


Abstract
CYP1A1, a major phase I enzyme, plays an important role in the metabolism of polycyclic aromatic hydrocarbons and in the chemical activation of xenobiotics to carcinogenic derivatives. The phenolic antioxidant tert-butylhydroquinone (tBHQ), often used as a food preservative, is generally considered to act only as a mono-functional inducer of phase II enzymes, thereby exerting chemo-protection. However, we recently observed that tBHQ elevated the activity of an aryl hydrocarbon receptor (AhR) response element (DRE)-driven luciferase reporter in human colon carcinoma cells (Caco-2). Therefore, we studied the effects of tBHQ on the activity of a DRE-driven reporter, CYP1A1 mRNA expression, and CYP1A enzyme activity in Caco-2 cells and human HepG2 hepatoma cells. We found tBHQ caused induction of reporter activity and CYP1A1 expression and activity in Caco-2 and HepG2 cells. Moreover, tBHQ combined with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) increased reporter activity and mRNA expression in Caco-2 cells in an additive manner. By contrast, tBHQ decreased TCDD-mediated induction of reporter activity and CYP1A1 mRNA expression in HepG2 cells. Resveratrol, an AhR antagonist, repressed the induction of CYP1A1 by tBHQ. Cotransfection of HepG2 cells with a dominant negative AhR nuclear translocator mutant abolished the tBHQ-induced CYP1A1 reporter activity. These findings indicate that CYP1A1 may be induced by the antioxidant tBHQ via an AhR-dependent mechanism.
The CYP1A subfamily catalyzes the metabolic activation of polycyclic aromatic hydrocarbons (PAHs), which generates genotoxic metabolites that bind to DNA, thus mediating PAH-induced carcino-genesis. Polyhalogenated PAHs, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), are also responsible for a variety of toxic responses, such as immune suppression, birth defects (cleft palate), and endocrine disruption (Poland and Knutson, 1982). Classically, transcription of several members of the CYP1A subfamily, such as CYP1A1/2 (Denison et al., 1989; Nebert and Jones, 1989; Quattrochi and Tukey, 1989) and CYP1B1 (Sutter et al., 1992), is induced by PAHs such as TCDD via activation of the aryl hydrocarbon receptor (AhR) (Poland et al., 1974), also called the dioxin receptor. TCDD binds to the cytosolic AhR ligand binding subunit, which undergoes an activation process involving several steps: translocation into the nucleus, release of 90 kDa heat-shock proteins, and heterodimerization with a partner protein, the AhR nuclear translocator (Arnt) (Denison and Nagy, 2003). This heterodimer interacts with a 5′-GCGTG-3′ DNA sequence, the core binding motif of the xenobiotic response element or dioxin response element (DRE) (Fujisawa-Sehara et al., 1987; Kubota et al., 1991), located and present in multiple copies in the upstream region of the CYP1A1 gene promoter. The human CYP1A1, the mouse Cyp1a2, and the mouse Cyp1b1 genes harbor 10, 12, and 11 DRE motifs (Zhang et al., 2003) in their respective upstream regions; however, not all the DREs are functional. Recently, we have identified four functional DREs within a 1.4-kilobase (kb) 5′-regulatory region of the human CYP1A1 gene enhancer (Kress et al., 1998).
AhR-mediated signaling pathways provide a first line of defense against potentially toxic environmental contaminants. However, induction of metabolic processes by the AhR can also produce highly carcinogenic metabolites, creating a link between AhR activation and chemical carcinogenesis. AhR-mediated induction can transcriptionally activate genes encoding enzymes of phase I and phase II and is therefore termed bifunctional, whereas antioxidants selectively induce phase II enzymes and are therefore termed monofunctional (Prochaska and Talalay, 1988). The antioxidant tert-butylhydroquinone (tBHQ), the major metabolite of 3-tert-butyl-hydroxyanisol in dogs, rats, and humans (Nakamura et al., 2003), has been found to be a prototype inducer of a novel, nonreceptor signaling pathway triggered by oxidative/electrophile stress. In the case of rodent glutathione S-transferases (GSTs) and rat and human NAD(P)H quinone oxidoreductase-1 (NQO1), antioxidant response elements (AREs) have been characterized in their 5′-regulatory regions mediating their transcriptional activation (Rushmore et al., 1990; Jaiswal, 1994). Oxidative/electrophile stress has also been proposed to activate via protein kinase (i.e., mitogen-activated protein kinases) transcription factors such as nuclear factor erythroid 2-related factor 2 (Nrf2), which binds to the ARE (Tony-Kong et al., 2001), thereby inducing the respective phase II enzyme.
Recent observations in Caco-2 cells (clone TC7) have shown induction of CYP1A activity (Münzel et al., 1999) and CYP1A1 reporter gene activity either by tBHQ alone or combined with TCDD. CYP1A1 transcriptional induction by oltipraz in Caco-2 cells was also recently described by Le Ferrec et al. (2002) as AhR- and calcium-dependent.
To study the mechanism of CYP1A1 induction by tBHQ, DRE-containing reporter vectors were transiently transfected into cells of two selected lines representing hepatocytes in liver (HepG2) and enterocytes in colon (Caco-2). These cell types should mimic the in vivo situation where environmental and food contaminants accumulate in the gastrointestinal tract, which expresses CYP1A metabolic capacity (de Waziers et al., 1990), and are further passed to the liver. In addition to DRE-containing reporter constructs, induction of CYP1A1 by tBHQ was investigated on mRNA and enzyme activity levels. The possible AhR/Arnt-DRE mechanism was examined using the AhR antagonist resveratrol, a dominant negative Arnt (dnArnt) mutant, and mutated DRE reporter constructs. Our findings provide strong evidence for an involvement of the AhR/Arnt-DRE mechanism in induction of CYP1A1 by the antioxidant tBHQ.
