


Abstract
Dapsone activates CYP2C9-mediated metabolism in various expression systems and is itself metabolized by CYP2C9 to its hydroxylamine metabolite. Studies were conducted with expressed CYP2C9 to characterize the kinetic effects of dapsone (0–100 μM) on (S)-flurbiprofen (2–300 μM), (S)-naproxen (10–1800 μM), and piroxicam (5–900 μM) metabolism in 6 × 6 matrix design experiments. The influence of (S)-flurbiprofen on dapsone hydroxylamine formation was also studied. Dapsone increased the Michaelis-Menten-derived Vmax of flurbiprofen 4′-hydroxylation from 12.6 to 20.6 pmol/min/pmol P450, and lowered its Km from 28.9 to 10.0 μM, suggesting that dapsone activates CYP2C9-mediated flurbiprofen metabolism without displacing flurbiprofen from the active site, supporting a two-site model describing activation. Similar results were observed with piroxicam 5′-hydroxylation, asVmax was increased from 0.08 to 0.20 pmol/min/pmol P450 and Km was decreased from 183 to 50 μM in the presence of dapsone. In addition, the kinetic profile for naproxen was converted from biphasic to hyperbolic in the presence of dapsone, while exhibiting similar decreases inKm and increases inVmax. Kinetic parameters were also estimated using the two-site binding equation, with α values <1 and β values >1, indicative of activation. Additionally, dapsone hydroxylamine formation was measured from incubations containing flurbiprofen, exhibiting a kinetic profile that was minimally affected by the presence of flurbiprofen. Overall, these results suggest that dapsone activates the metabolism of multiple substrates of CYP2C9 by binding within the active site and causing positive cooperativity, thus lending further support to a two-site binding model of P450-mediated metabolism.
It is becoming evident that many cytochrome P450 (P4501)-catalyzed reactions do not proceed with typical Michaelis-Menten kinetics. Observations of atypical kinetic events such as activation, autoactivation, and substrate inhibition are becoming increasingly common, with the term “cooperativity” often being used to describe these types of phenomena. To date, CYP3A4 has been the most studied isoform with respect to atypical kinetics due to the large number of reactions that it catalyzes and the complexity of its active site. However, other relevant P450s have been shown to participate in a number of these abnormal kinetic events (Shou et al., 1994; Ekins et al., 1998; Korzekwa et al., 1998), making analysis of in vitro data problematic. Consequently, it has been suggested that atypical kinetics must be considered during in vitro-in vivo scaling of kinetic data. Houston and Kenworthy (2000) have demonstrated that failure to properly assess atypical kinetics may result in under- or overestimation of in vivo clearance of drugs when based on in vitro kinetic parameter estimates. Therefore, it is imperative that the cause and mechanism of these events be determined, as well as the isoforms affected.
Our lab has previously demonstrated that CYP2C9-mediated flurbiprofen 4′-hydroxylation and naproxen demethylation are activated by dapsone in human liver microsomes and baculovirus-expressed CYP2C9 et al. (Korzekwa et al., 1998). However, kinetic effects of this activation have yet to be fully characterized. While flurbiprofen is exclusively metabolized by CYP2C9 (Tracy et al., 1995) and shows normal Michaelis-Menten kinetics (Tracy et al., 1996), naproxen is metabolized by CYP2C9 and CYP1A2 (Miners et al., 1996; Tracy et al., 1997) and has been shown to exhibit nonsaturable biphasic kinetics in microsomes (Tracy et al., 1997), as well as in expressed CYP2C9 (Korzekwa et al., 1998). The nonsaturable metabolic profile observed in expressed CYP2C9 suggests that there may be two binding components within the CYP2C9 active site, one with a low Km, lowVmax, the other with a highKm and highVmax, similar to naphthalene metabolism by CYP3A4 (Korzekwa et al., 1998). Piroxicam, another nonsteroidal anti-inflammatory drug reported to be metabolized by CYP2C9 (Zhao et al., 1992), has been shown to exhibit atypical kinetics as well (substrate inhibition) in baculovirus-expressed CYP2C9 (J. M. Hutzler and T. S. Tracy, unpublished results), and has a quite different structure than either flurbiprofen or naproxen.
Dapsone is a compound used to treat Pneumocystis cariniipneumonia in acquired immunodeficiency syndrome patients, and it is metabolized by CYP2C9 at therapeutic concentrations to its hydroxylamine metabolite (Gill et al., 1995; Winter et al., 2000). Thus, it appears that dapsone is also a substrate for the enzyme it activates, suggesting that dapsone may cause activation by binding in the active site of CYP2C9. This scenario is analogous to 7,8-benzoflavone, which activates CYP3A4-mediated phenanthrene metabolism, in addition to being a substrate for CYP3A4 (Shou et al., 1994).
