


Abstract
Valsartan is a highly selective angiotensin II AT1-receptor antagonist for the treatment of hypertension. Valsartan is mainly excreted into the bile in unchanged form. Because valsartan has an anionic carboxyl group, we hypothesized that a series of organic anion transporters could be involved in its hepatic clearance. In this study, to identify transporters that mediate the hepatic uptake and biliary excretion of valsartan and estimate the contribution of each transporter to the overall hepatic uptake and efflux, we characterized its transport using transporter-expressing systems, human cryopreserved hepatocytes, and Mrp2-deficient Eisai hyperbilirubinemic rats (EHBRs). Valsartan was significantly taken up into organic anion-transporting polypeptide (OATP) 1B1 (OATP2/OATP-C)- and OATP1B3 (OATP8)-expressing HEK293 cells. We also observed saturable uptake into human hepatocytes. Based on our estimation, the relative contribution of OATP1B1 to the uptake of valsartan in human hepatocytes depends on the batch, ranging from 20 to 70%. Regarding efflux transporters, the ratio of basal-to-apical transcellular transport of valsartan to that in the opposite direction in OATP1B1/MRP2 (multidrug resistance-associated protein 2) double transfected cells was the highest among the three kinds of double transfectants, OATP1B1/MRP2, OATP1B1/multi-drug resistance 1, and OATP1B1/breast cancer resistance protein-expressing MDCKII cells. We observed saturable ATP-dependent transport into membrane vesicles expressing human MRP2. We also found that the elimination of intravenously administered valsartan from plasma was markedly delayed, and the biliary excretion was severely impaired in EHBR compared with normal Sprague-Dawley rats. These results suggest that OATP1B1 and OATP1B3 as the uptake transporters and MRP2 as the efflux transporter are responsible for the efficient hepatobiliary transport of valsartan.
Valsartan is an antihypertensive drug acting on angiotensin II AT1 receptors. It has been reported that approximately 70% of the total clearance of valsartan is accounted for by hepatic clearance (Flesch et al., 1997). Valsartan undergoes a minor degree of metabolism involving 4-hydroxylation, whereas 85% of orally administered valsartan is excreted into feces in unchanged form (Waldmeier et al., 1997). Considering that the bioavailability is approximately 40% and the hepatic clearance is much less than the hepatic blood flow (Flesch et al., 1997), most of the drug passing into the bile is in the unmetabolized form. From these facts, valsartan is thought to be mainly excreted into the bile in unchanged form. However, the molecular mechanism of the hepatobiliary transport of valsartan has not been elucidated.
When valsartan is given orally to rats, the hepatic concentration is about 7 to 10 times higher than the plasma concentration, suggesting that valsartan is selectively distributed to the liver (interview form of Diovan tablet). Because valsartan is a hydrophilic compound with a log D value (pH = 7.0) of –0.34 and it has an anionic carboxyl group, it should have difficulty in crossing the plasma membrane. Therefore, a number of organic anion transporters could be involved in the hepatic transport of valsartan.
The organic anion-transporting polypeptide (OATP) family transporters play an important role in the transport of organic anions (Hagenbuch and Meier, 2004). Among them, OATP1B1 (OATP-C/OATP2) and OATP1B3 (OATP8) are thought to be responsible for the hepatic uptake of several organic anions in humans because of their selective expression in liver and broad substrate specificities (Hagenbuch and Meier, 2004). They can also accept a variety of clinically used drugs, such as 3-hydroxy-3-methylglutaryl CoA reductase inhibitors, rifampin, and methotrexate (Hsiang et al., 1999; Abe et al., 2001; Nakai et al., 2001; Vavricka et al., 2002; Shitara et al., 2003a; Hirano et al., 2004). OATP2B1 (OATP-B) is also expressed in the basolateral membrane of human liver. Its substrate specificity is relatively narrow compared with OATP1B1 and 1B3, but some of the OATP1B1 and OATP1B3 substrates can also be recognized by OATP2B1 (Tamai et al., 2000; Kullak-Ublick et al., 2001). Conversely, multidrug resistance 1 (MDR1), multidrug resistance-associated protein 2 (MRP2), and breast cancer resistance protein (BCRP) can be involved in the hepatic efflux transport of organic anions (Chandra and Brouwer, 2004).