Materials and Methods
Materials. tBHQ was purchased from Fluka (Buchs, Switzerland). TCDD was provided by Ökometric (Bayreuth, Germany). Resorufin, resveratrol, N-acetyl-l-cysteine (NAC), dicumarol, and Tris-(hydroxymethyl) aminomethane hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO). TRIzol reagent, nonessential amino acids, and trypsin/EDTA solution were purchased from Invitrogen (Carlsbad, CA). Oligonucleotide primers were purchased from MWG (Ebersberg, Germany). Dulbecco's modified Eagle's medium and penicillin/streptomycin were purchased from Biochrom (Berlin, Germany). Agarose peqGold Universal, DNA 1-kb ladder, reverse transcriptase peqGold AMV, and GenePorter were purchased from peqLab (Erlangen, Germany). The polymerase chain reaction (PCR) kit LightCycler FastStart DNA Master SYBR Green I was purchased from Roche Diagnostics (Mannheim, Germany). All the other chemicals were purchased from Merck (Darmstadt, Germany).
Cell Culture and Treatments. Human Caco-2 cells, clone TC7, were maintained as described previously (Münzel et al., 1999). Briefly, cells were cultured on 100-mm dishes in Dulbecco's modified Eagle's medium supplemented with 20% fetal calf serum and nonessential amino acids. The human hepatoblastoma cell line HepG2 (American Type Culture Collection, Manassas, VA) was cultured in α-minimum essential medium (Life Technologies, Eggenfelden, Germany) supplemented with 10% fetal calf serum, and cells were maintained at 37°C in a humidified atmosphere of 5% CO2/air. All the reagents for cell treatment were dissolved in dimethyl sulfoxide (DMSO). Cells were treated with the indicated substances and harvested after 24 or 48 h for RNA isolation or for measuring CYP1A activity as ethoxyresorufin-O-deethylase (EROD) activity, respectively. Transfected cells were treated after 24 h (electroporation) or 5 h (lipofection) two times for 24 h each. Solvent controls contained 0.2% DMSO. Treatment with dicumarol (10 μM), NAC (10 μM), or resveratrol (10 μM) was performed 24 h before addition of tBHQ or TCDD or both. Cells were washed with phosphate-buffered saline before harvest and stored at –80°C.
Plasmids, Generation of CYP1A1 Reporter Constructs, and Deletion Mutants.Recombinant Plasmid ArntΔb. ArntΔb/pcDNA3.1 lacking codons 66 through 87 (basic region of the bHLH motif) of native Arnt (Lindebro et al., 1995) was provided by Dr. L. Poellinger (Karolinska Institute, Stockholm, Sweden).
CYP1A1 Reporter Constructs. Reporter gene constructs containing CYP1A1 promoter were generated by PCR amplification of genomic DNA from HepG2 cells using forward primer 5′-ATGGGAGGTGAGGGGATTATTTTC-3′ and reverse primer 5′-CCGAGGAGGGGGCTTGAG-3′. The resulting 1.2-kb PCR fragment was cloned into vector pCR 2.1-TOPO (Invitrogen), and orientation and integrity were confirmed by sequencing. The resulting vector and the reporter vector pT81Luc were digested with XhoI and HindIII, and after purification and ligation of the fragments they resulted in plasmid pT81Luc/hCYP1A1-5′-wt. pT81 Luc was a gift of Dr. F. Gaunitz (Leipzig, Germany). The pT81/3xDRE reporter construct was generated using the motif of DRE3 from the CYP enhancer region (Kress et al., 1998) fused in triplicate tandem array into pT81Luc.
Generation of DRE Point Mutants by Site-Directed Mutagenesis. Point mutations in the DRE motifs (C, D, E, and F) (Kress et al., 1998) of the CYP1A1 reporter plasmid were generated according to the instructions of the manufacturer using the Chameleon, Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The constructs were sequenced in both directions to ensure that the mutations had been introduced at the correct sites during PCR and called pT81/CDEF.
Transfection of CYP1A1 Reporter Plasmids and Expression Assay. Electroporation was used for transfection of Caco-2 cells. Briefly, Caco-2 cells were trypsinized and diluted by culture medium. Caco-2 cells (107 cells/ml) were suspended in cytomix (60 mM KCl, 75 mM CaCl2, 5 mM KH2PO4, 1 mM EGTA, 2.5 mM MgCl2, 2 mM ATP, 5 mM glutathione, and 12 mM HEPES buffer, pH 7.6). Luciferase reporter constructs (16 pmol/ml) were added, and electroporation was carried out at 4°C with 659 V/cm and exponential pulse half-time of 35 ms. To control transfection efficiency (∼10%) and possible effects of inducers on cell growth, Renilla luciferase gene pTKRL (94 fmol/ml) (Promega, Madison, WI) was cotransfected. Cells were suspended immediately after the pulse in prewarmed culture medium and plated at approximately 70% confluence. The transfected cells (4 dishes/group) were then treated with the inducers and further cultivated for 42 h. Alternatively, chemical transfection with GenePorter (peqLab) was used for HepG2 cells according to the manufacturer's instructions. One microgram of luciferase reporter plasmid, 23.8 ng of pTKRL reference vector, and 5 μl of transfection reagent were used for 1 well of a 24-well plate. Firefly luciferase gene expression, normalized for Renilla luciferase activity, was analyzed using the Dual Luciferase Kit (Promega).
