


Abstract
Losartan, an angiotensin II receptor antagonist, is oxidized by hepatic cytochromes P450 to an active carboxylic acid metabolite, E-3174. The aim of the present investigation was to study the contribution of CYP2C9 and CYP3A4 in losartan oxidation in vitro and to evaluate the role of CYP2C9 polymorphism. Kinetic properties of different genetic CYP2C9 variants were compared both in a yeast expression system and in 25 different samples of human liver microsomes where all known genotypes of CYP2C9 were represented. Microsomes were incubated with losartan (0.05–50 μM), and the formation of E-3174 was analyzed by high-performance liquid chromatography to estimate Vmax,Km, and intrinsic clearance for all individual samples. Sulfaphenazole, a CYP2C9 inhibitor, blocked the formation of E-3174 at low losartan concentrations (<1 μM), whereas the inhibitory effect of triacetyloleandomycin, a CYP3A4 inhibitor, was significant only at high concentrations of losartan (>25 μM). In comparison to the CYP2C9.1 variant, oxidation of losartan was significantly reduced in yeast expressing the rare CYP2C9.2 or CYP2C9.3 variants. Moreover, the rate of losartan oxidation was lower in liver microsomes from individuals hetero- or homozygous for theCYP2C9*3 allele, or homozygous for theCYP2C9*2 allele. The difference between the common and rare CYP2C9 variants was mainly explained by a lowerVmax, both in yeast and human liver microsomes. In summary, these in vitro results indicate that CYP2C9 is the major human P450 isoenzyme responsible for losartan oxidation and that the CYP2C9 genotype contributes to interindividual differences in losartan oxidation and activation.
Losartan is the first, selective angiotensin II (AT1-subtype) receptor antagonist to be used in the treatment of hypertension and congestive heart failure (Timmermans et al., 1993). In vitro (Stearns et al., 1995; Yun et al., 1995) and in vivo (Kaukonen et al., 1998; McCrea et al., 1999; Meadowcroft et al., 1999) studies have demonstrated that losartan is metabolized by hepatic cytochrome P450 enzymes. Among seven oxidative and glucuronide metabolites, only the 5-carboxylic acid metabolite E-3174 has higher potency and longer half-life than losartan and is therefore responsible for most of the antihypertensive effect (Lo et al., 1995). In vitro experiments with human liver microsomes and specific inhibitors of different CYP1enzymes indicated a role for CYP2C9 (Stearns et al., 1995) and CYP3A4 (Yun et al., 1995) in the oxidation of losartan. Furthermore, in vivo studies revealed that fluconazole, an inhibitor of both CYP2C9 and CYP3A4, decreased the metabolism of losartan to E-3174 (Kazierad et al., 1997; Kaukonen et al., 1998). However, itraconazole, a CYP3A4-selective inhibitor, had no significant effect on losartan oxidation in vivo (Kaukonen et al., 1998). Studies in human volunteers have ruled out the importance of the polymorphic CYP2D6 and CYP2C19 enzymes in the metabolism of losartan (Sandwall et al., 1999).
CYP2C9 is a polymorphic enzyme responsible for the metabolism of a large number of clinically important drugs such asS-warfarin, phenytoin, tolbutamide, torsemide, and numerous nonsteroidal anti-inflammatory drugs (Miners and Birkett, 1998). TheCYP2C9*2 and CYP2C9*3 alleles include single nucleotide polymorphisms in exon 3 and exon 7 that cause amino acid substitutions Arg144Cys and Ile359Leu, respectively. The allele frequencies of CYP2C9*1, CYP2C9*2, andCYP2C9*3 have been reported to vary in the range 0.79 to 0.86, 0.08 to 0.125, and 0.03 to 0.085, respectively, in Caucasians (Miners and Birkett, 1998; Yasar et al., 1999, and references therein).