In general, the substrate specificity of each transporter is very broad and it is often very similar to that of other transporters, suggesting that a substrate can be recognized by multiple transporters. Now we can use human cryopreserved hepatocytes for the evaluation of transporter-mediated uptake. Shitara et al. (2003a) have succeeded in clarifying the importance of OATP1B1-mediated inhibition in the clinical drug-drug interaction between cerivastatin and cyclosporin A from in vitro experiments using human cryopreserved hepatocytes and transporter expression systems.
Hirano et al. (2004) have recently published their methodology for estimating the quantitative contribution of OATP1B1 and OATP1B3 to the hepatic uptake of compounds. In one of their approaches, they calculated the ratio of the uptake clearance of transporter-selective substrates in human cryopreserved hepatocytes to that in transporter-expression systems, and then estimated the hepatic uptake of test compounds mediated by certain transporters by multiplying that ratio by the clearance of the test compounds in transporter-expressing cells. We applied this method to the calculation of the contribution of OATP1B1 and OATP1B3 to the human hepatic uptake. To check the involvement of efflux transporters in the biliary excretion of compounds, Matsushima et al. (2005) have shown that OATP1B1/MDR1, OATP1B1/MRP2, and OATP1B1/BCRP double transfectants can be used for the rapid identification of anionic bisubstrates of OATP1B1 and each efflux transporter by measuring the vectorial transport of substrates across each monolayer. If valsartan is a substrate of OATP1B1, these cell lines should help us to identify which transporters are involved in its biliary excretion in humans.
In this study, we analyzed the involvement and relative contribution of OATP1B1 and OATP1B3 to the hepatic uptake of valsartan using human cryopreserved hepatocytes and transporter-expressing cells and identified the transporters responsible for the biliary excretion of valsartan using double transfectants and transporter-expressing vesicles. We also checked the involvement of MRP2 in the pharmacokinetics of valsartan in vivo using Eisai hyperbilirubinemic rats (EHBR), in which Mrp2 is deficient.
Materials and Methods
Materials. [3H]Valsartan (80.9 Ci/mmol) and unlabeled valsartan were kindly donated by Novartis Pharma K.K. (Basel, Switzerland). [3H]Estradiol-17β-glucuronide (E217βG) (45 Ci/mmol) and [3H]estrone-3-sulfate (46 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA), and [3H]cholecystokinin octapeptide (CCK-8) (77 Ci/mmol) was purchased from GE Healthcare Bio-Sciences (Buckinghamshire, UK). Unlabeled E217βG, estrone-3-sulfate, and CCK-8 were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were of analytical grade and commercially available.
Cell Culture. Transporter-expressing or vector-transfected HEK293 cells and MDCKII cells were grown in Dulbecco's modified Eagle's medium low glucose (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B at 37°C with 5% CO2 and 95% humidity. LLC-PK1 cells were cultured in Medium 199 (Invitrogen) supplemented with 10% fetal bovine serum (Sigma), 100 U/ml penicillin, and 100 μg/ml streptomycin.
Transport Study Using Transporter Expression Systems. Cells were seeded in 12-well plates coated with poly-l-lysine/poly-l-ornithine at a density of 1.5 × 105 cells/well 72 h before transport assay. For the transport study, the cell culture medium was replaced with culture medium supplemented with 5 mM sodium butyrate 24 h before transport assay to induce the expression of OATP1B1, OATP1B3, or OATP2B1.
