


Abstract
Transformation of escitalopram (S-CT), the pharmacologically active S-enantiometer of citalopram, to S-desmethyl-CT (S-DCT), and ofS-DCT to S-didesmethyl-CT (S-DDCT), was studied in human liver microsomes and in expressed cytochromes (CYPs). Biotransformation of theR-enantiomer (R-CT) was studied in parallel. S-CT was transformed to S-DCT by CYP2C19 (Km = 69 μM), CYP2D6 (Km = 29 μM), and CYP3A4 (Km = 588 μM). After normalization for hepatic abundance, relative contributions to net intrinsic clearance were 37% for CYP2C19, 28% for CYP2D6, and 35% for CYP3A4. At 10 μM S-CT in liver microsomes,S-DCT formation was reduced to 60% of control by 1 μM ketoconazole, and to 80 to 85% of control by 5 μM quinidine or 25 μM omeprazole. S-DDCT was formed fromS-DCT only by CYP2D6; incomplete inhibition by quinidine in liver microsomes indicated participation of a non-CYP pathway. Based on established index reactions, S-CT andS-DCT were negligible inhibitors (IC50> 100 μM) of CYP1A2, -2C9, -2C19, -2E1, and -3A, and weakly inhibited CYP2D6 (IC50 = 70–80 μM).R-CT and its metabolites, studied using the same procedures, had properties very similar to those of the correspondingS-enantiomers. Thus S-CT, biotransformed by three CYP isoforms in parallel, is unlikely to be affected by drug interactions or genetic polymorphisms. S-CT andS-DCT are also unlikely to cause clinically important drug interactions via CYP inhibition.
Previous studies have evaluated the properties of biotransformation of racemic citalopram (CT) to monodesmethylcitalopram (DCT) in vitro by human liver microsomes and by heterologously expressed individual human cytochromes (Kobayashi et al., 1997; Rochat et al., 1997; von Moltke et al., 1999b). These studies indicate that CYP3A4, -2C19, and -2D6 all contribute to this reaction, with approximate relative contributions at low concentrations of CT estimated at 34% for CYP3A, 36% for CYP2C19, and 30% for CYP2D6. Stereoselective clearance has also been suggested, in that intrinsic clearance of S-citalopram (approved generic name of Escitalopram) may exceed that ofR-citalopram (R-CT; Olesen and Linnet, 1999). Clinical studies indicate that steady-state concentrations ofR-CT may exceed those of S-CT during chronic administration of racemic CT to humans, and that the elimination half-life of R-citalopram exceeds that ofS-citalopram (Bondolfi et al., 1996; Foglia et al., 1997;Voirol et al., 1999). These differences are of potential clinical importance, since the S-isomers of CT and DCT have greater intrinsic antidepressant activity than their respectiveR-isomers (Hyttel et al., 1992). S-Citalopram itself is currently under investigation as an antidepressant agent.
The present study evaluated the biotransformation of the individualR- and S-isomers of CT to DCT, and ofR- and S-DCT to didesmethylcitalopram (DDCT), by human liver microsomes, and by microsomes containing pure human cytochromes expressed by cDNA-transfected human lymphoblastoid cells. The inhibitory actions of R- and S-CT, -DCT, and -DDCT versus index reactions representing activity of specific human cytochromes were also evaluated.
Materials and Methods
In Vitro Procedures.
Liver samples from individual human donors with no known liver disease were provided by the International Institute for the Advancement of Medicine, Exton, PA, the Liver Tissue Procurement and Distribution System, University of Minnesota, Minneapolis, MN, and the National Disease Research Interchange, Philadelphia, PA. All samples were of the CYP2D6 and CYP2C19 normal metabolizer phenotype based on prior in vitro phenotyping studies.
Microsomes were prepared by ultracentrifugation; microsomal pellets were suspended in 0.1 M potassium phosphate buffer containing 20% glycerol and stored at −80°C until use. Microsomes containing individual human cytochromes expressed by cDNA-transfected human lymphoblastoid cells (Gentest, Woburn, MA) were similarly stored at −80°C until use. Pure samples of R- and S-CT, -DCT, and -DDCT were provided by H. Lundbeck A/S (Copenhagen-Valby, Denmark) through Forest Laboratories (New York, NY). Other chemical reagents and drug entities were purchased from commercial sources or kindly provided by their pharmaceutical manufacturers.