The Arg144Cys substitution in CYP2C9.2 has been suggested to affect the interaction between the P450 enzyme molecule and P450 reductase (Crespi and Miller, 1997), which might explain a slower metabolism of some CYP2C9 substrates such as S-warfarin and tolbutamide (Sullivan-Klose et al., 1996; Miners and Birkett, 1998; Aithal et al., 1999). It appears, however, that the Ile359Leu substitution in CYP2C9.3 is of greater importance in terms of slower drug metabolism, at least according to several in vitro studies (Miners and Birkett, 1998;Yamazaki et al., 1998a,b; Takanashi et al., 2000). Residue 359 is located in the CYP2C9 active site, where it participates in substrate recognition (Gotoh, 1992). However, for a given CYP2C9 substrate it is difficult to predict whether, and to what extent, the CYP2C9 polymorphism is of clinical significance. This is particularly evident when there are other P450s catalyzing the metabolism of the same drug.
The objective of the present study was to clarify the roles of CYP2C9 and CYP3A4, and in particular, the importance of the CYP2C9polymorphism in the oxidation of losartan in vitro.
Materials and Methods
Losartan and E-3174 were kindly provided by Merck Sharp and Dohme (West Point, PA). Diclofenac, 3′- hydroxy (OH)-diclofenac, 4′-OH-diclofenac, 5′-OH-diclofenac, and 3′-OH-4′-methoxy-diclofenac were kindly supplied by Novartis (Basel, Switzerland). Ketoconazole was purchased from Janssen Biotech NV (Olen, Belgium); restriction enzymesEcoRI, SalI, and SacI from New England Biolabs (Herts, UK); and primers from Life Technologies (Gaithersburg, MD).N,N,N′,N′-Tetramethylethylenediamine, ammonium persulfate, sulfaphenazole (SPZ), triacetyloleandomycin (TAO), NADPH, antihuman IgG-horseradish peroxidase conjugate,p-coumaric acid, and luminol were purchased from Sigma (St. Louis, MO). Acetonitrile, ortho-phosphoric acid, and potassium phosphate were from Merck; SDS from Bio-Rad (Hercules, CA); bis-acrylamide from Severn Biotech (Worcestershire, UK); acrylamide from Scotlab (Luton, UK); and bactopeptone, casamino acids, nitrogen base, and tryptophan from Becton Dickinson (Sparks, MD).
Site-Directed Mutagenesis
Arg144Cys (CYP2C9*2) and Ile359Leu (CYP2C9*3) mutants of CYP2C9 were constructed from aCYP2C9*1 cDNA (generous gift from Charlotta Otter, AstraZeneca R&D, Umeå, Sweden) with a USE mutagenesis kit (Amersham Pharmacia Biotech, Uppsala, Sweden). Mutations were introduced into a CYP2C9 cDNA cloned into a pBlue/script/KS vector (Stratagene, La Jolla, CA) using mutagenic primers (mutations in bold) 5′-GCATTGAGGACTGTGTTCAAGAGG-3′ for CYP2C9*2 and 5′-CCAGAGATACCTTGACCTTCTCCCCACC-3′ for CYP2C9*3(Oscarson et al., 1997). Sequencing of mutant cDNAs was performed using an ABI Prism BigDye terminator kit and analyzed on an ABI Prism 377 DNA sequencer. The variant cDNAs were later subcloned into pYeDP60 (V60) yeast expression vector (Urban et al., 1990, kindly provided by Dr. Denis Pompon, Gif-sur-Yvette, France) using the restriction enzyme sites EcoRI and SacI. Correct mutagenesis was finally confirmed by DNA sequencing.
Expression of Variant cDNAs in Yeast Cells
CYP2C9*1, *2, and *3 cloned in the V60 vector were expressed in Saccharomyces cerevisiae strain W(R) that overexpresses yeast reductase kindly provided by Dr. Denis Pompon. Culturing conditions for the yeast and preparation of microsomes were performed essentially as described previously (Oscarson et al., 1997). Following mechanical disruption of cell walls, microsomes were isolated by ultracentrifugation at 100,000gfor 60 min, and resuspended in TEG buffer (50 mM Tris-HCl, 1 mM EDTA, 20% glycerol, pH 7.4).