The transport study was carried out as described previously (Hirano et al., 2004, 2006). Uptake was initiated by adding Krebs-Henseleit buffer containing radiolabeled and unlabeled substrates after cells had been washed twice and preincubated with Krebs-Henseleit buffer at 37°C for 15 min. The Krebs-Henseleit buffer consisted of 118 mM NaCl, 23.8 mM NaHCO3, 4.8 mM KCl, 1.0 mM KH2PO4, 1.2 mM MgSO4, 12.5 mM HEPES, 5.0 mM glucose, and 1.5 mM CaCl2 adjusted to pH 7.4. The uptake was terminated at designated times by adding ice-cold Krebs-Henseleit buffer after removal of the incubation buffer. Then, cells were washed twice with 1 ml of ice-cold Krebs-Henseleit buffer, solubilized in 500 μl of 0.2 N NaOH, and kept overnight at 4°C. Aliquots (500 μl) were transferred to scintillation vials after adding 250 μl of 0.4 N HCl. The radioactivity associated with the cells and incubation buffer was measured in a liquid scintillation counter (LS6000SE; Beckman Coulter, Inc., Fullerton, CA) after adding 2 ml of scintillation fluid (Clear-sol I; Nacalai Tesque, Kyoto, Japan) to the scintillation vials. The remaining 50 μl of cell lysate was used to determine the protein concentration by the method of Lowry et al. (1951) with bovine serum albumin as a standard.
Transport Study Using Human Cryopreserved Hepatocytes. This experiment was performed as described previously (Hirano et al., 2004). Cryopreserved human hepatocytes were purchased from In Vitro Technologies (Baltimore, MD) (lot 094 and OCF) and from the Research Institute for Liver Disease (Shanghai, China) (lot 03-013). Immediately before the study, the hepatocytes (1-ml suspension) were thawed at 37°C, then quickly suspended in 10 ml of ice-cold Krebs-Henseleit buffer and centrifuged (50g) for 2 min at 4°C, followed by removal of the supernatant. This procedure was repeated once more to remove cryopreservation buffer, and then the cells were resuspended in the same buffer to give a cell density of 1.0 × 106 viable cells/ml for the uptake study. The number of viable cells was determined by trypan blue staining. Before the uptake studies, the cell suspensions were prewarmed in an incubator at 37°C for 3 min. The uptake studies were initiated by adding an equal volume of buffer containing labeled and unlabeled substrates to the cell suspension. After incubation at 37°C for 0.5, 2, or 5 min, the reaction was terminated by separating the cells from the substrate solution. For this purpose, an aliquot of 80-μl incubation mixture was collected and placed in a centrifuge tube (450 μl) containing 50 μl of 2 N NaOH under a layer of 100 μl of oil (density, 1.015; a mixture of silicone oil and mineral oil; Sigma-Aldrich), and subsequently, the sample tube was centrifuged for 10 s using a tabletop centrifuge (10,000g; Beckman Microfuge E; Beckman Coulter, Inc.). During this process, hepatocytes passed through the oil layer into the alkaline solution. After an overnight incubation in alkali to dissolve the hepatocytes, the centrifuge tube was cut and each compartment was transferred to a scintillation vial. The compartment containing the dissolved cells was neutralized with 50 μl of 2 N HCl and mixed with scintillation cocktail, and the radioactivity was measured in a liquid scintillation counter.
Transcellular Transport Study Using Double Transfected Cells. The protocol has been described in detail previously (Matsushima et al., 2005). In brief, transfected MDCKII cells were seeded in a Transwell membrane insert (6.5-mm diameter, 0.4-μm pore size; Corning Costar, Cambridge, MA) at a density of 1.4 × 105 cells per well 96 h before the transport study. Among a series of cell lines we used in this experiment, human MDR1, MRP2, and OATP1B1 were stably transfected into MDCKII cells as shown previously (Evers et al., 1998; Matsushima et al., 2005). Human BCRP cDNA was transduced into MDCKII cells by the infection of recombinant adenovirus 48 h before the transport study. The cell culture medium was replaced with culture medium supplemented with 5 mM sodium butyrate 24 h before the transport assay. For uptake studies, cells were washed three times and preincubated with Krebs-Henseleit buffer. The experiment was initiated by replacing the medium at either the apical or the basal side of the cell layer with complete medium containing 3H-labeled and unlabeled valsartan or E217βG (0.1 μM). The cells were incubated at 37°C and aliquots of medium were taken from each compartment at several time points. Radioactivity in 100 μl of medium was measured in a liquid scintillation counter after addition of 2 ml of scintillation fluid. At the end of the experiments, the cells were washed three times with 1.5 ml of ice-cold Krebs-Henseleit buffer and solubilized in 500 μl of 0.2 N NaOH. After addition of 100 μl of 1 N HCl, 400-μl aliquots were transferred to scintillation vials. Then, 50-μl aliquots of cell lysate were used to determine protein concentrations by the method of Lowry et al. (1951) with bovine serum albumin as a standard.