Incubation mixtures contained 50 mM phosphate buffer, 5 mM Mg2+, 0.5 mM NADP+, and an isocitrate/isocitric dehydrogenase regenerating system. Appropriate substrates, with and without inhibitor in methanol solution, were added to a series of incubation tubes. The solvent was evaporated to dryness at 40°C under conditions of mild vacuum. Reactions were initiated by addition of microsomal protein (0.25 mg/ml for liver microsomes, 1.0 mg/ml for expressed CYPs). After an appropriate incubation duration at 37°C, reactions were stopped by cooling on ice and addition of 100 μl of acetonitrile. Internal standard was added, the incubation mixture was centrifuged, and the supernatant was transferred to an autosampling vial for high performance liquid chromatography analysis. The mobile phase consisted of a combination of acetonitrile and 50 mM phosphate buffer as appropriate for each assay. The analytical column was stainless steel, 30 cm × 3.9 mm, containing reverse-phase C-18 μBondapak, or 15 cm × 3.9 mm, containing reverse-phase C-18 Novapak (Waters Associates, Milford, MA). Column effluent was monitored by ultraviolet absorbance at the appropriate wavelength. All biotransformation reactions were verified to be in the linear range with respect to incubation duration and protein concentration.
Inhibition Studies.
The potential inhibitory effect of the stereoisomers of CT, DCT, and DDCT on the activity of six human cytochromes was evaluated using index reactions and methods as follows (Table1): CYP1A2, phenacetin (100 μM) to acetaminophen (von Moltke et al., 1996a; Venkatakrishnan et al., 1998b); CYP2C9, tolbutamide (100 μM) to hydroxytolbutamide (Venkatakrishnan et al., 1998c); CYP2C19, S-mephenytoin (25 μM) to 4′-OH-mephenytoin (Venkatakrishnan et al., 1998c); CYP2D6, dextromethorphan (25 μM) to dextrorphan (Schmider et al., 1997; von Moltke et al., 1998c); CYP2E1, chlorzoxazone (50 μM) to 6-OH-chlorzoxazone (Court et al., 1997); CYP3A, triazolam (250 μM) to α-OH-triazolam (von Moltke et al., 1996b, 1998a, 2001; Perloff et al., 2000). Substrate specificity in the potency of CYP3A inhibitors has been suggested (Kenworthy et al., 1999); triazolam is an index CYP3A substrate reported to be representative of the most common substrate category.
In vitro systems for evaluating CYP inhibitory activity of citalopram stereoisomers and metabolites
Varying concentrations of R- and S-CT,R- and S-DCT, and R- andS-DDCT (0–250 μM) were coincubated with fixed concentrations of the index substrate. Also studied in each system were “positive control” index inhibitors, as well as SSRIs previously established as inhibitory for that specific reaction (Table 1).
Reaction velocities with coaddition of inhibitor were expressed as a percentage ratio (Rv) of the control velocity with no inhibitor present. The relation of Rv to inhibitor concentration was analyzed by nonlinear regression to determine the inhibitor concentration producing a 50% decrement in reaction velocity (IC50) (von Moltke et al., 1998b).
Enzyme Kinetic Studies.
Varying concentrations of R-CT, S-CT,R-DCT, and S-DCT (0–1000 μM) were added to incubation tubes, and the solvent evaporated to dryness. Incubations with human liver microsomes or with heterologously expressed human cytochromes (Crespi, 1995; Crespi and Penman, 1997; Crespi and Miller, 1999) were performed as described above and as reported previously (von Moltke et al., 1999b). Reactions were stopped by addition of acetonitrile and by cooling on ice. Internal standard (phenacetin) was added, the mixtures were centrifuged, and supernatants were transferred to high performance liquid chromatography autosampling vials. The mobile phase was 70% 50 mM KH2PO4 and 30% acetonitrile at 2 ml/min. Column effluent was monitored by ultraviolet absorbance at 230 nm. Concentrations of DCT (following incubation withR- or S-CT) or of DDCT (following incubation withR- or S-DCT) were determined using calibration curves containing known amounts of DCT or DDCT. Reaction velocities were calculated in units of picomoles per minute per milligram of microsomal protein for liver microsomal studies, or picomoles per minute per picomole of CYP in expressed cytochrome studies.
Liver Microsomes.