Determination of CYP2C9 Holoenzyme and P450 Reductase
The CYP2C9 content in yeast microsomes was determined by measuring the reduced CO/spectrum (Omura and Sato, 1964). Total protein content (Lowry et al., 1951), and reductase levels (Yasukochi and Masters, 1976) were determined according to previously described methods.
Preparation of Human Liver Microsomes
Microsomes of 25 healthy organ donor livers were prepared from the liver bank (approved by the Ethical Review Board) established at the Department of Clinical Pharmacology in Huddinge University Hospital as described earlier (von Bahr et al., 1980). The protein content was estimated according to Lowry et al. (1951) using bovine serum albumin as standard and human albumin as a quality control. The microsomes were stored in potassium phosphate buffer (50 mM, pH 7.4) at −80°C until use.
Genotyping of DNAs Isolated from Human Liver Tissues
QIAamp Tissue DNA preparation kit (Qiagen GmbH, Hilden, Germany) was used to isolate genomic DNA from human liver tissue. DNA samples from the livers used in the study were genotyped for theCYP2C9*2 and CYP2C9*3 alleles using a previously validated method (Sullivan-Klose et al., 1996; Yasar et al., 1999).
Analysis of Enzyme Kinetics
Diclofenac 4′-Hydroxylation in Yeast Microsomes.
Yeast microsomes corresponding to 10 pmol of CYP2C9 were incubated with diclofenac in the presence of NADPH (1 mM) at 37°C for 6 min in a total volume of 500 μl of potassium phosphate buffer (50 mM) at pH 7.4. Diclofenac was dissolved in water and 10 different concentrations ranging from 1 to 100 μM were used. The reactions were terminated by addition of 100 μl of 30% acetonitrile and freezing in dry ice. The chromatographic system consisted of a UV detector operating at 280 nm, two pumps, and a dual valve sampling injector for on-line column switching. After centrifugation at 5000g for 10 min, 100 μl of the sample was directly injected onto a Biotrap column for on-line extraction using a Biotrap C18 column (20 × 4 mm; ChromTech, Cheshire, UK) (Hermansson et al., 1998). The extraction mobile phase (A) consisted of 20 mM phosphoric acid, pH 2.1, and the flow rate was 1 ml/min with an extraction time of 3 min. Chromatographic separation was achieved isocraticly with a mobile phase of acetonitrile/20 mM phosphoric acid, pH 2.1 (42:58, v/v) on a Zorbax SP-phenyl column (250 × 4.6 mm) connected with a precolumn. The flow rate was 1.0 ml/min and after 13 min under isochratic conditions, a linear gradient started where the acetonitrile increased to 80%. The total run time was 20 min. 4′-OH-Diclofenac was dissolved in water with addition of 0.2% ammonia and a standard curve was prepared in the concentration range 50 to 2000 nM. The limit of quantification was 50 nM and the interday coefficient of variation was 7 and 5% at a concentration of 0.3 and 1 μM, respectively. Formation of 4′-OH-diclofenac was quantified using pure 4′-OH metabolite as standard and was in the linear range between 2 and 15 min and 5 and 30 pmol of CYP2C9.
Losartan Oxidation in Yeast Microsomes.
Yeast microsomes corresponding to 20 pmol of CYP2C9 were incubated with losartan in the presence of NADPH (1 mM) at 37°C for 10 min in a total volume of 500 μl of potassium phosphate buffer (50 mM) at pH 7.4. Ten different losartan concentrations were used in the range of 0.05 to 50 μM. Reactions were terminated by the addition of 50 μl of ortho-phosphoric acid (5 M), followed by centrifugation at 15,000g for 10 min. The resulting supernatant (450 μl) was mixed with 50 μl of isopropanol and injected (without any further extraction) into an HPLC system including a fluorescence detection method essentially as described by Ritter et al. (1997). The limit of detection was 5 nM. Formation of E-3174 was in the linear range between 2 and 15 min and 5 and 40 pmol of enzyme. The identity of E-3174 and losartan peaks in HPLC was confirmed by direct collection from HPLC followed by liquid chromatography/mass spectrometry analysis (data not shown).