Real-Time PCR. Cells were washed twice with ice-cold 0.9% NaCl solution and scraped off the plate. RNA was isolated using TRIzol (Invitrogen) and quantified spectrophotometrically at 260 nm and 280 nm. Five hundred nanograms of total RNA was heated at 95°C for 5 min and cooled on ice. RNA-derived synthesis of CYP1A1 cDNA was carried out with the following reaction conditions: 0.5 μg of RNA, 5 mM MgCl2, 1 mM dNTP, 0.5 U RNasin (Promega), 12.5 U peqGold avian myeloblastosis virus reverse transcriptase (peqLab), and 0.25 μg of oligo(dT18) and 0.25 μg of random hexamers as primers in a final volume of 20 μl of 1× GeneAmp PCR buffer II (Applied Biosystems, Foster City, CA). The samples were incubated at 42°C for 60 min, and reverse transcriptase was inactivated by heating at 95°C for 5 min. PCR was carried out in the LightCycler instrument using the FastStart DNA Master SYBR Green I kit (Roche). PCR reactions contained 2.4 mM MgCl2 and 0.5 μM concentration of both forward and reverse primers: human CYP1A1 (GenBank: NM_000499) forward primer, 5′-GTCTTTCTCTTCCTGGCTATC-3′ and human CYP1A1 reverse primer, 5′-TACCTGTTGTCTCTGGAGGGT-3′. PCR temperature program consisted of heating to 95°C for 5 s, followed by annealing at 55°C for 5 s and elongation at 72°C for 14 s. The PCR product after 45 cycles resulted in a 353-bp fragment. The relative expression ratio of the target gene CYP1A1 in the samples was computed based on the crossing point difference of a sample versus a control and its real-time PCR efficiency, according to Pfaffl (2001). Cyclophilin B (peptidyl-prolyl isomerase B) was used as reference gene to account for any variance in the quality of mRNA and the amount of input cDNA. To determine real-time PCR efficiencies, standard curves constructed with cloned standards of CYP1A1 and peptidyl-prolyl isomerase B cDNA were generated for each transcript.
Determination of Enzyme Activities. Cells were washed twice and harvested by scraping off in cold NaCl solution (0.9%). The cells were then centrifuged for 10 min at 500g and at 4°C. The supernatant was removed; 0.25 M sucrose/0.01 M Tris-HCl, pH 7.4 (buffered sucrose), was added; and the cells were homogenized by sonication for 2 × 5 s. CYP1A activity was measured as EROD activity according to Lubet et al. (1985). Protein concentration was determined in homogenates using bovine serum albumin as a standard (Lowry et al., 1951).

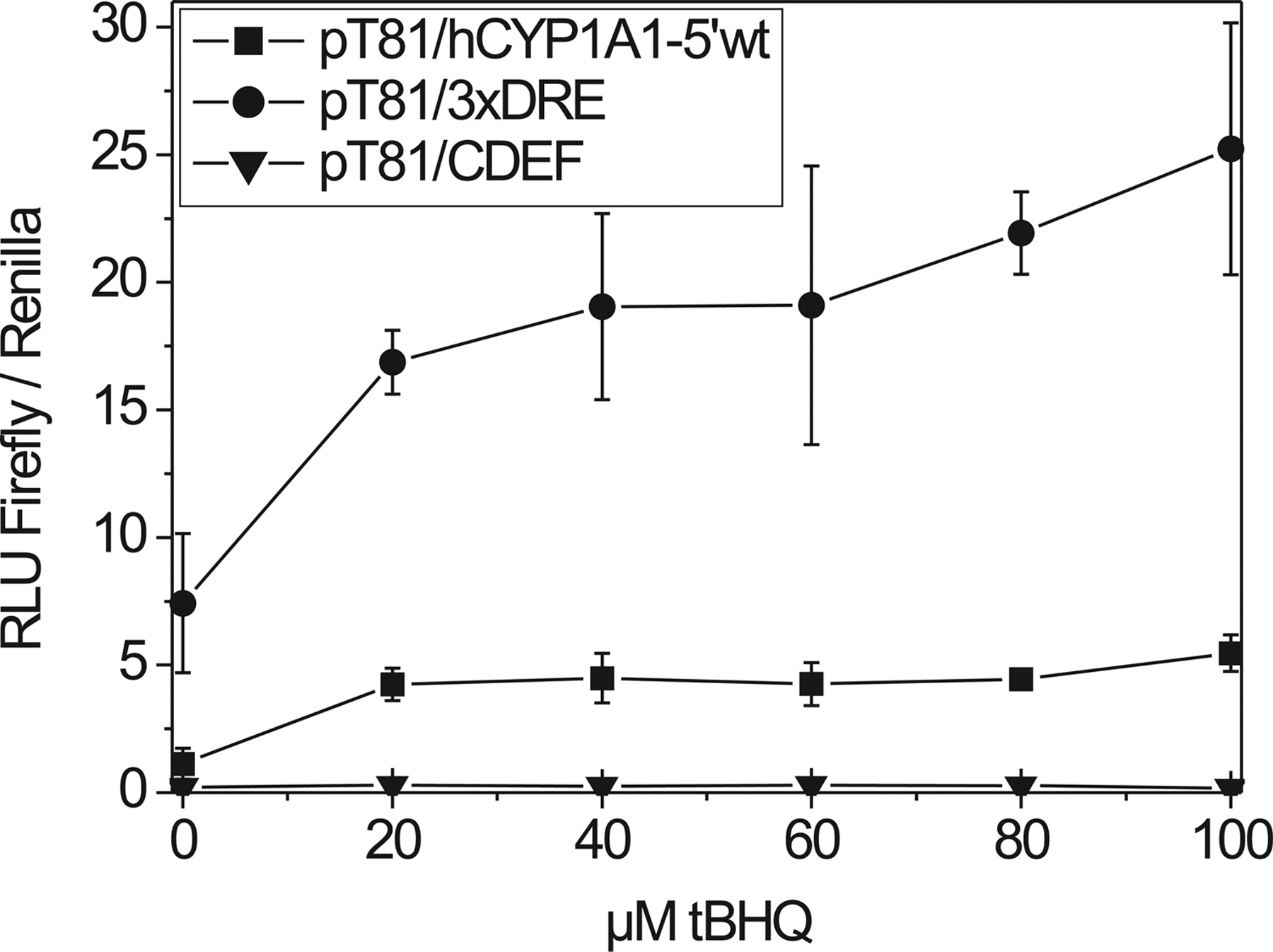
Effects of increasing concentrations of tBHQ on the activity of pT81/hCYP1A1-5′-wt-, 3xDRE-, and CDEF mutant-driven luciferase reporters. HepG2 cells were transiently transfected with one of the three reporter constructs and incubated with vehicle (0.2% DMSO) or tBHQ (20, 40, 60, 80, and 100 μM). Mean ± S.D. of four independent experiments is shown (RLU, relative luminescence units).