Vesicle Transport Assay. The preparation procedure of the membrane vesicles expressing human MRP2 was described previously (Hirouchi et al., 2004). The transport medium (10 mM Tris, 250 mM sucrose, and 10 mM MgCl2, pH 7.4) contained the labeled and unlabeled valsartan, 5 mM ATP, and an ATP-regenerating system (10 mM creatine phosphate and 100 μg/μl creatine phosphokinase). An aliquot of transport medium (15 μl) was mixed rapidly with the vesicle suspension (5 μg of protein in 5 μl). The transport reaction was stopped by the addition of 1 ml of ice-cold buffer containing 250 mM sucrose, 0.1 M NaCl, and 10 mM Tris-HCl buffer (pH 7.4). The stopped reaction mixture was passed through a 0.45-μm HA filter (Millipore Corp., Billerica, MA) and then washed twice with 5 ml of stop solution. The radioactivity retained on the filter was measured in a liquid scintillation counter after the addition of scintillation cocktail. Ligand uptake was normalized in terms of the amount of membrane protein.
In Vivo Pharmacokinetic Study. Male Sprague-Dawley (SD) rats and EHBRs (7–8 weeks old) were purchased from Nippon SLC (Shizuoka, Japan). All animals were maintained under standard conditions with a reverse dark-light cycle and were treated humanely. Food and water were available ad libitum. This study was carried out in accordance with the guidelines provided by the Institutional Animal Care Committee (Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan). SD rats and EHBRs were anesthetized by inhalation of diethyl ether. The abdomen was opened with a midline incision and the common bile duct was cannulated with a polyethylene tube (Becton Dickinson Primary Care Diagnostics, Sparks, MD). The phosphate-buffered saline containing [3H]valsartan (8 μCi/ml) and unlabeled valsartan (1 mg/ml) was injected into a femoral vein (1 ml/kg body weight). Blood samples were collected from a femoral artery and bile samples were collected in preweighed tubes at designated times. The total radioactivity in plasma and bile samples was measured in a liquid scintillation counter.
Kinetic Analyses of Uptake Transporters. Ligand uptake was expressed as the uptake volume [μl/mg protein], given as the amount of radioactivity associated with the cells [dpm/mg protein] divided by its concentration in the incubation medium [dpm/μl]. Specific uptake was obtained by subtracting the uptake into vector-transfected cells from the uptake into cDNA-transfected cells. Kinetic parameters were obtained using the following equation:  where v is the uptake velocity of the substrate (pmol/min/mg protein), S is the substrate concentration in the medium (μM), Km is the Michaelis constant (μM), Vmax is the maximum uptake rate (pmol/min/mg protein), and Pdif is the nonsaturable uptake clearance (μl/min/mg protein). Fitting was performed by the nonlinear least-squares method using a MULTI program (Yamaoka et al., 1981), and the Damping Gauss-Newton Method algorithm was used for curve fitting. The input data were weighted as the reciprocal of the observed values.
where v is the uptake velocity of the substrate (pmol/min/mg protein), S is the substrate concentration in the medium (μM), Km is the Michaelis constant (μM), Vmax is the maximum uptake rate (pmol/min/mg protein), and Pdif is the nonsaturable uptake clearance (μl/min/mg protein). Fitting was performed by the nonlinear least-squares method using a MULTI program (Yamaoka et al., 1981), and the Damping Gauss-Newton Method algorithm was used for curve fitting. The input data were weighted as the reciprocal of the observed values.

Time profiles of the uptake of valsartan by OATP1B1- and OATP1B3-expressing HEK293 cells. Squares, triangles, and circles represent the uptake in OATP1B1- and OATP1B3-expressing cells and vector-control cells, respectively. Each point represents the mean ± S.E. (n = 3).