Biotransformation of R- or S-CT to DCT was consistent with Michaelis-Menten kinetics in four of eight instances. The following equation was fitted to data points using nonlinear regression:
Biotransformation of R- and S-DCT to DDCT was consistent with a two-enzyme process, with the high-affinity (lowKm) component consistent with a Michaelis-Menten process. The low-affinity process required approximation by a linear function. The following equation was fitted to data points.
Heterologously Expressed Cytochromes.
Initial screening studies were performed to evaluate the possible formation of DCT from R- and S-CT, and of DDCT from R- and S-DCT, using expressed CYP1A2, -1A6, -2B6, -2C9, -2C19, -2D6, -2E1, and -3A4. Formation of DCT from the enantiomers of CT was mediated by CYP2C19, -2D6, and -3A4; other CYPs did not yield detectable metabolite formation. Transformation of the enantiomers of DCT to DDCT was mediated only by CYP2D6, with no detectable metabolic activity from other cytochromes.
Accordingly, transformation of R- and S-CT to DCT was characterized as described above using varying concentrations ofR- and S-CT incubated with heterologously expressed CYP2C19, -2D6, and -3A4 in separate experiments. Transformation of R- and S-DCT to DDCT was characterized using heterologously expressed CYP2D6.
The relative activity factor approach was used to estimate relative contributions of CYP2C19, -2D6, and -3A4 to net clearance ofR-CT and S-CT. Average relative in vivo abundances, equivalent to the relative activity factors, were estimated using methods described in detail previously (Crespi, 1995;Venkatakrishnan et al., 1998a,c, 1999, 2000, 2001; von Moltke et al., 1999a,b; Störmer et al., 2000). These were 13.1% for CYP2C19, 3.8% for -2D6, and 83.1% for -3A4. Vmaxand intrinsic clearance values for the individual cytochromes were multiplied by the average relative abundances and the values were normalized to 100%. The resulting values were used to estimate the contribution of each cytochrome to net reaction velocity in relation to concentration of R-CT or S-CT.
Chemical Inhibition of R-CT, S-CT,R-DCT, and S-DCT Biotransformation in Microsomes.
Human liver microsomes were incubated with fixed concentrations (10 μM or 100 μM) of R-CT or S-CT. These were performed in the control condition (without inhibitors), or with coaddition of index inhibitors corresponding to the three CYP isoforms participating in CT biotransformation (Table 1). These were omeprazole (25 μM) versus CYP2C19, quinidine (5 μM) versus CYP2D6, and ketoconazole (1 μM) versus CYP3A. We recognize that 25 μM omeprazole may not be fully specific as a CYP2C19 inhibitor (Ko et al., 1997; Giancarlo et al., 2001). The effect of quinidine on biotransformation of R-CT and S-CT to DDCT was studied using similar methods. Reaction velocities with coaddition of inhibitor were expressed as a percentage ratio of the control velocity with no inhibitor present.
Results
Inhibition Studies.
In all systems the positive control inhibitors produced the expected degree of inhibition of their respective index reactions (Table 1).
CYP1A2.
R- and S-CT and metabolites all were negligible inhibitors of phenacetin O-deethylation, the index reaction for CYP1A2. None of these compounds produced 50% inhibition. The mean IC50 for α-naphthoflavone was 0.2 μM, and the mean IC50 for fluvoxamine was 0.3 μM.
CYP2C9.
R-CT, S-CT, R-DCT, andS-DCT were weak inhibitors of CYP2C9, represented by tolbutamide hydroxylation, with less than 50% inhibition produced even at 250 μM. R-DDCT and S-DDCT produced a moderate degree of inhibition, with IC50 values of 30.7 (±6.3) μM and 25.7 (±8.0) μM, respectively. Sulfaphenazole was a strong inhibitor (IC50 = 1.3 μM), and the SSRI fluvoxamine also was a moderately strong inhibitor (IC50 = 9.4 μM).
CYP2C19.
R- and S-CT were very weak inhibitors, with less than 50% inhibition of S-mephenytoin hydroxylation even at 100 μM. R- and S-DCT also were weak inhibitors.R- and S-DDCT were moderate inhibitors, with mean IC50 values of 18.7 and 12.1 μM, respectively. Omeprazole was a strong inhibitor of CYP2C19, as was the SSRI fluvoxamine (see Table 2).
IC50 values versus S-mephenytoin hydroxylation (CYP2C19) or dextromethorphan O-demethylation (CYP2D6)
CYP2D6.