Losartan Oxidation in Human Liver Microsomes.
Microsomes, corresponding to 600 μg of protein per 0.5 ml of phosphate buffer, from 25 different human livers with characterized CYP2C9 genotypes were incubated with losartan at 10 different concentrations from 0.05 to 50 μM at 37°C for 15 min. Formation of E-3174 was in the linear range between 5 and 30 min and 0.1 and 1.5 mg of protein. Inhibition experiments were carried out by pretreatment with the CYP3A4-specific inhibitor TAO (10 μM; Chang et al., 1994;Yamazaki and Shimada, 1998) or the CYP2C9-specific inhibitor SPZ (5 μM; Bloomer et al., 1994) in the presence of NADPH (1 mM) for 4 to 5 min at 37°C. The rate of E-3174 formation in different genotypes was compared at a losartan concentration of 0.5 μM in the absence of inhibitors, whereas the determination of Kmand Vmax in each sample of human liver microsomes was based on results obtained in the presence of TAO.
Immunoblotting of CYP2C9
A serum sample from a patient with tienilic acid hepatitis was used in CYP2C9 immunoblotting. As shown previously, this serum (PIJ, kindly provided by P. Beaune, Paris, France) specifically recognizes CYP2C9, but not CYP2C8, CYP2C18, or CYP2C19 (despite a >80% amino acid identity with CYP2C9) when used in a dilution of 1:20,000 (Lecoeur et al., 1994). In the present study, the anti-CYP2C9 specificity was confirmed by including yeast-expressed CYP2C19 as a negative control in immunoblots. Polyacrylamide gel electrophoresis was performed under standard conditions loading 5 and 10 μg of protein/well, and proteins were transferred to a nitrocellulose membrane with a conventional method (Laemmli, 1970;Towbin et al., 1979). After primary (PIJ serum) and secondary (antihuman IgG horseradish peroxidase conjugate) antibody incubations, immunoblots were developed in p-coumaric acid, luminol, and 3% hydrogen peroxide mixture and exposed in Chem Doc (Bio-Rad). Immunoquantification was based on comparisons with internal blotting standards (consisting of yeast-expressed CYP2C9) that showed a linear relationship to the amount of protein applied on gels. Because of an undetermined efficiency of the S. cerevisiae strain to express CYP2C9 holoenzyme (Imaoka et al., 1996), CYP2C9 apoprotein levels in human liver microsomes were expressed in arbitrary units. Bio-Rad Chem Doc software was used for visualization and quantification.
Data Analysis and Statistics
Kinetic data were applied to a Michaelis-Menten (one-enzyme) kinetic model in GraFit 4.03 (Erithacus Software Limited, Surrey, UK) a curve-fitting program based on nonlinear regression analysis, whereby Km,Vmax, and intrinsic clearance (Vmax/Km) could be estimated. Differences in kinetic parameters between differentCYP2C9 genotypes were evaluated for statistical significance by unpaired Student's t test.
Results
Analysis of Losartan Oxidation and Diclofenac 4′-Hydroxylation in CYP2C9 Variants Expressed in Yeast.
The yeast preparations with different variants of CYP2C9 showed similar levels of expression of both CYP2C9 apoprotein, holoenzyme (148 ± 33, 141 ± 10, and 114 ± 10 pmol of cytochrome P450/mg of protein for CYP2C9.1, CYP2C9.2, and CYP2C9.3, respectively) and cytochrome P450 reductase (data not shown). Kinetic parameters for both diclofenac 4′-hydroxylation and losartan oxidation are presented in Table 1. The apparentKm of diclofenac 4′-hydroxylation in the yeast system was 3.3 times higher for CYP2C9.3 compared with CYP2C9.1, resulting in an overall 70% reduction of intrinsic clearance. In contrast, the CYP2C9.2 enzyme differed from CYP2C9.1 mainly by a lowerVmax, and a 40% reduction of intrinsic clearance.