To determine the saturable hepatic uptake clearance in human hepatocytes, we first determined the hepatic uptake clearance (CL(2 min-0.5 min))(μl/min/106 cells) by calculating the slope of the uptake volume (Vd)(μl/106 cells) between 0.5 and 2 min (eq. 2). The saturable component of the hepatic uptake clearance (CLhep) was determined by subtracting CL(2 min-0.5 min) in the presence of 100 μM substrate (excess) from that in the presence of 1 μM substrate (tracer quantity) (eq. 3). 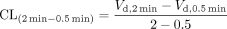
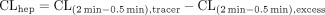 where CL(2 min-0.5 min), tracer and CL(2 min-0.5 min), excess represent the CL(2 min-0.5 min) values estimated in the presence of 1 and 100 μM substrate, respectively.
where CL(2 min-0.5 min), tracer and CL(2 min-0.5 min), excess represent the CL(2 min-0.5 min) values estimated in the presence of 1 and 100 μM substrate, respectively.
Estimation of the Relative Contribution of Each Transporter to the Hepatic Uptake. This method for estimating the contribution of OATP1B1 and OATP1B3 to the overall hepatic uptake has been used previously (Hirano et al., 2004). In this analysis, estrone-3-sulfate and CCK-8 were chosen as transporter-selective substrates of OATP1B1 and OATP1B3, respectively. The ratio of the uptake clearance of the reference compounds in human hepatocytes to that in the expression system was calculated and defined as Ract, OATP1B1 and Ract, OATP1B3. The uptake clearance mediated by OATP1B1 and OATP1B3 in human hepatocytes was separately calculated by multiplying the uptake clearance of valsartan in transporter-expressing cells (CLOATP1B1, test and CLOATP1B3, test) by Ract, OATP1B1 and Ract, OATP1B3, respectively, as described in the following equations: 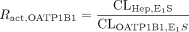

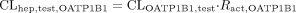

Kinetic Analyses of Efflux Transporters. The basal-to-apical transcellular clearance (CLtrans) was calculated by dividing the steady-state efflux velocity for the transcellular transport (Vapical) by the ligand concentration in the incubation buffer on the basal side, whereas the efflux clearance across the apical membrane (PSapical) in double transfected cells was obtained by dividing Vapical by the intracellular concentration of ligand at 120 min. In the vesicle transport assay, ATP-dependent transporter-specific uptake was calculated by subtracting the uptake in the presence of AMP from that in the presence of ATP. The saturation kinetics of CLtrans, PSapical, and ATP-dependent uptake into vesicles were calculated using eq. 1 by the curve-fitting procedure described above.
Pharmacokinetic Analysis. The plasma concentration-time profile was fitted to a biexponential equation and the AUC0-∞ was estimated by integration up to infinity. The initial distribution volume (V1) was calculated by dividing the dose by the initial plasma concentration estimated from the fitted biexponential equation. The plasma clearance (CLp) was calculated as Dose/AUC0-∞. The biliary clearance (CLbile) was calculated as the ratio of the cumulative excreted amount in bile over 120 min to the AUC over 120 min (AUC0–120 min).
Results
Uptake of Valsartan by OATP Transporter-Expressing Cells. Valsartan was significantly taken up into OATP1B1- and OATP1B3-expressing HEK293 cells compared with vector-transfected cells in a time-dependent manner (Fig. 1). The saturation kinetics of valsartan by OATP1B1- and OATP1B3-expressing cells and vector-transfected HEK293 was evaluated by the uptake for 5 min, over which time the uptake of valsartan remained linear, and shown as Eadie-Hofstee plots (Fig. 2). The concentration dependence of the uptake of valsartan could be explained by a one-saturable and one-nonsaturable component. Their kinetic parameters are summarized in Table 1. In contrast, valsartan was not significantly transported in OATP2B1-expressing HEK293 cells [0.627 ± 0.092 μl/min/mg protein (OATP2B1-expressing HEK293 cells) versus 0.552 ± 0.081 μl/min/mg protein (vector-control cells); mean ± S.E.].