The purpose of the current study was to determine the effect of dapsone on the kinetics of (S)-flurbiprofen, (S)-naproxen, and piroxicam, three prototypical substrates of CYP2C9 with differing structures and kinetic profiles (Fig.1). It was hypothesized that a kinetic model that suggests that both the substrate and effector may be present in the active site simultaneously, both having access to the active oxygen, was applicable (Shou et al., 1994, 1999; Korzekwa et al., 1998). Thus, kinetic data from expressed CYP2C9 were fit to the two-site model (eq. 3) in addition to either the Michaelis-Menten equation (eq. 1) or a two-site biphasic model (eq. 2). If our hypothesized two-site model for CYP2C9 is accurate, and the activator (dapsone) also serves as a substrate then theoretically, near-simultaneous metabolism should occur, and metabolites from both compounds should be measurable. Therefore, the effect of flurbiprofen on dapsone hydroxylation kinetics was also examined by measuring dapsone hydroxylamine formation and fitting data to the Michaelis-Menten equation.
Structures of the three CYP2C9 substrates studied for activation by dapsone.
Arrows indicate site of metabolism.
Materials and Methods
Chemicals.
Acetonitrile and dibasic potassium phosphate were obtained from Fisher Scientific (Pittsburgh, PA). (S)-Flurbiprofen, 4′-hydroxy flurbiprofen, and 2-fluoro-4-biphenyl acetic acid (internal standard) were gifts from Pharmacia Corp. (Kalamazoo, MI). (S)-naproxen and desmethylnaproxen were gifts from Syntex Laboratories Inc. (Palo Alto, CA). Piroxicam and dapsone were obtained from Sigma Chemical Co. (St. Louis, MO), while 5′-hydroxy piroxicam was a gift from Pfizer Inc. (Groton, CT). Dapsone hydroxylamine was synthesized by the method of Uetrecht et al. (1984) and was a gift from Robert Branch at the University of Pittsburgh, Pittsburgh, PA. All other chemicals were obtained from commercial sources and were of the highest purity available.
Incubation Conditions.
Microsomal preparations resulting from the coexpression, mediated by baculovirus delivery, of CYP2C9, NADPH oxidoreductase, and cytochromeb5 in BTI-TN-5B1-4 cells were used as the enzyme source and were a gift from Camitro Corp. (Menlo Park, CA). Incubation mixtures, in a 6 × 6 matrix design, contained 1 pmol of expressed CYP2C9 (5 pmol for piroxicam) with either (S)-flurbiprofen (2–300 μM), (S)-naproxen (10–1800 μM), or piroxicam (5–900 μM) incubated with dapsone (0–100 μM) in 50 mM potassium phosphate buffer at pH 7.4. Final incubation volume was 0.2 ml and reactions were initiated with 1 mM NADPH and allowed to incubate at 37°C for 20 min (45 min for piroxicam). Incubations with flurbiprofen were quenched by adding 200 μl of acetonitrile containing internal standard (180 ng/ml 2-fluoro-4-biphenylacetic acid), followed by addition of 40 μl of half-strength H3PO4. Incubations with naproxen were quenched by adding 200 μl of acetonitrile followed by addition of 40 μl of half-strength H3PO4, while piroxicam reactions were quenched by adding 20 μl of perchloric acid, followed by directly placing incubation tubes on ice. Samples from all incubations were then centrifuged at 10,000 rpm for 5 min, placed into autosampler vials, and 10 to 100 μl was injected onto HPLC system. In experiments examining flurbiprofen effect on dapsone hydroxylamine formation, flurbiprofen concentrations were 0, 2, 5, and 10 μM, with dapsone concentrations ranging from 1 to 300 μM. To prevent degradation of the dapsone hydroxylamine metabolite, 1 mM ascorbic acid was included in the 50 mM potassium phosphate buffer in those cases where this metabolite was being measured. The reaction was stopped by the addition of 0.1 ml of cold methanol. Samples were then spun at 20,000g for 5 min at 3°C in an Eppendorf 5417R centrifuge and 15 μl was injected onto an liquid chromatography/mass spectrometry system for immediate dapsone hydroxylamine analysis to prevent instability issues. All other conditions were as described above.
Analysis of 4′-Hydroxy Flurbiprofen.
4′-Hydroxy flurbiprofen was analyzed by HPLC with fluorescence detection and comparison to a standard dissolved in 50:50 (v/v), acetonitrile/50 mM potassium phosphate buffer, pH 7.4. The HPLC system consisted of a Waters 501 HPLC pump, a Waters 717 autosampler, and a Waters 470 fluorescence detector set at an excitation wavelength of 260 nm and an emission wavelength of 320 nm. The mobile phase consisted of 45:55 (v/v), acetonitrile/20 mM K2HPO4, pH 3.0, pumped at 1 ml/min through a Brownlee Spheri-5 C18 4.6- × 100-mm column. 4′-Hydroxy flurbiprofen and internal standard eluted at approximately 2.2 and 4.8 min, respectively.