Statistical Analysis. For statistical analysis, the Student's t test was used. The differences were considered significant when P < 0.05.
Results
tBHQ-Mediated Induction of CYP1A1 Reporter Constructs in HepG2 Cells. HepG2 cells were transiently transfected with one of the following reporter constructs: 1) pT81/hCYP1A1-5′-wt, containing nine DREs, including the four functional DREs, 3, 4, 5, and 6 (also termed C, D, E, and F); 2) a mutant construct derived thereof by site-directed mutagenesis harboring knockouts of the four functional DREs, 3, 4, 5, and 6 (termed pT81/CDEF); and 3) an artificial construct containing three DRE motifs (pT81/3xDRE). Cells were incubated in the presence of varying concentrations of tBHQ (20–100 μM) for 48 h. The activities of the pT81/hCYP1A1-5′-wt and pT81/3xDRE reporters were inducible by tBHQ: in a concentration-dependent manner in the case of pT81/3xDRE (up to 3-fold at 80 μM tBHQ), whereas in the case of pT81/hCYP1A1-5′ no concentration dependence was observed (Fig. 1). For all the further experiments, a concentration of 80 μM tBHQ was chosen, at which significant induction of luciferase reporters and CYP1A1 mRNA was seen (Münzel et al., 1999), and no cytotoxic effects were observed in HepG2 and Caco-2 cells. Reporter activity was almost completely abolished in the CDEF mutant (Fig. 1), indicating that DRE motifs in the enhancer region of CYP1A1 contribute to transcriptional activation of the gene by the antioxidant tBHQ. Comparable effects on reporter activity were obtained in Caco-2 cells when using the same reporter constructs and tBHQ concentrations (data not shown).
Effects of tBHQ, TCDD, and Resveratrol on CYP1A1 in HepG2 Cells. Treatment of HepG2 cells with tBHQ (80 μM) or TCDD (10 nM) increased the activity of a transiently transfected pT81Luc/3xDRE reporter 7-fold and 25-fold, respectively, compared with DMSO-treated controls (Fig. 2A). However, combined treatment with TCDD and tBHQ reduced the TCDD-mediated induction by 30% (Fig. 2A). After 24 h of treatment with tBHQ, TCDD, or