Only R-DCT had potentially important inhibiting potency versus CYP2D6, represented by dextromethorphanO-demethylation. The mean IC50 was 25.5 (± 2.1) μM. This is very close to the inhibitory potency of sertraline and is consistent with clinical data suggesting that racemic citalopram and sertraline have comparably weak CYP2D6 inhibitory potency. The SSRI paroxetine was at least an order of magnitude more potent (IC50 = 2.6 μM) than R-DCT as a CYP2D6 inhibitor (see Table 2). Fluoxetine and norfluoxetine (mean IC50 = 2.0 and 2.7 μM, respectively) also were strong CYP2D6 inhibitors.
CYP3A.
CT and metabolites all were very weak or negligible inhibitors of CYP3A, as indicated by triazolam hydroxylation. None of the compounds (at 100 μM) produced more than 50% inhibition. Fluvoxamine and nefazodone were moderately strong inhibitors (von Moltke et al., 1996b,1999c).
CYP2E1.
CT and metabolites were weak or negligible inhibitors of chlorzoxazone 6-hydroxylation, producing less than 20% inhibition at 250 μM.
Enzyme Kinetic Studies: Liver Microsomes.
The mean Km for biotransformation ofR-CT to R-DCT was higher than for S-CT (256 versus 165 μM) (Fig. 1). Using theVmax/Km ratio (intrinsic clearance) as an approximation of metabolic activity at low substrate concentrations, the mean ratio for S-CT exceeded that for R-CT (6.1 versus 4.9 μl/min/mg protein), but the difference was not statistically significant based on Student's pairedt test.
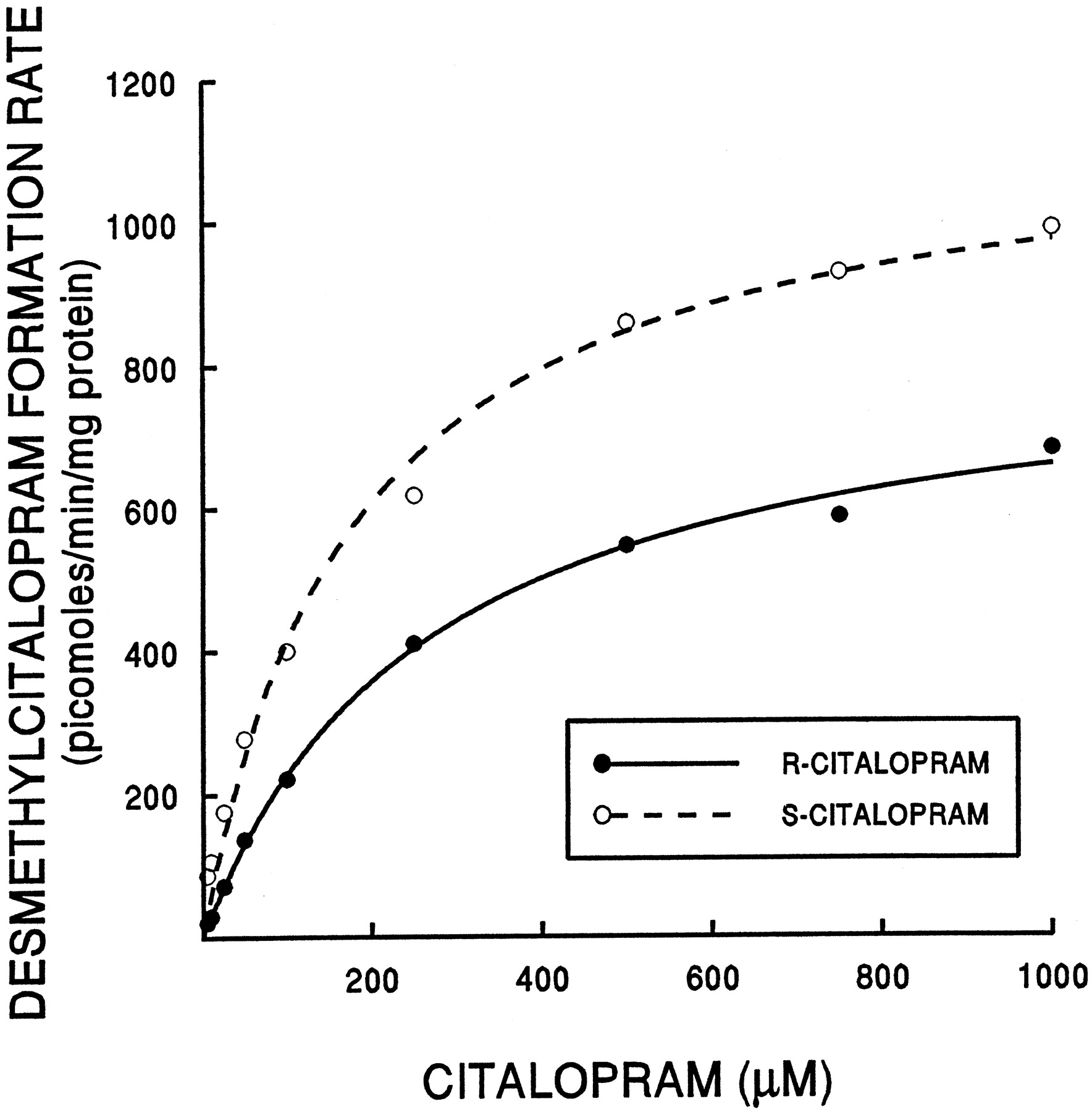
Formation of DCT from R- and S-CT by liver microsomes from a representative human liver sample.