Kinetic analysis of 4′-OH-diclofenac and E-3174 formation by recombinant CYP2C9 variants expressed in yeast
As shown in Table 1, the kinetics of losartan oxidation differed from diclofenac hydroxylation. A 7-fold lower intrinsic clearance of losartan by CYP2C9.3, compared with CYP2C9.1, was mainly explained by a 5-fold lower Vmax without major differences in Km. The intrinsic clearance of losartan by CYP2C9.2 was at an intermediate level compared with the other allelic variants.
Analysis of Losartan Oxidation in Human Liver Microsomes.
The quality of different human liver microsomes was assessed by spectral P450 determinations, showing that the mean (±S.D.) P450 content was 479 ± 196 pmol/mg protein (n = 25, range 106–965 pmol/mg protein). A similar level of spectral P450 was found in all different variants of human liver microsomes (Table2). Levels of CYP2C9 apoprotein were compared by immunoquantification (Table 2) using yeast-expressed CYP2C9 as internal blotting standard. The results from immunoblotting showed a 6-fold variability of CYP2C9 apoprotein levels in the 25 different samples of human liver microsomes. Importantly, there was no significant difference in the CYP2C9 apoprotein level between the common and rare CYP2C9 genotypes (p > 0.2). However, we have not been able to determine the absolute amount of catalytically active CYP2C9 holo-enzyme in each sample (underDiscussion).
Total P450 content (pmol/mg of protein) and CYP2C9 apoprotein levels (arbitrary units) in human liver microsomes with different CYP2C9 genotypes (mean ± S.D.)
The rate of losartan oxidation to E-3174 versus substrate concentration in human liver microsomes is shown in Fig.1A. By using a wide range of substrate concentrations (0.05–200 μM), it was possible to detect a biphasic Eadie-Hofstee plot, indicative of the involvement of two different enzymes (Fig. 1B). Furthermore, a slight inhibition of E-3174 formation was evident at concentrations of losartan higher than 100 μM (Fig.1A).
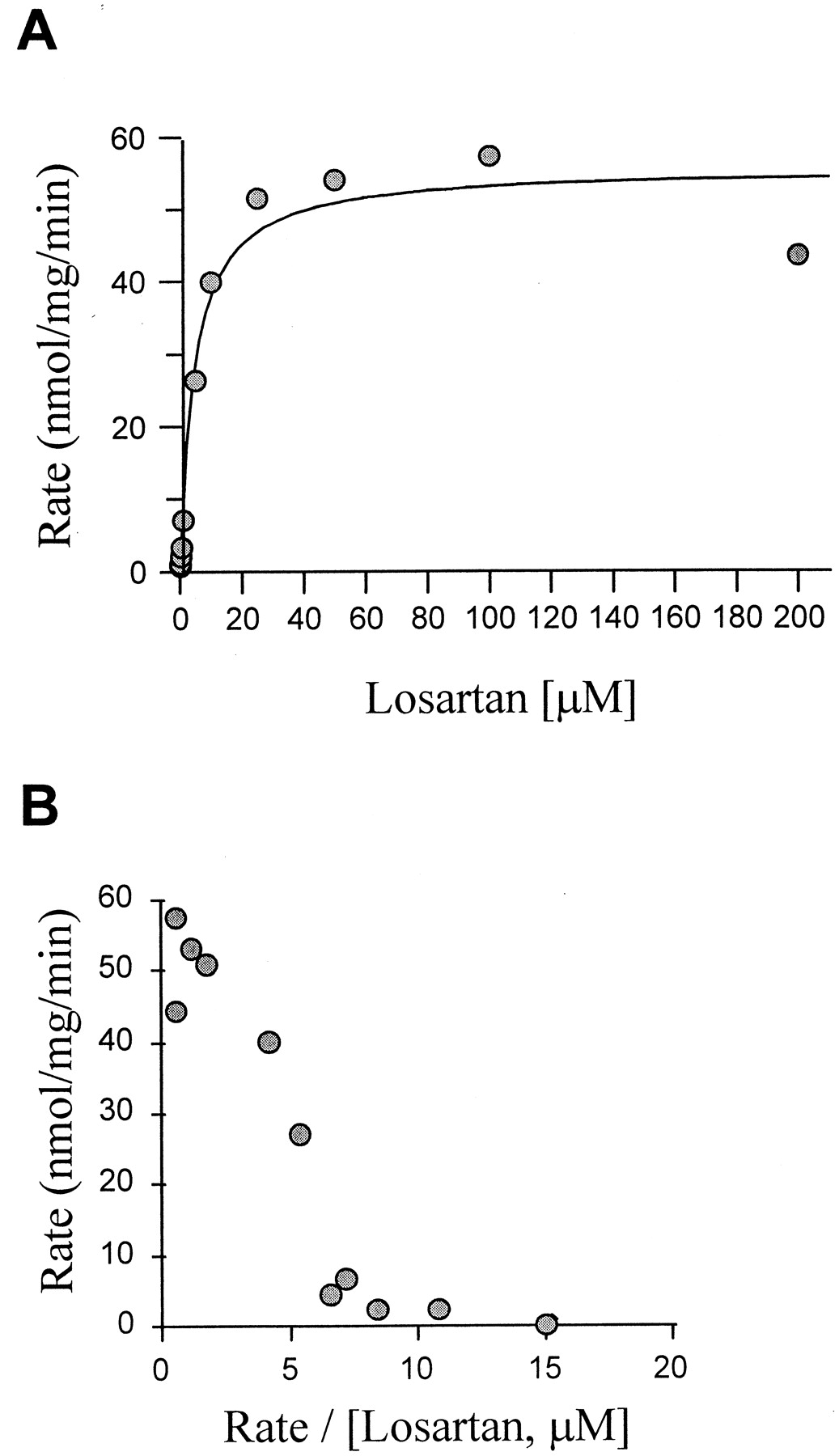
A, Michaelis-Menten plot of E-3174 formation from losartan in HL 60 microsomes (CYP2C9.1/1) in the absence of P450 inhibitors; B, Eadie-Hofstee plot of E-3174 formation from losartan in HL 60 microsomes (CYP2C9.1/1) in the absence of P450 inhibitors.