both, total RNA was isolated and CYP1A1 mRNA expression was determined using real-time PCR. tBHQ induced CYP1A1 mRNA expression 22-fold compared with the level seen in DMSO-treated cells (Fig. 2B). TCDD elevated CYP1A1 mRNA about 800-fold. Only a small decline in the TCDD-elicited response could be detected by combined treatment with tBHQ. Finally, CYP1A activity was measured as EROD activity after treatment of HepG2 cells with tBHQ, TCDD, or a combination of both compounds for 48 h (Fig. 2C). tBHQ increased EROD activity 3-fold compared with DMSO control, whereas an 840-fold increase was obtained with TCDD. tBHQ decreased the TCDD-induced activity of CYP1A1 (>600 pmol/min/mg protein) by more than 40%.
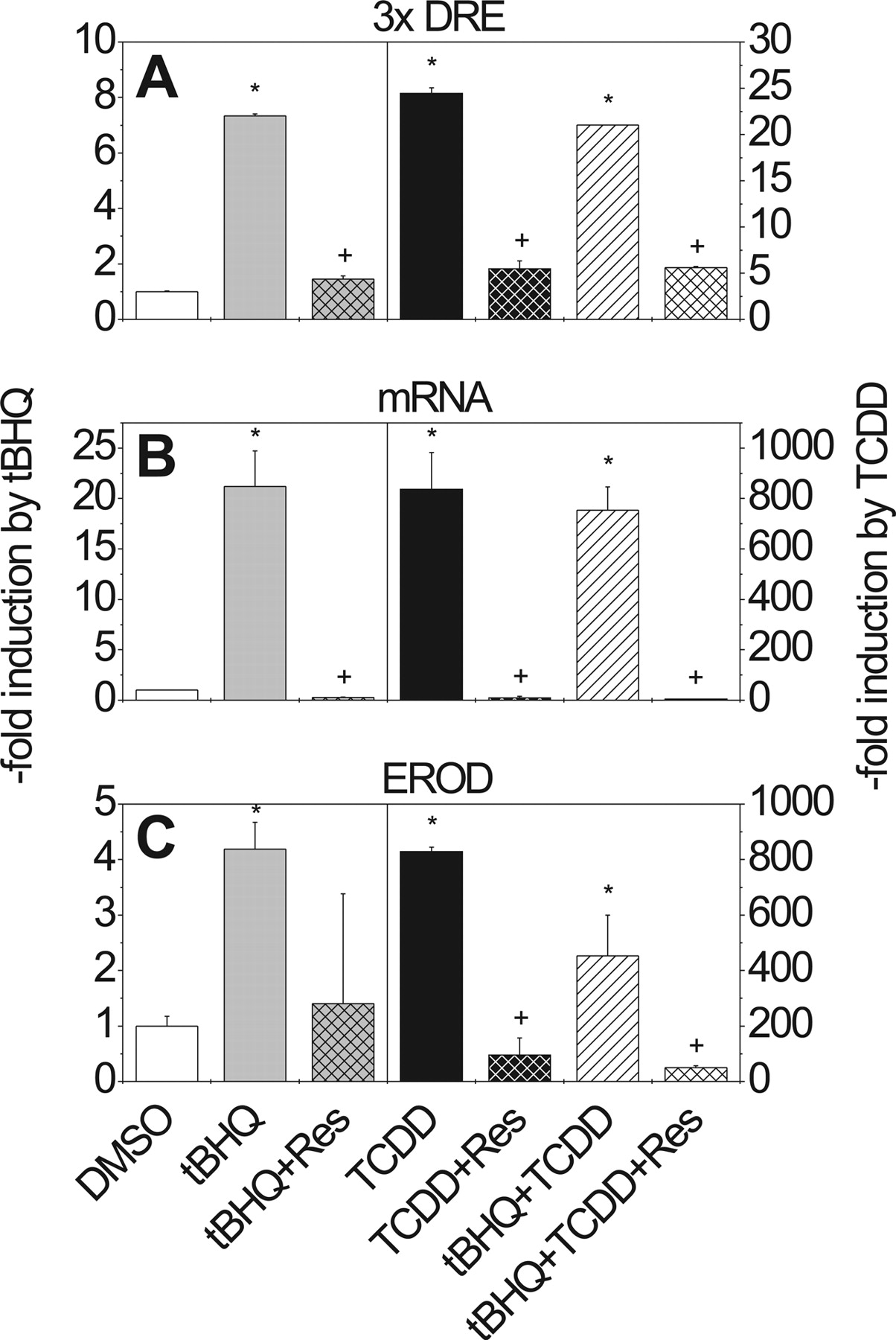
Effect of tBHQ, TCDD, and resveratrol on DRE-driven luciferase reporter activity, CYP1A1 expression, and EROD activity in HepG2 cells. A, reporter assay: HepG2 cells were transiently transfected with pT81/3xDRE and incubated for 42 h with tBHQ (80 μM) or/and TCDD (10 nM), minus/plus resveratrol (100 μM); values indicate -fold changes in luciferase reporter activity relative to DMSO controls. B, changes in expression of CYP1A1 mRNA in HepG2 cells incubated for 24 h with the indicated chemicals (relative to the DMSO control). C, changes in EROD activity in HepG2 cells incubated with the indicated chemicals. The values are given relative to DMSO-treated controls with different scales on the y-axis. Mean ± S.D. of four experiments is shown. Significant effects of induction by tBHQ/TCDD or repression by resveratrol are indicated by * and +, respectively (P < 0.05).
We subsequently investigated the effect of the AhR inhibitor resveratrol on CYP1A1 reporter pT81/3xDRE activity, CYP1A1 mRNA expression, and EROD activity in HepG2 cells. Cells were transiently transfected with pT81/3xDRE and treated with tBHQ (80 μM) and/or TCDD (10 nM), as well as with resveratrol (100 μM). Resveratrol significantly reduced the TCDD-mediated increase in pT81/3xDRE reporter activity in HepG2 cells (Fig. 2A). Resveratrol also decreased the tBHQ- or TCDD-mediated increase in expression of CYP1A1 mRNA (Fig. 2B). EROD activation, provoked by the two compounds, was also inhibited (Fig. 2C). This suggests the participation of the AhR in the induction mechanism of CYP1A1 by tBHQ.
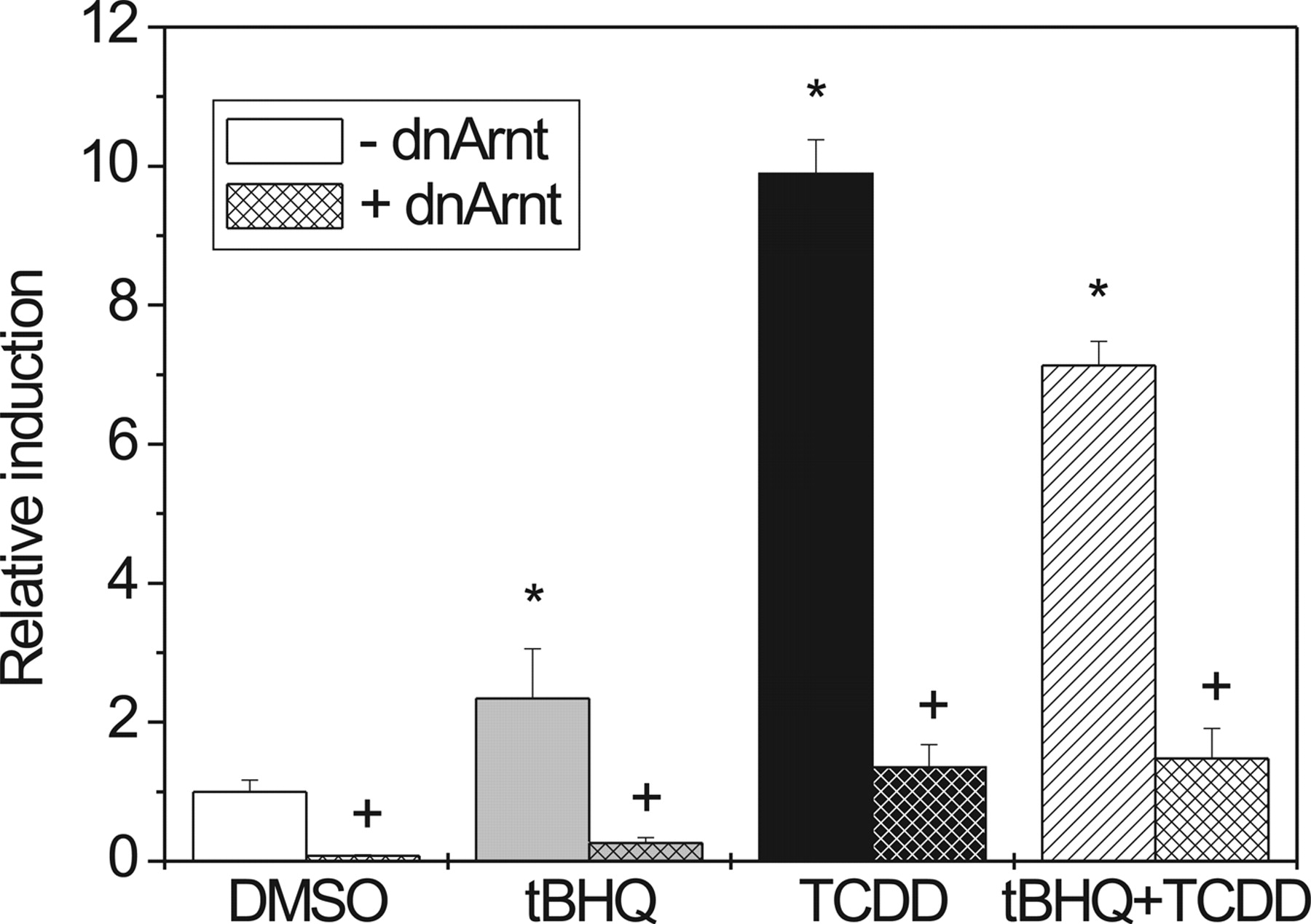
Effect of dnArnt on Induction of pT81/3xDRE by tBHQ and TCDD in HepG2 Cells. HepG2 cells were transiently cotransfected with pT81/3xDRE and dnArnt or a control vector (pcDNA3.1) and treated with tBHQ, TCDD, or both compounds. Expression of dnArnt (harboring a deleted DNA binding domain) results in the formation of an inactive AhR/dnArnt complex that fails to bind specifically to DNA. Figure 3 illustrates the effect of dnArnt on luciferase activity of pT81Luc/3xDRE after treatment of cells with tBHQ, TCDD, or both compounds compared with mock (pcDNA3.1) cotransfected controls. In the presence of dnArnt, the activity of the pT81/3xDRE reporter was almost totally blocked, regardless of whether cells were treated with tBHQ, TCDD, or a combination of both.
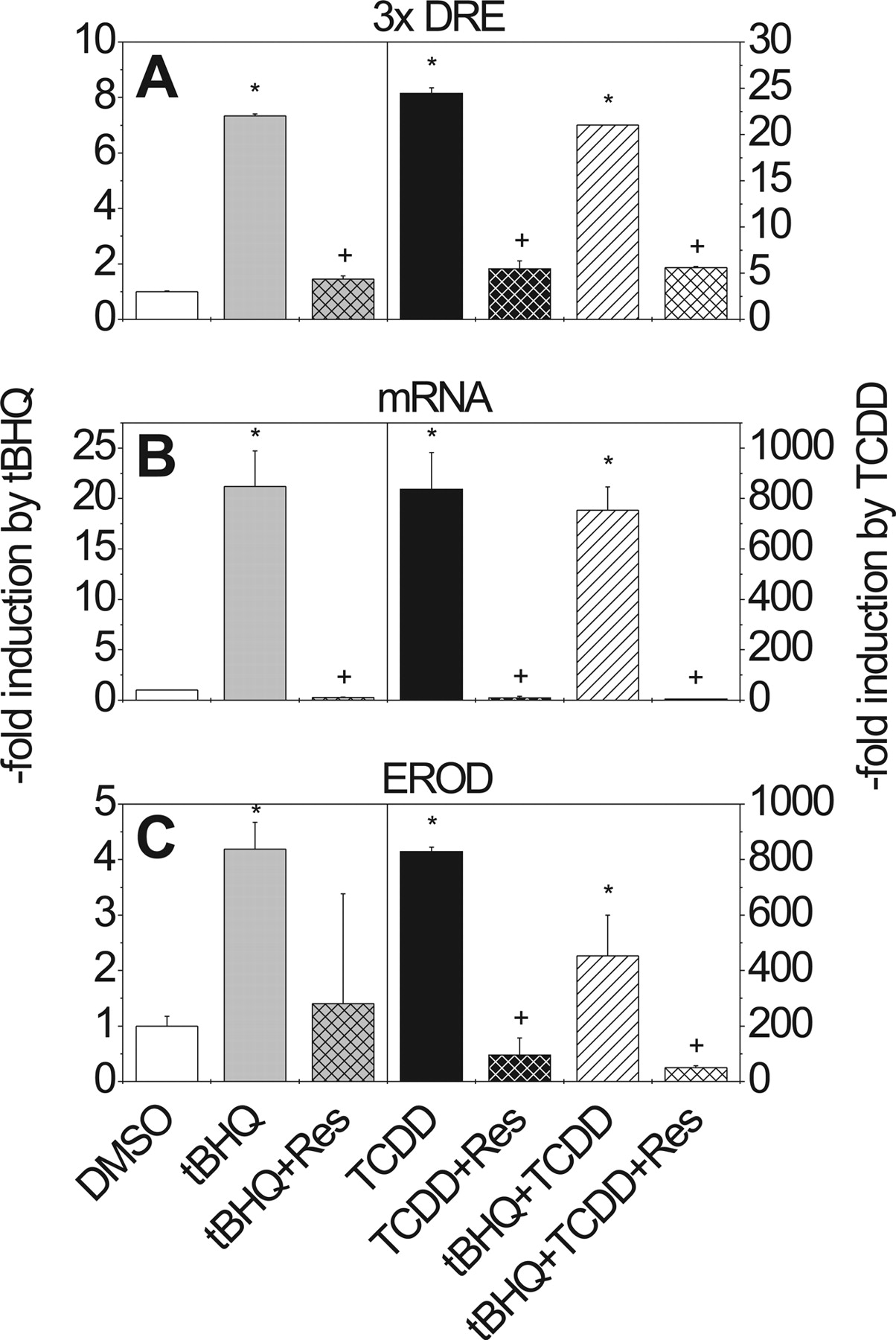
Effects of tBHQ, TCDD, and Resveratrol on pT81/3xDRE Reporter Activity, CYP1A1 mRNA Expression, and EROD Activity in Caco-2 Cells. To compare the results obtained in the liver-derived HepG2 cell line with a second cell system in which xenobiotic metabolizing enzymes play an important role in chemoprotection and activation of carcinogens, the gastrointestinal colon-derived cell line Caco-2 was chosen. Caco-2 cells, transiently transfected with pT81/3xDRE and treated with tBHQ, exhibited a 6-fold elevated reporter activity when compared with the DMSO control (Fig. 4A). Treatment of cells with TCDD resulted in a lower increase in reporter activity (7-fold) (Fig. 4A) than the one that was obtained in HepG2 cells (22-fold) (Fig. 2A). In contrast to what was seen in HepG2 cells, combined treatment with tBHQ and TCDD resulted in a 22-fold induction of reporter activity, suggesting a more than additive effect (Fig. 4A).

A dnArnt suppresses induction of a 3xDRE-driven luciferase reporter by tBHQ or TCDD. HepG2 cells were transiently cotransfected with the pT81/3xDRE reporter construct and dnARNT/pcDNA3.1 or the respective control vector (pcDNA3.1). Cells were treated with 80 μM tBHQ, 10 nM TCDD, or both for 42 h. Mean ± S.D. of three experiments is shown. Significant effects of induction by tBHQ/TCDD or repression by dnArnt are indicated by * and +, respectively (P < 0.05).
Treatment of Caco-2 cells with tBHQ significantly increased CYP1A1 mRNA levels, an effect that could be completely abolished by simultaneous treatment with resveratrol (Fig. 4B). The same inhibitory effect of resveratrol was observed when it was administered to Caco-2 cells along with TCDD. CYP1A1 activity, measured as EROD activity, was also reduced to control values when tBHQ or TCDD was administered together with resveratrol (Fig. 4C). Simultaneous tBHQ and TCDD treatment of Caco-2 cells led to a reduction in EROD activity by almost 50% when compared with values obtained by incubation of cells with TCDD alone. Resveratrol was again strongly inhibitory in these treatment regimens (Fig. 4C).
Comparability between HepG2 and Caco-2 Cells. Caco-2 cells express a nonfunctional form of NQO1 (NQO1*2 allele), which plays a role in detoxification of quinones (Bonnesen et al., 2001). Therefore, we incubated HepG2 cells with the NQO1 inhibitor dicumarol (Ernster, 1967) to mimic the situation in Caco-2 cells. No difference excluding a crucial role of NQO1 possibly elicited by tBHQ was observed between untreated and dicumarol-treated HepG2 cells (data not shown). The glutathione precursor NAC is known as an antioxidant scavenging reactive oxygen species. NAC was used in a set of experiments to test whether tBHQ-produced reactive oxygen species are important in the HepG2 system. No effect of NAC on CYP1A1 inducibility by tBHQ was observed (data not shown).
Discussion
In our present study, we have investigated the effect of the antioxidant tBHQ on expression and activity of CYP1A1 in two human cell systems, HepG2 and Caco-2 cells, representing models for liver and gastrointestinal cells. This study was initiated because we had previously observed that phenolic antioxidants can induce human UDP-glucuronosyltransferase 1A6 by a mechanism in which DRE binding of the AhR plays some role (Münzel et al., 2003). Moreover, two recent studies report on an inducing effect of tBHQ on CYP1A1 in HepG2 cells (Sugatani et al., 2004) and Hepa1c1c7 cells (Gharavi and El-Kadi, 2005).

Effect of tBHQ, TCDD, and resveratrol on DRE-driven luciferase reporter activity (A), CYP1A1 mRNA expression (B), and EROD activity (C) in Caco-2 cells. For experimental details, see legend to Fig. 2. Mean ± S.D. of four experiments is shown. Significant effects of induction or repression are indicated by * and +, respectively (P < 0.05); n.d., not determined.
Phenolic antioxidants act through activation of Nrf2, and interaction between Nrf2 and AhR signal transduction has been reported to occur during induction of NQO1 (Ma et al., 2004). Therefore, we were interested to examine whether and, if so, how antioxidants such as tBHQ could regulate CYP1A1 expression. The results of our study now clearly show that higher concentrations of tBHQ can induce CYP1A1 both in HepG2 and Caco-2 cells. CYP1A promoters/enhancers do not contain any sequences that would match the ARE consensus sequence, suggesting that their transcriptional activation by anti-oxidants involves an ARE-independent pathway. To examine the possible involvement of DREs in CYP1A1 induction by tBHQ, transfection experiments with three different DRE-containing reporter constructs were performed: pT81/hCYP1A1-5′ containing 1.2 kb of the upstream region with four functional DRE core motifs (Kress et al., 1998), pT81/3xDRE with three functional DREs in tandem-like array, and a pT81/CDEF reporter in which the four functional DREs were mutated. Treatment of HepG2 cells with tBHQ (10–100 μM) caused induction of the DRE-driven reporters, whereas no such effect was seen when using the construct pT81/CDEF containing the four mutated DREs (Fig. 1). These results strongly suggested that induction of the reporters containing the correct DRE sequences by tBHQ is DRE-mediated. The missing concentration dependence with the pT81/hCYP1A1-5′ construct may indicate that additional factors are involved in regulation of the wild-type promoter.