Actual data points and functions of best fit are shown.
The mean Km for biotransformation ofR-DCT to R-DDCT was higher than forS-DCT (108 versus 72 μM) (Fig.2). Based on theVmax/Km ratio, intrinsic clearance for S-DCT was slightly higher than forR-DCT (1.32 versus 1.17 μl/min/mg protein). Furthermore, intrinsic clearances for DCT formation from R- orS-CT both were higher than clearances for transformation ofR- or S-DCT to DDCT.

Formation of DDCT from R- and S-DCT by human liver microsomes from the same human liver sample as in Fig. 1.
Actual data points and functions of best fit are shown.
Enzyme Kinetic Studies: Individual Human Cytochromes.
Table 3 shows enzyme kinetic values for formation of DCT from R- and S-CT by heterologously expressed human CYP2C19, -2D6, and -3A4 (Fig.3). Consistent with prior studies of racemic CT (von Moltke et al., 1999b), CYP2D6 had the highest affinity (lowest Km), CYP3A4 had the lowest affinity, and CYP2C19 fell in between. This was true for bothR-CT and S-CT.Vmax/Km ratios corresponding to each of the isoforms were higher for S-CT than for R-CT.
Kinetic analysis of biotransformation of citalopram and desmethycitalopram enantiomers by heterologously expressed human cytochromes

Formation of DCT from R- or S-CT by heterologously expressed CYP2D6, -2C19, and -3A4.
See Table 3 for kinetic parameters. Contributions have not been normalized for relative abundance of the three CYP isoforms. Left,R-CT as substrate (open square is an outlying data point for CYP2C19; this was not used in analysis); right, S-CT as substrate.
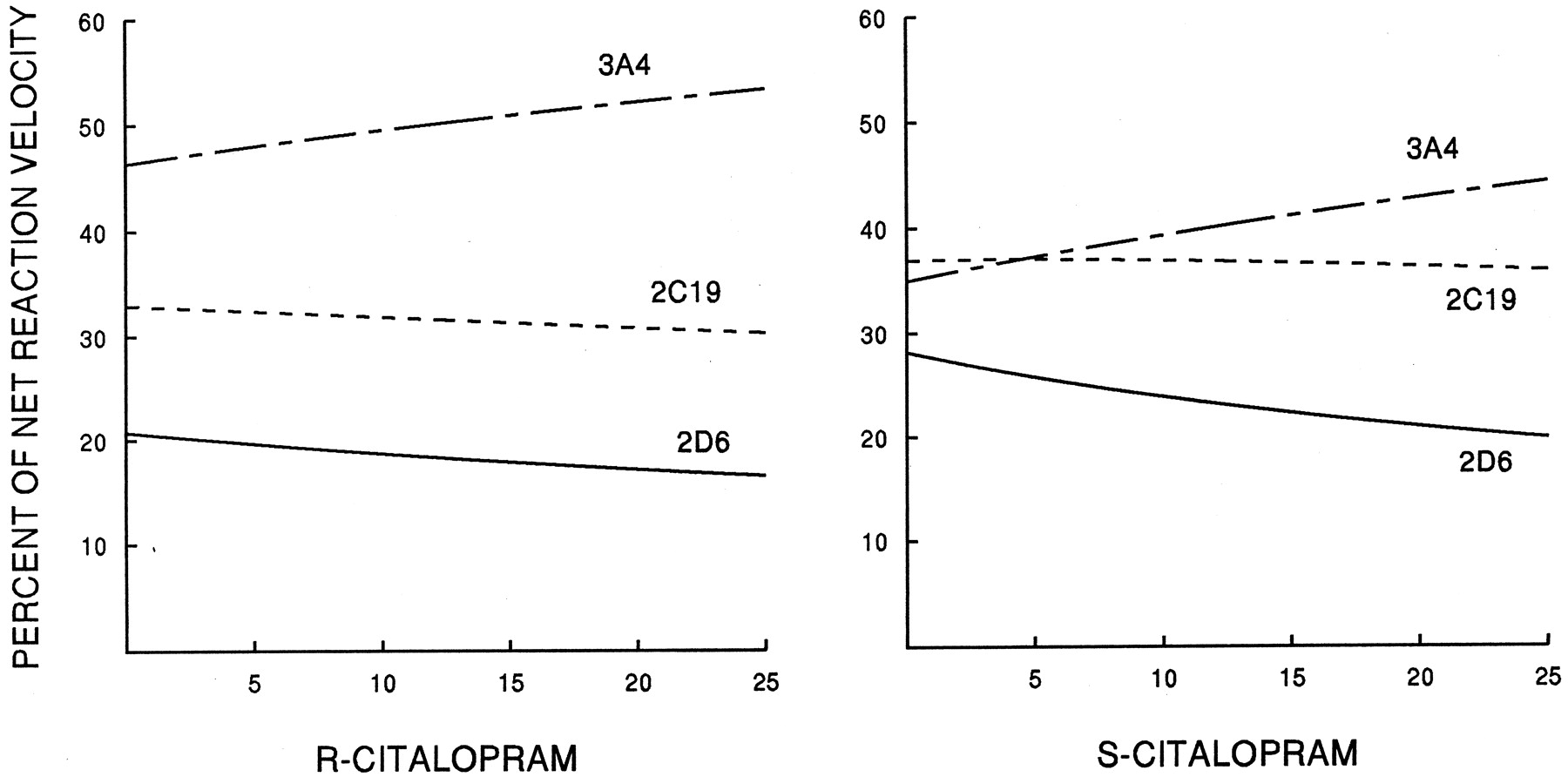
After normalization for estimated relative abundance of the three individual CYP isoforms, CYP3A accounted for 46% of net intrinsic clearance of R-CT, CYP2C19 for 33%, and CYP2D6 for 21%. For S-CT, CYP3A accounted for 35% of net clearance, CYP2C19 for 37%, and CYP2D6 for 28% (Fig. 4) (Table 3).