Inhibition experiments were performed using the CYP2C9-specific inhibitor SPZ and the CYP3A4-specific inhibitor TAO. When used in combination, essentially all formation of E-3174 was blocked in human liver microsomes (90–100%). The major part of inhibition was exerted by SPZ both at low and high concentrations of losartan, as SPZ alone inhibited more than 90% of losartan oxidation at 0.5 μM losartan, and by 74 ± 16% (mean ± S.D., n = 22) at 50 μM losartan. At low substrate concentrations, there was no significant inhibition by TAO alone and the average inhibitory effect by TAO at 50 μM losartan was only 16 ± 3% (n = 22).
The rate of E-3174 formation was compared between human liver microsomes with different genotypes and the results are summarized in Table 3A. Statistically significant 2- to 3-fold lower rates of E-3174 formation were evident in microsomes fromCYP2C9*2/*2 and CYP2C9*1/*3 individuals compared with CYP2C9*1/*1. Almost a 9-fold lower rate was observed in liver microsomes from a single CYP2C9*3/*3 individual.
Kinetics of losartan oxidation in different CYP2C9 genotypes
To further explore the impact of CYP2C9 polymorphism on losartan oxidation, complete one-enzyme kinetics of E-3174 formation, including determinations of Vmax,Km, and intrinsic clearance, was characterized in all 25 individual samples of human liver microsomes in the presence of TAO. The results are presented in Table 3B, where it is evident that the intrinsic clearance of losartan was significantly lower in CYP2C9.1/3 liver samples compared with CYP2C9.1/1 livers (p < 0.01), and almost 20-fold lower in the single CYP2C9.3/3 sample (Table 3B). Both in this latter sample, and in the group of samples from heterozygous CYP2C9*1/*3individuals, the lower intrinsic clearance was mainly explained by a lower Vmax compared with CYP2C9.1/1 samples. No statistically significant differences were found comparing CYP2C9.1/2 or CYP2C9.2/2 samples with CYP2C9.1/1 samples, but there was an apparent trend that the CYP2C9.2/2 livers showed a lower intrinsic clearance (p = 0.07).