Analysis of Desmethylnaproxen.
Desmethylnaproxen was analyzed identically to 4′-hydroxy flurbiprofen except that the fluorescence detector was set at an excitation wavelength of 230 nm and an emission wavelength of 340 nm, and no internal standard was used. Desmethylnaproxen eluted at approximately 2.1 min.
Analysis of 5′-Hydroxy Piroxicam.
5′-Hydroxy piroxicam was analyzed by HPLC with UV detection (365 nm). The HPLC system was as described above, but was fitted to a Waters 486 UV detector. The mobile phase consisted of 50:50 (v/v), acetonitrile/50 mM K2HPO4, pH 3.0, pumped at 1 ml/min through a Phenomenex Luna C18 5-μm, 4.6- × 150-mm column. Retention time for 5′-hydroxy piroxicam was approximately 4.3 min.
Analysis of Dapsone Hydroxylamine.
HPLC/ESI-mass spectrometry was performed on a PerkinElmer Sciex API 150ex single quadrupole mass spectrometer connected to a PerkinElmer Series 200 LC pump and autosampler. The mass spectrometer was fitted with a turbo ion-spray source and operated in positive-ion selective ion monitoring electrospray mode, with an ESI spray voltage of 5.0 kV and source temperature of 450°C. The turbo ion-spray gas flow rate was set at 7000 ml/min. Tuning of the ESI source and mass spectrometry was accomplished by infusing a solution containing 20 μg/ml dapsone hydroxylamine into the mobile phase flow path post column via a T-connector using a Harvard Apparatus (Holliston, MA) manual injector. The instrument was controlled by a Dell Optiplex GX1 computer running Analyst version 1.1 software. Dapsone hydroxylamine formation was confirmed by appearance of the MH+ion at m/z of 265.1 and chromatographic retention time corresponding to that of a synthetic standard dissolved in dimethyl sulfoxide. Stability of dapsone hydroxylamine metabolite was exhibited over the time required to analyze all of the incubation samples. Analytical separation