Predicted relative contributions of CYP2D6, -2C19, and -3A4 to net rate of formation of DCT from R- or S-CT.
Contributions have been normalized for relative abundance of the three CYP isoforms. Left, R-CT as substrate; right,S-CT as substrate.
Figure 5 shows the biotransformation ofR-DCT and S-DCT to DDCT by heterologously expressed CYP2D6. Km values (12.4 and 16.8 μM, respectively; Table 3) were lower than the high-affinity components of the reaction in liver microsomes.

Formation of DDCT from R- or S-DCT by heterologously expressed CYP2D6.
Left, R-DCT as substrate; right, S-DCT as substrate.
Chemical Inhibition of CT and DCT Biotransformation in Microsomes.
At 10 μM R- or S-CT, ketoconazole reduced reaction velocity to 55 to 60% of control, quinidine to 80% of control, and omeprazole to 80 to 85% of control (Fig.6). When the R- andS-CT concentration was increased to 100 μM, the degree of inhibition by ketoconazole increased, while inhibition by quinidine decreased (Fig. 6). These findings are consistent with the data from heterologously expressed CYP isoforms.

Inhibition of formation of DCT from R- or S-CT by ketoconazole, quinidine, or omeprazole.
Reaction velocities are expressed as a percentage of the control velocity without inhibitor. Each bar represents the mean (± S.E.) value for four separate human liver samples. Left, R- orS-CT concentration, 10 μM; right, R- orS-CT concentration, 100 μM.
Quinidine (5 μM) reduced formation of DDCT from R- orS-DCT to only 70 to 80% of control (Fig.7), whereas the same concentration of quinidine reduced formation of R- and S-DCT to less than 10% of control values in heterologously expressed CYP2D6.