Kinetic parameters of the uptake of valsartan by OATP1B1- and OATP1B3-expressing HEK293 cells
Data shown in Fig. 2 were used to determine these parameters calculated by nonlinear regression analysis as described under Materials and Methods. Each parameter represents the mean ± computer-calculated S.D.
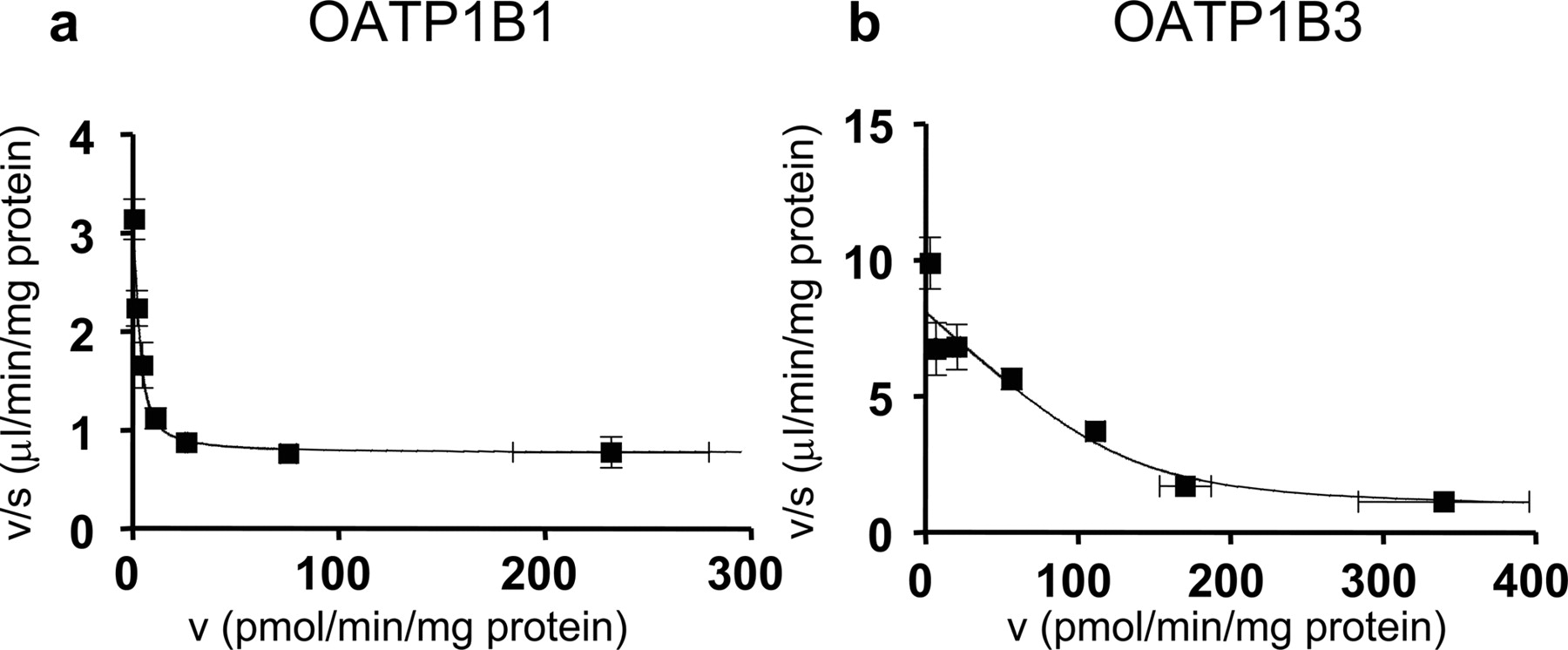
Eadie-Hofstee plots of the uptake of valsartan by OATP1B1- and OATP1B3-expressing HEK293 cells. The concentration dependence of OATP1B1 (a)- and OATP1B3 (b)-mediated uptake of valsartan is shown as Eadie-Hofstee plots. The uptake of valsartan for 5 min was determined at various concentrations (0.1–300 μM). Each point represents the mean ± S.E. (n = 3).