was accomplished using a Zorbax SB-C8 2.1 × 50 mm, 5-μm microbore column with a gradient elution profile. Solvent flow through the column was 0.7 ml/min with 30% of the flow being diverted to the mass spectrometer. Gradient elution consisted of mobile phase A [methanol] and mobile phase B [100 mM ammonium acetate, pH 4.38]. The gradient was initiated at a ratio of 10% A:90% B. This ratio was altered in a linear manner to 80% A:20% B over 1 min and held for 2.4 min to allow elution of dapsone hydroxylamine. The system was then returned to initial conditions over 0.1 min and allowed to equilibrate for 4.4 min prior to the next injection. The flow was diverted to waste for 1 min post-injection to prevent fouling of the interface from polar compounds in the solvent front. Dapsone hydroxylamine eluted at approximately 2.9 min.
Data Analysis and Equations.
Kinetic parameters for flurbiprofen and piroxicam hydroxylation were estimated using the nonlinear regression function in Sigma Plot (version 6.0), and the following standard Michaelis-Menten velocity equation:
The two-site model equation (Korzekwa et al., 1998) used for surface plot analysis of flurbiprofen 4′-hydroxylation, naproxen demethylation, and piroxicam 5′-hydroxylation in the presence of dapsone was as follows:
Results
Dapsone Activation of Purified CYP2C9.
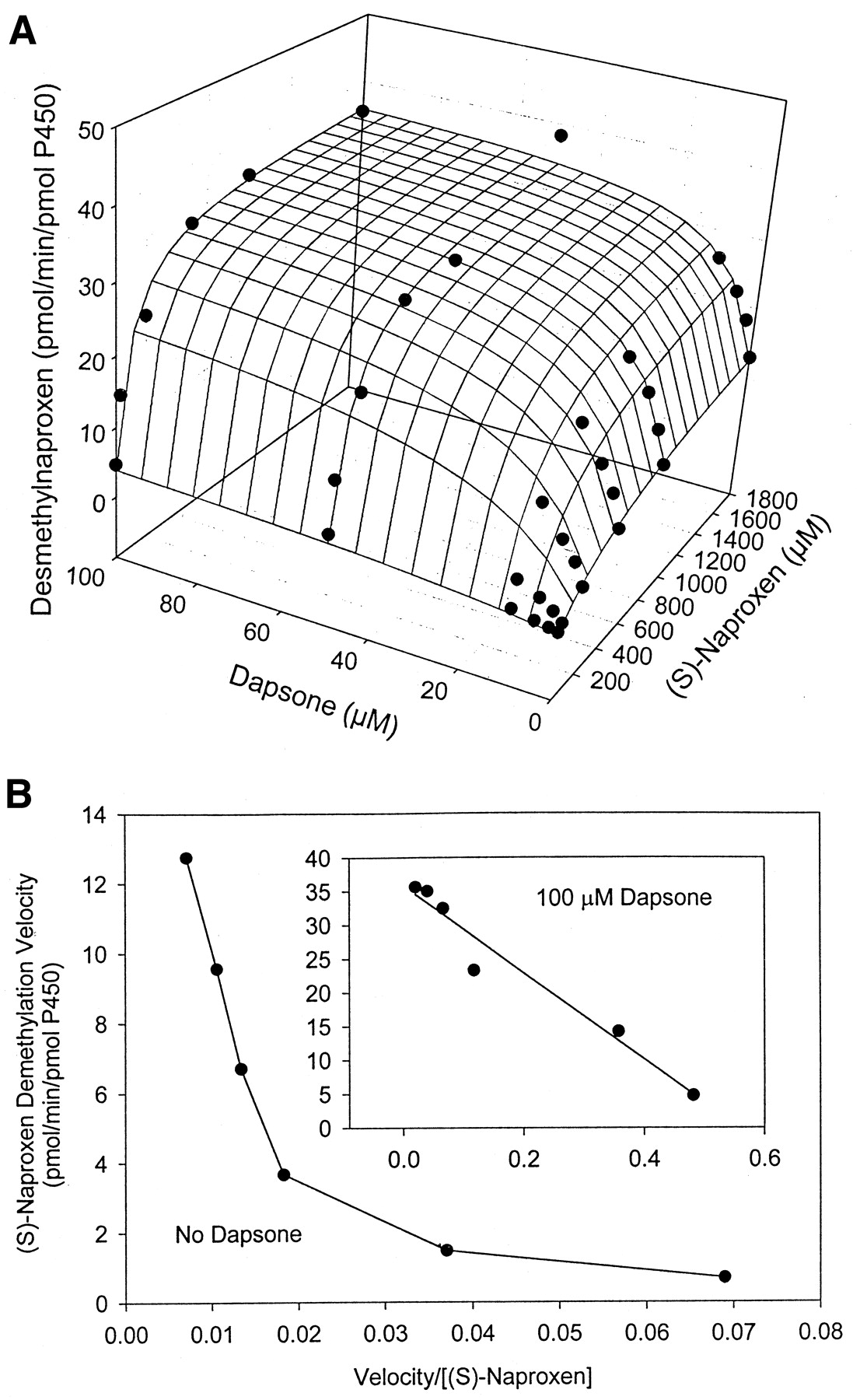
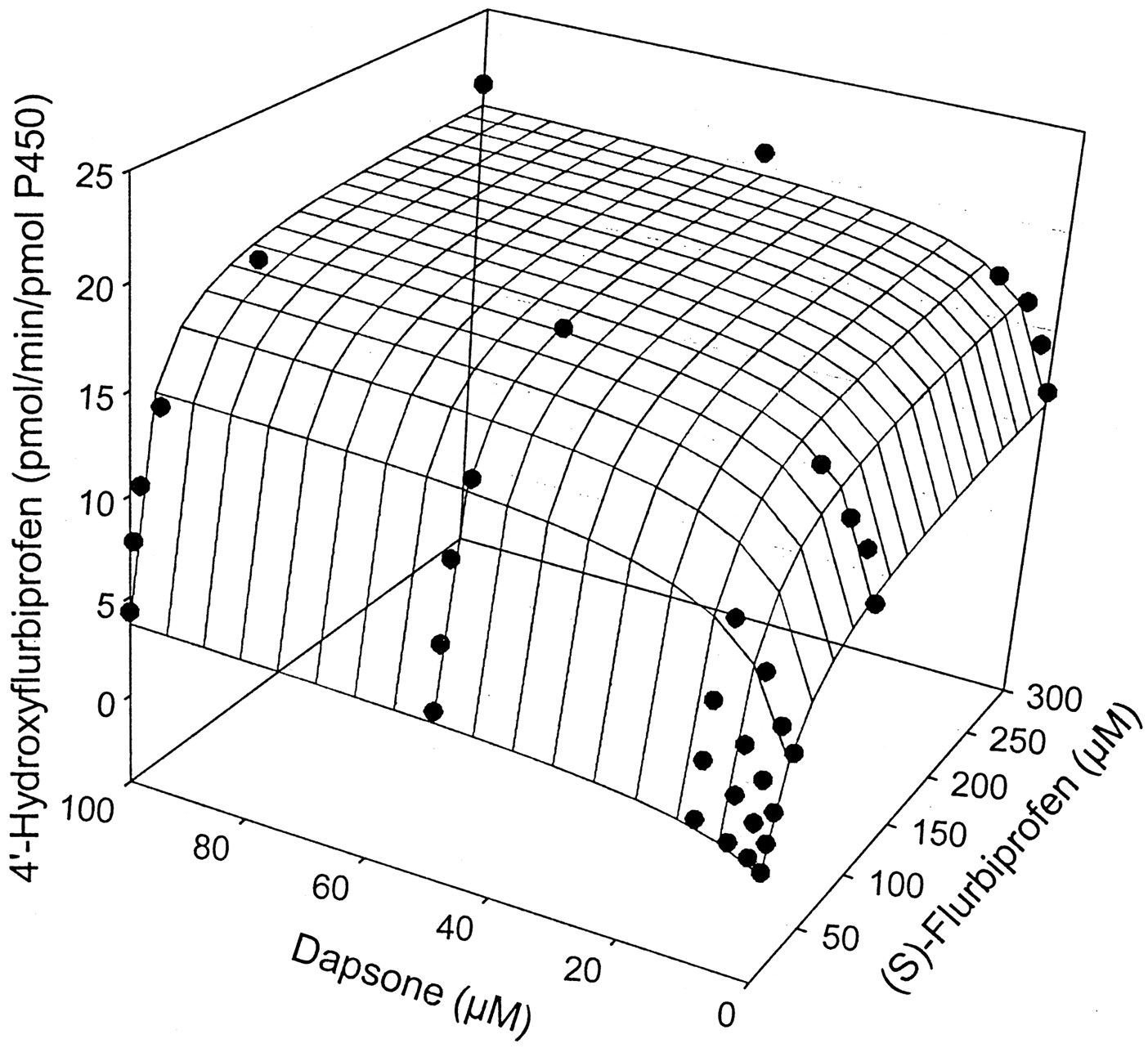
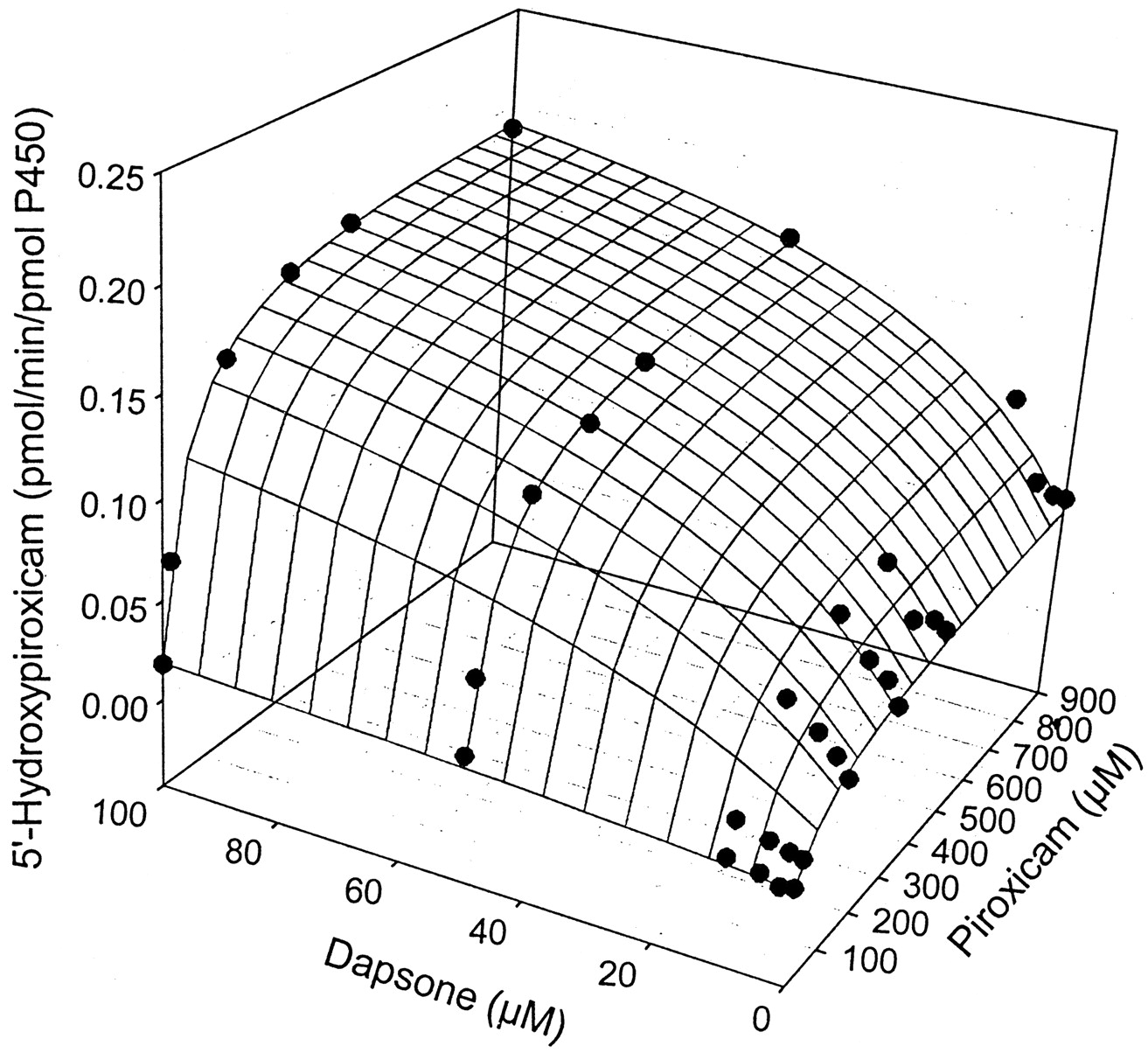
The kinetic parameters for flurbiprofen and piroxicam hydroxylation were determined by fitting data to the Michaelis-Menten equation (eq.1), while parameters for naproxen demethylation were estimated by fitting data to a two-site biphasic model equation (eq. 2). Dapsone activated flurbiprofen 4′-hydroxylation in purified CYP2C9, increasingVmax from 12.6 to 20.6 pmol/min/pmol P450, while lowering the Km from 28.9 to 10.0 μM (Table 1). Meanwhile, theVmax for piroxicam hydroxylation was increased from 0.08 to 0.20 pmol/min/pmol P450, with theKm reduced from 183 to about 50 μM (Table1). Likewise, naproxen demethylation was activated by the presence of dapsone with the data fit to a biphasic equation reflecting the kinetic profile of the reaction. The Vmax for naproxen demethylation was increased from 11.7 to 34.7 pmol/min/pmol P450, while the Km was decreased from 433 to 67.4 μM (Table 2), a greater fold change than observed with either flurbiprofen or piroxicam. Interestingly, the kinetic profile for naproxen demethylation, which was biphasic, became hyperbolic in nature in the presence of 100 μM dapsone (Fig. 4, A and B). The CLint of the first naproxen binding site (Vmax1/Km1) increased from 0.027 in the absence of dapsone to 0.52 in the presence of 100 μM dapsone, suggesting that the CLint of the second site (Vmax2/Km2) [presumably the linear portion of the profile] contributes a smaller percentage to total naproxen demethylation when dapsone is present (Table 2). Kinetic parameters for flurbiprofen, naproxen, and piroxicam metabolism were also estimated (Table 3) using the two-site model equation (eq. 3). Data from this table further suggests that dapsone activates the metabolism of these three compounds, as all α values are below 1, and all β values are above 1. It is also interesting to note that the binding constant for dapsone varies according to the substrate being activated (Table 3). The three-dimensional activation plot for flurbiprofen can be seen in Fig.2, demonstrating activation of flurbiprofen hydroxylation by dapsone. However, flurbiprofen appeared to not substantially affect the formation of dapsone hydroxylamine, as similar velocity curves were obtained in the presence of 0, 2, 5, and 10 μM flurbiprofen (Fig. 3).Vmax values for dapsone hydroxylation ranged from 