Discussion
The results indicate that CYP2C9 is the major catalyst of losartan oxidation over a wide range of different substrate concentrations, whereas the contribution by CYP3A4 is significant only at very high concentrations of losartan. The importance of CYP2C9 in losartan oxidation was first proposed by Stearns et al. (1995), who found that SPZ inhibited the formation of E-3174 in human liver microsomes by 81%, at a losartan concentration of 20 μM, whereas the CYP3A4 inhibitor ketoconazole inhibited maximally 51% of E-3174 formation. Similar inhibitory effects were found using anti-CYP2C9 IgG and anti-CYP3A4 IgG, respectively (Stearns et al., 1995). It should be pointed out that the sensitivity of E-3174 detection in our experimental system made it possible to use lower concentrations of losartan than previously tested (<1 μM), down to levels where CYP2C9-dependent catalysis appears to dominate completely. Importantly, these levels correspond to the expected plasma concentrations in losartan-treated subjects (Lo et al., 1995). However, using a very high concentration of losartan (100 μM), it was reported that losartan oxidation in human liver microsomes correlated very well with nifedipine oxidation mediated by CYP3A4, but not with tolbutamide hydroxylation, a marker of CYP2C9 activity (Yun et al., 1995). Clearly, the choice of substrate concentration is critical in systems where different enzymes have overlapping substrate specificity.
In subsequent experiments, the role of the CYP2C9polymorphism in losartan oxidation was evaluated. Our results show that the intrinsic clearance of losartan was dramatically reduced in human liver microsomes obtained from a CYP2C9*3/*3 homozygous individual. Furthermore, a significantly lower activity was also found in the larger group of microsomal samples from CYP2C9*1/*3heterozygous individuals. The rate of E-3174 formation was also significantly reduced in microsomes from CYP2C9*2/*2, but not from heterozygous CYP2C9*1/*2 individuals. Two samples of microsomes with the CYP2C9*2/*3 genotype differed widely in their activities, allowing no conclusion to be drawn about this particular genotype. In general however, it appears that liver microsomes with rare variants of CYP2C9 differed mainly by exhibiting a lower Vmax of losartan oxidation, a finding supported by the yeast data (Tables 1 and 3B). This is similar to some (e.g., piroxicam, phenytoin, and tenoxicam), but not all other CYP2C9 substrates, as illustrated by diclofenac in Table 1, and in agreement with previous reports (Yamazaki et al., 1998a; Takanashi et al., 2000). Given that CYP2C9.3 differs from CYP2C9.1 by an Ile359Leu substitution in substrate recognition site-5 of the CYP2C9 molecule (Gotoh, 1992), it is not surprising that substrate turnover is affected in a substrate-specific manner by this amino acid substitution.
In the present collection of human liver microsomes, total P450 contents, apoprotein levels, and activities of CYP2C9 varied about 6-fold within the CYP2C9.1/1 group. A similar range of interindividual variability of spectral P450, and isozyme-specific catalytic activities, has been reported previously in human liver microsomes (Shimada et al., 1994; Transon et al., 1996; Westlind et al., 1999).
Immunoblotting of human liver microsomes was primarily carried out to test the possibility that low CYP2C9 catalytic activity in microsomal samples from subjects with rare alleles was a result of lower expression of CYP2C9 enzyme. This turned out not to be the case. However, it is important to bear in mind that the SDS-polyacrylamide gel electrophoresis/immunoblotting technique only allows for quantification of apoprotein and not catalytically active holo-enzyme. Interestingly, there was no obvious relationship between the individual apoprotein level and activity of losartan oxidation in our different human liver microsomes (data not shown). Apparently, this could not simply be explained by genotype-related variability since no significant correlation between apoprotein and activity was found even within the group of genetically homogenous *1/*1 microsomes. A very similar finding has been reported previously, where the microsomal CYP2C9 apoprotein levels did not correlate to any of several different CYP2C9-specific activities (Shimada et al., 1994; Transon et al., 1996; Westlind et al., 1999). The reason for this in vitro observation is not clear but might relate to sample differences in holo-enzyme/apoprotein ratios, or variable amounts of other forms of inactivated CYP2C9. In conclusion, immunoquantification of CYP2C9 does not appear to be reliable in predictions of catalytic activity.