To further elucidate the possible involvement of the AhR in tBHQ-mediated induction of CYP1A1, experiments were conducted in which HepG2 and Caco-2 cells were simultaneously exposed to tBHQ and TCDD. Combined TCDD/tBHQ treatment of HepG2 cells led to a decrease in all the parameters measured (DRE reporter activity, CYP1A1 mRNA expression, and CYP1A activity) when compared with values obtained with TCDD alone. tBHQ has been shown to bind and activate the AhR (Gharavi and El-Kadi, 2005). However, it may behave as a partial agonist that, when present at a very high concentration compared with TCDD, may lower the maximal achievable inducing effect of the latter. One could also speculate that tBHQ activates a transcription factor that competes with the AhR for DRE binding. Evidence in favor for this latter mechanism comes from experiments in which oltipraz was combined with 3-methylcholanthrene and was found to inhibit induction of CYP1A1 by the latter, even though oltipraz alone was able to induce CYP1A1 (Cho and Kim, 2003). Oltipraz interacts with the transcription factor C/EBPα, and it was shown that the binding sites for C/EBPα and the AhR in the promoter region of rat GST are nearly identical (Pimental et al., 1993). Therefore, the effects of tBHQ/TCDD cotreatment of HepG2 on CYP1A1 expression resemble those of oltipraz/3-methylcholanthrene cotreatment in H4IIE cells. We do not know why Caco-2 cells behaved somewhat differently: whereas the activity of CYP1A (EROD test) was also lowered in cells of these lines on combined treatment with tBHQ and TCDD, when compared with the effect produced with TCDD alone, additive effects were seen on DRE reporter activity and CYP1A1 mRNA. This discrepancy of magnitude between transcriptional activation and induction of EROD activity by tBHQ (Figs. 2 and 4) may result from inhibition of CYP1A enzymes by high concentrations of tBHQ, which still could be present in the cells 48 h after treatment.
There are two strong lines of evidence that are in favor of the hypothesis for an involvement of the AhR in CYP1A1 induction by tBHQ. 1) Addition of resveratrol produced inhibitory effects in both HepG2 and Caco-2 cells as it strongly suppressed luciferase activity of the pT81/3xDRE reporter, CYP1A1 mRNA, and CYP1A enzyme activity (EROD). Resveratrol is an antagonist of the AhR, able to inhibit the PAH-induced CYP1A1 transcription in HepG2 cells (Ciolino et al., 1998). 2) Cotransfection of HepG2 cells with dnArnt resulted in a dramatic loss of inducibility of pT81/3xDRE reporter activity by tBHQ and/or TCDD. dnArnt may serve as heterodimerization partner of the AhR, but because of the deleted DNA binding domain fails to support transcriptional activation of AhR target genes (Lindebro et al., 1995). Therefore, the observed suppression of tBHQ-mediated activation of the DRE-driven luciferase reporter by coexpression of dnArnt strongly suggests the participation of the AhR/Arnt complex in the induction mechanism of tBHQ and supports the idea that the heterodimer is involved in CYP1A1 induction by tBHQ. This is also substantiated by findings of others, who showed, using gel electrophoretic mobility shift assays, that tBHQ causes AhR/Arnt binding to mouse DRE3 when using extracts from Hepa1c1c7 cells incubated with the antioxidant (Gharavi and El-Kadi, 2005).
Strong evidence exists that kinases are also involved in the induction of drug metabolizing enzymes of phase I and II by tBHQ. Yu et al. (1997) showed that tBHQ activates extracellular signal-regulated protein kinase 2 via the formation of phenoxyl radicals, which results in induction of phase II enzymes. The ARE-mediated increase in expression of rat GSTA2 in rat hepatoma cells H4IIE by tBHQ depends on the activation of two kinases, namely, phosphatidylinositol-3-kinase and Akt (Kang et al., 2001). TCDD-mediated activation of the AhR requires phosphorylation reactions by the mitogen-activated protein kinases extracellular signal-regulated protein kinase and c-Jun NH2-terminal kinase (Tan et al., 2002). Furthermore, transcription of CYP1A1 depends on protein kinase C (Carrier et al., 1994; Long et al., 1998) and calcium ions (Puga et al., 2002), and phosphorylation of the AhR is required for its DNA binding and transcriptional activity (Chen and Tukey, 1996). Therefore, activation of kinase-dependent signaling pathways by tBHQ may contribute to AhR-dependent CYP1A1 induction by the antioxidant.
Antioxidants like tBHQ are believed to represent monofunctional inducers that selectively induce phase II enzymes of xenobiotic metabolism, thereby potentially acting chemoprotective. Our present findings now indicate that tBHQ may also induce CYP1A1 via a DRE-dependent mechanism involving the AhR and Arnt. This finding indicates that the antioxidant may act as a bifunctional inducer, at least at higher concentrations.
Acknowledgments
We thank Birgit Kaltschmitt, Ingrid Voith, and Silvia Vetter for expert technical assistance.
Footnotes
-
This work was supported by the Deutsche Forschungsgemeinschaft (DFG).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.009662.
-
ABBREVIATIONS: PAH, polycyclic aromatic hydrocarbon; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; AhR, aryl hydrocarbon receptor; Arnt, aryl hydrocarbon receptor nuclear translocator; DRE, dioxin response element; tBHQ, tert-butylhydroquinone; GST, glutathione S-transferase; NQO1, NADPH quinone oxidoreductase-1; ARE, antioxidant response element; Nrf2, nuclear factor erythroid 2-related factor 2; dnArnt, dominant negative aryl hydrocarbon receptor nuclear translocator; NAC, N-acetyl-l-cysteine; PCR, polymerase chain reaction; DMSO, dimethyl sulfoxide; EROD, ethoxyresorufin O-deethylase.
-
↵1 T.D.S. and C.K. contributed equally to this work and should both be considered first authors.
- Received February 6, 2006.
- Accepted March 28, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}