36 to 44 pmol/min/pmol P450, whileKm values ranged from 81 to 144 μM (data not shown). Similarly, a three-dimensional plot exhibiting activation of naproxen demethylation is shown in Fig.4A, while in Fig. 4B, an Eadie-Hofstee plot shows the effect of dapsone on the kinetic profile of naproxen. In the absence of dapsone, a biphasic plot is observed, whereas the presence of 100 μM dapsone causes the biphasic profile of naproxen to become linear. As a result, naproxen demethylation in the presence of 100 μM dapsone was also fit to the Michaelis-Menten curve, with a Vmax of 37.0 pmol/min/pmol P450 and a Km of 80 μM (Table 2). Last, a three-dimensional plot in Fig. 5 shows that piroxicam, a structurally different CYP2C9 substrate, is also activated by dapsone.
Kinetic parameter estimates for flurbiprofen 4′-hydroxylation and piroxicam 5′-hydroxylation in the presence of dapsone
Kinetic parameter estimates for naproxen demethylation in the presence of dapsone

A, three-dimensional surface plot and B, Eadie-Hofstee plot.
A, three-dimensional surface plot showing activation of naproxenO-demethylation by dapsone in expressed CYP2C9. The surface is the fit to eq. 2. B, Eadie-Hofstee plot for (S)-naproxen demethylation in the absence of dapsone showing biphasic kinetic profile. Inset figure shows Eadie-Hofstee plot for (S)-naproxen in the presence of 100 μM dapsone, resulting in a linear kinetic profile.
Kinetic parameter estimates for flurbiprofen hydroxylation, naproxen demethylation, and piroxicam hydroxylation in the presence of dapsone

Three-dimensional surface plot showing activation of flurbiprofen 4′-hydroxylation by dapsone in expressed CYP2C9.
The surface is the fit to eq. 2.

Two-dimensional plots demonstrating minimal change in dapsone hydroxylamine formation in the presence of 2, 5, and 10 μM flurbiprofen.
Kinetic parameters were estimated using eq. 1.

Three-dimensional surface plot showing activation of piroxicam 5′-hydroxylation by dapsone in expressed CYP2C9.
The surface is the fit to eq. 2.
Stability of Dapsone Hydroxylamine.
Dapsone hydroxylamine is known to be an unstable compound in potassium phosphate buffer (Vage et al., 1994; Vage and Svensson, 1994). A standard solution was prepared in potassium phosphate buffer containing 1 mM ascorbic acid and a 15-μl injection was made from the same vial every 20min at room temperature and analyzed according to methods described under Materials and Methods. When subjected to our incubation conditions over 5 h, the peak area coefficient of variation was 2.3% (data not shown), demonstrating that quantitation would not be affected by metabolite instability.
Discussion
Results from this study suggest that dapsone activates multiple substrates of CYP2C9, being described by a model in which dapsone (effector) and substrate may fit in the active site simultaneously. The increase in Vmax and decrease inKm for flurbiprofen 4′-hydroxylation while dapsone hydroxylamine metabolite is also measurable provide additional evidence for this theory. In addition, the naproxen metabolic profile was changed from biphasic to hyperbolic in the presence of dapsone, and piroxicam, a third CYP2C9 substrate with a distinct structure, was also metabolically activated by dapsone.
It was once believed that most P450-mediated reactions obeyed normal Michaelis-Menten enzyme kinetics, allowing for relatively simple prediction of kinetic parameters. However, atypical cytochrome P450 kinetics is now being recognized more frequently in the area of drug metabolism, which complicates scaling kinetic data from the in vitro to the in vivo situation. To date, CYP3A4 has been the most studied isoform in terms of atypical kinetic behavior. For example, studies bySchwab et al. (1988) and Shou et al. (1994) have shown that purified CYP3A4 enzymes are directly activated by 7,8-benzoflavone. This is particularly interesting because like dapsone, 7,8-benzoflavone is a substrate for the enzyme that it activates. More recent data suggest that P450 enzymes other than CYP3A4 may behave in an atypical manner.Korzekwa et al. (1998) suggest that CYP1A1-mediated naphthalene metabolism shows a sigmoidal saturation curve and that Hep G2-expressed CYP2B6, CYP2C8, CYP2C9, and CYP3A5 may also exhibit non-Michaelis-Menten kinetics, depending on the substrate. Likewise,Ekins et al. (1998) have shown that CYP1A2 displays autoactivation kinetics for ethoxyresorufin metabolism, although results appear to be enzyme source-dependent. Despite these studies describing activation, no clear mechanism has been determined, although many models have been proposed. Ueng et al. (1997) have described a model that suggests that an allosteric site is involved in the cooperativity of certain CYP3A4 reactions, but the proximity of this site is not defined. Harlow and Halpert (1998) suggest that effector binding is most likely in the active site along with the substrate, but go on to propose that oxidation only occurs at one binding region within the active site. Another possibility that has been put forth is the idea of kinetically distinguishable conformers of CYP3A4 (Koley et al., 1995), which was proposed based on kinetic studies of carbon monoxide binding. However, the model we believe that most accurately describes the activation phenomenon is that of Shou and Korzekwa (Shou et al., 1994, 1999;Korzekwa et al., 1998), which suggests that both substrate and effector are bound in the active site simultaneously with or without distinctive binding regions and access to the reactive oxygen.
From our experiments, we have observed near-simultaneous metabolism of flurbiprofen and dapsone, which provides support to the primary hypothesis that substrate and activator may bind in the active site simultaneously, potentially ruling out the theory of an allosteric site causing cooperativity. The main basis for this hypothesis is that dapsone is also a substrate for CYP2C9. Although the CYP2C9 active site is not as large as CYP3A4, it still may be able to accommodate two rather small compounds. In fact, NMR studies by Banci et al. (1994)have shown that both pyridine and imidazole can fit into the P450cam active site simultaneously. Furthermore, flurbiprofen 4′-hydroxylation, naproxen demethylation, and piroxicam 5′-hydroxylationKm values were all decreased (i.e., increased affinity) in the presence of dapsone (Tables 1 and 2), suggesting that dapsone does not displace these compounds from the active site. An interesting discovery that provides further evidence to the two-site hypothesis is the kinetic profile of naproxen. In the absence of dapsone, naproxen exhibits a biphasic kinetic profile, as shown by the Eadie-Hofstee plot in Fig. 4B, which suggests in itself that CYP2C9 may have a low Km, lowVmax binding component, and a highKm, high Vmaxbinding component. As a result, data were fit to a two-site biphasic equation instead of the Michaelis-Menten equation. However, naproxen demethylation in the presence of 100 μM dapsone was best fit with the Michaelis-Menten equation (Table 2). The inset in Fig. 4B demonstrates that addition of 100 μM dapsone alters naproxen kinetics, resulting in a linear Eadie-Hofstee plot and supporting a fit using the standard Michaelis-Menten equation. This change in kinetic profile is most likely due to dapsone binding to the highKm site when present in high concentrations, occupying that site such that only one binding region of CYP2C9 is operable in naproxen metabolism. Piroxicam metabolism is apparently also activated by dapsone, which is interesting considering its structural differences and larger size compared with naproxen and flurbiprofen. It is worthy to note that piroxicam has been shown to exhibit substrate inhibition in microsomes and baculovirus-expressed CYP2C9 (J. M. Hutzler and T. S. Tracy, unpublished data). However, this was not observed in the expression system used for this study, confirming previous reports that many of these kinetic events are expression system-dependent (Ekins et al., 1998).
Fitting of data from all three substrates to the two-site model (eq. 3) was done to estimate a single Km andVmax, along with the effector binding constant (KB) and α and β values. Results show α values (change in Kmresulting from effector binding) less than 1, and β values (change inVmax resulting from effector binding) greater than 1 for all substrates (Table 3), suggesting thatKm was reduced andVmax increased in all cases. It has been demonstrated previously that an α < 1 and/or a β > 1 is indicative of activation (Korzekwa et al., 1998). It should be realized that eq. 3 does not consider substrates that bind twice, which is not the case for naproxen. However, the decrease in the naproxenKm is readily apparent from the data in Table 2.
Another component that can be determined when using the two-site model is the binding constant of dapsone. It is apparent from Table 3 that this constant varies considerably depending on the substrate being activated, which argues for different binding orientations in the active site or changes in binding efficiency. Other results from this study show that the presence of flurbiprofen does not substantially influence the formation of dapsone hydroxylamine, although conditions were optimized for flurbiprofen hydroxylation, as activation conditions were desired. Close examination of data points in Fig. 3 indicates an inadequate estimation of Vmax, which may be the cause for minimal differences in Kmestimates (81–144 μM). In addition, dapsone has been shown previously to have a sigmoidal kinetic profile (Korzekwa et al., 1998), suggesting two binding regions for dapsone, one of which is cooperative. However, the sigmoidal portion of dapsone's kinetic profile was present at very low dapsone concentrations, so either not enough low concentration data points were taken in this study to observe sigmoidal kinetics or perhaps the presence of flurbiprofen altered dapsone kinetics, resulting in a hyperbolic profile. Nonetheless, Michaelis-Menten fits for dapsone hydroxylation in the presence of flurbiprofen appear to be relatively superimposable, suggesting similar kinetic parameters for each condition.
One factor that may affect activation is the presence of cytochromeb5, which has been shown to affect the catalytic activity of certain P450 reactions (Yamazaki et al., 1996). However, our results with baculovirus-expressed CYP2C9 show that activation of flurbiprofen 4′-hydroxylation is notb5-dependent, as activation still occurs in the absence of cytochrome b5 (data not shown). It is also possible that dapsone somehow affects the coupling of NADPH-CYP reductase with CYP to increase catalytic activity of CYP2C9, as was suggested by Shou et al. (1994) with 7,8-benzoflavone and CYP3A4. Another potential contributor to the activation mechanism may be the interaction with or displacement of water molecules from the enzyme active site, thus affecting hydrogen bonding within the active site. The exit of water molecules from the active site could possibly change the conformation of the enzyme if the hydrogen bonds were in positions essential for stabilizing the protein structure (Ekins et al., 1998). It is possible that one, all or even additional factors are contributing to some degree to the activation mechanism of dapsone.
Overall, evidence from this study suggests that dapsone activates the metabolism of multiple substrates of CYP2C9 by binding within the active site in a region that causes positive cooperativity. Thus, studies exploring the CYP2C9 active site will be important, beginning with determining the effects of allelic variants and mutants of CYP2C9 on dapsone activation, which may more conclusively prove that active site binding interactions are of significance. With respect to CYP2C9, the ∗2 (R144C) and ∗3 (I359L) allelic variants and F114L mutant have been shown to variably affect the metabolism of certain compounds (Bhasker et al., 1997; Yamazaki et al., 1998), and thus are currently being studied to help elucidate the mechanism of activation. In addition, more extensive studies exploring the effects of flurbiprofen, naproxen, and piroxicam on dapsone hydroxylamine formation, as well as examining analogs of dapsone to determine structural requirements for activation have begun.
Acknowledgments
We gratefully acknowledge R. J. Armstrong and M. A. Gore of Camitro Corp. (Menlo Park, CA) for providing expressed CYP2C9 microsomes.
Footnotes
-
J.M.H. was supported in part by a fellowship from the American Foundation for Pharmaceutical Education.
- Abbreviations used are::
- P450
- cytochrome P450
- HPLC
- high-performance liquid chromatography
- ESI
- electrospray ionization mass spectrometry
- CLint
- intrinsic clearance
- Received January 31, 2001.
- Accepted March 30, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}