Further control experiments were carried out with a few samples showing exceptionally low losartan metabolism, using omeprazole as substrate. The low activity of losartan oxidation detected in the CYP2C9.3/3 human liver microsomes was not associated with low activity in general, since omeprazole 5′-hydroxylation, as a measure of CYP2C19 activity, and omeprazole sulfon formation, as a measure of CYP3A4 activity (Tybring et al., 1997), were within normal range (data not shown).
In conclusion, the present study for the first time clarifies the role of CYP2C9.2 and CYP2C9.3 variants in losartan metabolism. Interestingly, the results are consistent with a recent case report describing that conversion of losartan to E-3174 in vivo, was reduced more than 90% in a subject homozygous for CYP2C9*3/*3(Spielberg et al., 1996). Similarly, we have recently shown (this study) significantly higher plasma AUClosartan/AUCE-3174ratios not only in subjects homozygous for CYP2C9*2 orCYP2C9*3 compared with CYP2C9*1 (approximately 4- and 30-fold higher, respectively) but also in CYP2C9*1/*3and CYP2C9*2/*3 genotypes after a single oral dose of losartan (Lo et al., 1995). Thus, our in vitro findings are consistent with in vivo data with regard to the functional importance of bothCYP2C9*2 and CYP2C9*3 in losartan metabolism. Establishing the role of CYP2C9 polymorphism in the metabolism of losartan is of importance for two major reasons. First, E-3174 is the metabolite responsible for the major antihypertensive effect of losartan (Lo et al., 1995). It is possible that individuals with slow CYP2C9 metabolism might show an impaired therapeutic response to the drug, but this remains to be studied. Second, it is necessary to establish a safe, simple, and specific phenotyping procedure for CYP2C9, considering the general importance of CYP2C9 in drug metabolism, as well as the connection between rare geneticCYP2C9 variants and risk of bleeding complications during warfarin therapy (Aithal et al., 1999). Because of its safety, losartan can be an alternative to phenytoin (Aynacioglu et al., 1999) and tolbutamide (Miners and Birkett, 1996), which have been suggested earlier as probes for CYP2C9 phenotyping.
Acknowledgments
We thank Birgit Eiermann for genotyping of human liver samples, Anna Nordmark for Western blotting assistance, and Karl Bodin for identification of losartan and E-3174 by liquid chromatography/mass spectrometry.
Footnotes
-
The study was supported financially by The Swedish Medical Research Council (3902 and 5949) and The Swedish Society of Medicine. E.E. is a recipient of a Merck Sharp and Dohme fellowship in Clinical Pharmacology. Ü.Y. is a recipient of a Turkish Higher Education Council Ph.D. scholarship in clinical pharmacology. This study was partly presented in 12th International Symposium on Microsomes and Drug Oxidations Stresa, Italy, July 10–14, 2000.
- Abbreviations used are::
- CYP
- cytochrome P450
- SPZ
- sulfaphenazole
- TAO
- triacetyloleandomycin
- HPLC
- high-performance liquid chromatography
- Received March 7, 2001.
- Accepted April 5, 2001.
- The American Society for Pharmacology and Experimental Therapeutics
References




{kind=link}
Jump to section
- Article
- Abstract
- Materials and Methods
- Site-Directed Mutagenesis
- Expression of Variant cDNAs in Yeast Cells
- Determination of CYP2C9 Holoenzyme and P450 Reductase
- Preparation of Human Liver Microsomes
- Genotyping of DNAs Isolated from Human Liver Tissues
- Analysis of Enzyme Kinetics
- Immunoblotting of CYP2C9
- Data Analysis and Statistics
- Results
- Discussion
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters