolism, indicating a potential lack for clinically significant findings. Three compounds could inhibit all pathways of terbinafine metabolite formation at higher concentrations. Disulfiram, an inhibitor of several CYPs (Chang et al., 1994), reduced terbinafine metabolite formation ∼50% at 10 to 25 μM and 90% at >100 μM. Nifedipine inhibited CYP1A2- and CYP2C9-mediated dihydrodiol formation (IC50 ∼15 μM) and inhibited total metabolite formation (90%) at 100 μM. High concentrations of terfenadine (200 μM) inhibited terbinafine metabolite formation (77%), and the inhibition could not be attributed to a specific enzyme. Azole type antifungals inhibited total terbinafine metabolism (30–72%) only at the highest concentrations investigated. The azole antifungals had little or no effect on dihydrodiol formation or the side chain oxidation, moderate effects on N-demethylation, and were potent inhibitors for the CYP3A-mediated deamination pathway. Fluconazole also exhibited a 50% inhibition of the CYP3A-mediated deamination pathway at 50 μM. Other known CYP3A inhibitors were also effective in the inhibition of the deamination pathway, CsA (IC50 ∼12 μM) and troleandomycin (IC50 ∼2 μM). In general, specific CYP inhibitors were found to inhibit a particular pathway of terbinafine metabolite formation but not to substantially reduce total metabolite formation. The CYP1A2 mechanism-based inhibitor furafylline effectively inhibited formation of the dihydrodiols (IC50 <1 μM), but had little effect on the other terbinafine pathways. Sulfaphenazole, a specific inhibitor of CYP2C9 at low micromolar concentrations (Baldwin et al., 1995), resulted in ∼50% inhibition of all pathways at or above 100 μM. These results are consistent with multiple enzymes involved in terbinafine metabolism. When the specific inhibitors were used with the corresponding single CYP, they inhibited terbinafine (20 μM) metabolite formation at the expected potency: furafylline (IC50 ∼2 μM, CYP1A2), sulfaphenazole (IC50 ∼0.1 μM, CYP2C9), ketoconazole (IC50 ∼0.5 μM, CYP3A4), and CsA (IC50 ∼3 μM, CYP3A4).
Inhibition of terbinafine metabolism
Effect of Terbinafine on CYP Probe Substrates.
Terbinafine (1–150 μM) had little or no effect on the metabolism of many characteristic CYP substrates (Table6). CYP1A2 catalyzed phenacetinO-deethylation, or theophylline metabolism was inhibited ∼50% at 30 and 50 μM terbinafine, respectively. Additional CYP-dependent pathways investigated, including (S)-mephenytoin 4′-hydroxylation (CYP2C19), paclitaxel 6α-hydroxylation (CYP2C8), tolbutamide 4-hydroxylation (CYP2C9), chlorzoxazone 6-hydroxylation (CYP2E1), and CsA metabolism (CYP3A), exhibited IC50 values of ∼100 μM or greater. Of these substrates only CsA metabolite formation was reduced (40% at 50 μM terbinafine) but not sufficiently to attain an IC50 value.
Effect of terbinafine on the metabolism of characteristic CYP substrates in human liver microsomes
Terbinafine proved to be a potent inhibitor of the CYP2D6-mediated dextromethorphan O-demethylation and bufuralol 1-hydroxylation with IC50 values of 0.2 and 0.25 μM, respectively (Fig. 5). The potential of CYP2D6 inhibition by the primary metabolites D and OL was also investigated because each possess a basic nitrogen, which is a requirement for binding to CYP2D6 (Koymans et al., 1992). Both D and OL displayed similar IC50 values toward the CYP2D6 substrates as terbinafine.

Inhibition of dextromethorphan O-demethylation by terbinafine.
Dextromethorphan (5 μM) was incubated with human liver microsomes (HHM0011) in the absence or presence of 0 to 5 μM either terbinafine or metabolites D or OL. Data are presented as the percentage of control incubations in the absence of terbinafine or its metabolites.
The mode of terbinafine inhibition on CYP2D6 was characterized by determining the kinetic parameters of dextromethorphanO-demethylation in the presence of a fixed concentration of the inhibitor terbinafine (0.5, 2, 5, 10 μM). Terbinafine was a competitive inhibitor of dextromethorphan O-demethylation. The Km of dextromethorphanO-demethylation was increased ∼30-fold in the presence of 10 μM terbinafine. The Ki value of 0.03 μM indicates that terbinafine is a very potent inhibitor for CYP2D6 (Fig. 6).

Inhibition of dextromethorphan O-demethylation by terbinafine.
Dextromethorphan (1, 2, 5, 10, 20, 40, and 80 μM) was incubated with human liver microsomes (HHM0011) in the absence or presence of 0 to 10 μM terbinafine. Data are presented according to Lineweaver-Burk.
Discussion
The metabolites of terbinafine formed by human liver microsomes, including products of N-demethylation, deamination, alkyl side chain oxidation, and dihydrodiol formation, were found previously in human plasma and urine (Jensen, 1989; Humbert et al., 1995). Terbinafine metabolites are formed by at least seven human CYP isoenzymes. This explains why no changes in terbinafine concentrations were found upon coadministration of other compounds, with the exception of the general inhibitor cimetidine (Jensen, 1990).
The human in vitro findings from this study support the known in vivo results. Only compounds that can inhibit several enzymes such as disulfiram and most azole antifungals were relatively potent inhibitors of all pathways. A specific inhibition of a particular pathway was only found for the dihydrodiol formation by furafylline (CYP1A2) and for the inhibition of the deamination reaction by CYP3A inhibitors such as ketoconazole and clotrimazole. Nevertheless, for these compounds, the inhibition (IC50) was less pronounced than expected based on the potential of these compound to inhibit CYP1A2- and CYP3A-catalyzed reactions. For example, for ketoconazole,Ki values of 0.01 to 0.04 μM have been reported for the inhibition of specific CYP3A-catalyzed reactions (Fischer et al., 1998; Gibbs et al., 1999). For most other inhibitors, the difference of the observed inhibitory constants to the potency of the compounds toward inhibition of specific enzymes was even greater. The reason for this difference is most likely the involvement of multiple enzymes in all pathways of terbinafine metabolism. In individual, recombinant enzyme systems the inhibitory constants were consistent with literature values for the specific isoenzymes.
The involvement of multiple human CYP isoenzymes in terbinafine metabolism was further confirmed by using recombinant single human CYPs. The contributions of the individual enzymes to in vitro human liver microsomal intrinsic clearance was estimated using the RAFs of enzyme-specific substrates, except for CYP2B6, for which a specific substrate has only recently been identified (Ekins et al., 1998). CYP2B6, however, has a low abundance in human liver (Shimada et al., 1994) and is, therefore, not expected to be a major contributor to terbinafine metabolism. The recombinant enzyme data predicted CYP2C9 to be the major enzyme in terbinafine metabolism in contrast to the lack of chemical inhibition by the specific CYP2C9 inhibitor sulfaphenazole (Baldwin et al., 1995). Dihydrodiol formation by baculovirus insect cell-expressed CYP1A2 was consistent with the inhibition of this pathway by the CYP1A2 inhibitor furafylline in human liver microsomes. Also CYP3A4 was predicted from recombinant enzymes to be the major enzyme catalyzing deamination and typical CYP3A inhibitors such as ketoconazole or troleandomycin inhibited deamination.
Terbinafine does not inhibit most major CYP enzymes at clinically relevant concentrations. However, terbinafine inhibits CYP2D6 competitively. Furthermore, the two metabolites tested (D and OL), which contain the basic nitrogen required for metabolism by CYP2D6 (Koymans et al., 1992), were also capable of inhibiting CYP2D6. These data could explain the case report whereby a reduction of dose was required for the CYP2D6 substrate nortriptyline in the presence of terbinafine (Van der Kuy et al., 1998). In another study, phenotypically extensive metabolizers for dextromethorphan were converted into poor metabolizers in the presence of terbinafine (Leeder et al., 1998). However terbinafine and its metabolites are ∼99% plasma protein-bound (Jensen, 1989), which could reduce the clinical relevance of these findings as indicated by the low number of reports indicating potential interactions with CYP2D6 in spite of more than 10 million patients treated with oral terbinafine.
In summary, terbinafine is metabolized by multiple enzymes but inhibits only CYP2D6 at relatively low concentrations. Based on these data, which are consistent with clinical observations, terbinafine elimination is not likely to be affected by comedication. The clinical relevance for the inhibition of elimination of CYP2D6 substrates will be determined in ongoing clinical trials.
Footnotes
-
Send reprint requests to: Dr. Volker Fischer, Novartis Institute for Biomedical Research, 59 Route 10, East Hanover, NJ 07936. E-mail: volker.fischer{at}pharma.novartis.com
- Abbreviations used are::
- CYP
- cytochrome P-450
- CsA
- cyclosporine A
- ESI
- electrospray ionization
- RAF
- relative activity factor
- LC-MS
- liquid chromatography-mass spectrometry
- D
- desmethylterbinafine
- OL
- hydroxyterbinafine
- DOL
- desmethylhydroxyterbinafine
- CA
- carboxyterbinafine
- NA
- 1-naphthoic acid
- NM
- 1-naphthalenemethanol
- NAL
- 1-naphtaldehyde
- Received February 17, 1999.
- Accepted May 20, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}