Inhibition of formation of DDCT from R-DCT or S-DCT by quinidine at substrate concentrations of 10 μM (n = 3, left) or 100 μM (n = 7, right).
Reaction velocities are expressed as a percentage of the control value without inhibitor. Each point represents the mean (± S.E.) value for a series of human liver samples.
Discussion
R-CT, S-CT, R-DCT, andS-DCT are weak or negligible inhibitors of human CYP1A2, -2C9, -2C19, -2E1, and -3A in human liver microsomes. R- andS-CT also are weak or negligible inhibitors of CYP2D6. TheR-isomer of DCT is a weak to moderate inhibitor of CYP2D6, comparable in potency to sertraline. R- andS-DDCT are moderate inhibitors of CYP2C9 and -2C19. However, this is unlikely to be of clinical importance due to the low plasma levels (less than 0.05 μM) of DDCT achieved clinically (Baumann and Larsen, 1995).
Transformation clearance of S-CT to DCT in liver microsomes is higher than that of R-CT, accounting for the trend to higher plasma levels of R-CT during clinical use of racemic citalopram and the shorter elimination half-life of S-CT (Bondolfi et al., 1996; Foglia et al., 1997; Voirol et al., 1999). Formation clearance of DCT from CT exceeds elimination clearance of DCT to DDCT. Since plasma levels of DCT do not exceed those of CT during clinical use of racemic citalopram, the findings suggest that another metabolic pathway (in addition to formation of DDCT) or another mechanism of elimination may contribute to DCT clearance (Rochat et al., 1998; Dalgaard and Larsen, 1999).
Studies with heterologously expressed human CYP isoforms indicate that CYP2D6, -2C19, and -3A all contribute to formation of DCT fromR- or S-CT, with CYP3A accounting for 35 to 46% of net intrinsic clearance. The contribution of CYP3A is predicted to increase at higher concentrations of CT, while the contribution of CYP2D6 is predicted to decrease. This was verified by studies of chemical inhibition of this reaction in liver microsomes by index inhibitors. As in the case of liver microsomes, intrinsic clearance ofS-CT by the three CYP isoforms was higher than that ofR-CT.
CYP2D6 was the only identified CYP isoform mediating formation of DDCT from R- or S-DCT. However, these were high-affinity (low Km) reactions in expressed CYP2D6, with Km values lower than the high-affinity component in liver microsomes. Furthermore, the reaction was incompletely inhibited by quinidine (5 μM) in liver microsomes but was extensively inhibited by quinidine in expressed CYP2D6. This again suggests participation of some other process mediating net biotransformation of DCT in liver microsomes and in vivo.
Thus S-CT is biotransformed to its principal demethylated metabolite by three distinct human CYP isoforms in parallel. As such, impaired activity of any one of these isoforms due to a drug interaction or a genetic “poor metabolizer” polymorphism is unlikely to have a large effect on net metabolic clearance. As examples, clearance of racemic CT among individuals identified as phenotypic poor metabolizers of CYP2D6 or CYP2C19 was only modestly lower than in normal metabolizer controls (Sindrup et al., 1993). Coadministration of ketoconazole with racemic CT produced a reduction in CT clearance of approximately 10%, which was not statistically significant (Gutierrez and Abramowitz, 2001). Cimetidine, an inhibitor of multiple CYP isoforms, reduced clearance of racemic CT by 30% (Priskorn et al., 1997a). S-CT and its metabolites themselves are weak or negligible inhibitors of human CYP isoforms, indicating a low likelihood of clinically important drug interactions due to impaired CYP activity (Gram et al., 1993; Priskorn et al., 1997b; Avenoso et al., 1998; Taylor et al., 1998; Nolting and Abramowitz; 2000; von Bahr et al., 2000).
Acknowledgments
We are grateful for the collaboration and assistance of Karthik Venkatakrishnan, Ph.D.
Footnotes
-
This work was supported by Grants MH-01237, MH-58435, MH-34223, DA-13209, DK/AI-58496, and DA-05258 from the Department of Health and Human Services, and by a grant from Forest Laboratories, New York, NY.
- Abbreviations used are::
- CT
- citalopram
- DCT
- desmethylcitalopram
- DDCT
- didesmethylcitalopram
- CYP
- cytochrome P450
- SSRI
- selective serotonin reuptake inhibitor
- Received January 2, 2001.
- Accepted April 16, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}