Uptake of Estrone-3-Sulfate, CCK-8, and Valsartan by Human Cryopreserved Hepatocytes. Typical time profiles of the uptake of estrone-3-sulfate, CCK-8, and valsartan in one batch of human hepatocytes (lot OCF) are shown in Fig. 3. The uptake of labeled valsartan by human hepatocytes was inhibited by unlabeled 100 μM valsartan in a concentration-dependent manner (Fig. 4). Time-dependent uptake of all ligands was observed at 1 μM, and this was reduced in the presence of 100 μM unlabeled ligands in all batches of hepatocytes examined in the present study (data not shown). The uptake clearance of these substrates in each batch is listed in Table 2. Based on the data in Table 2, following the method for estimating the contribution of OATP1B1 and OATP1B3 to the overall hepatic uptake described previously (Hirano et al., 2004), we calculated the estimated clearance of valsartan mediated by OATP1B1 and OATP1B3 and the quantitative contribution of these transporters to the hepatic uptake in three batches of human hepatocytes. Our estimation indicated that the relative contribution of OATP1B1 and OATP1B3 depends on the batch of human hepatocytes, ranging from 20% to 70% (as the contribution of OATP1B1) (Table 3).
Uptake clearance of reference compounds (E1S and CCK-8) and valsartan in expression systems and human hepatocytes
Contribution of OATP1B1 and OATP1B3 to the hepatic uptake of valsartan in each batch of human hepatocytes
In the column `Estimated Clearance of Valsartan,' the lower row shows the percentage of OATP1B1- or OATP1B3-mediated uptake clearance relative to the sum of the estimated clearance mediated by OATP1B1 and OATP1B3. The details of this estimation are described under Materials and Methods.
Transcellular Transport of Valsartan across MDCKII Monolayers. To identify the efflux transporters involved in the biliary excretion of valsartan, we investigated the transcellular transport of valsartan across the MDCKII monolayers expressing uptake and efflux transporters. We could not see any significant vectorial transcellular transport of valsartan in single transfected MDCKII cells expressing OATP1B1, MDR1, MRP2, and BCRP, and vector-transfected control cells. In contrast, as shown in Fig. 5, the basal-to-apical transcellular transport of valsartan in OATP1B1/MRP2 double transfected cells was the largest among three kinds of double transfectants, OATP1B1/MRP2, OATP1B1/MDR1, and OATP1B1/BCRP. In parallel, we also checked the transcellular transport of E217βG, whose basal-to-apical transport was reported to be observed in three kinds of double transfectants we tested (Fig. 5, a–c). The basal-to-apical transport of E217βG was 36, 8.9, and 6.1 times larger than that in the opposite direction in OATP1B1/MRP2, OATP1B1/MDR1, and OATP1B1/BCRP double transfectants, respectively, which is almost comparable to the previous results (Matsushima et al., 2005). Then, we studied the concentration dependence of the transcellular transport of valsartan in OATP1B1/MRP2-expressing cells (Fig. 6a), and the efflux clearance across the apical membrane (PSapical) was determined by measuring the cellular accumulation of valsartan at 120 min (Fig. 6b). The Km value of transcellular transport of valsartan (27.5 μM) was smaller than that for PSapical (99.0 μM) (Table. 4).
Kinetic parameters of the transcellular transport of valsartan in OATP1B1/MRP2 double transfectant
Data shown in Fig. 6 were used to determine these parameters calculated by nonlinear regression analysis as described under Materials and Methods. Each parameter represents the mean ± computer-calculated S.D.

Typical time profiles of the uptake of estrone-3-sulfate, CCK-8, and valsartan by human hepatocytes (lot OCF). The uptake of estrone-3-sulfate (a), CCK-8 (b), and valsartan (c) for 0.5, 2, and 5 min was determined at two concentrations (squares, 1 μM; triangles, 100 μM) at 37°C. Each point represents the mean ± S.E. (n = 3).

Concentration dependence of uptake of valsartan by human hepatocytes. The uptake of valsartan for 0.5 and 2 min was determined at three concentrations (1, 10, and 100 μM) at 37°C. The uptake clearance was obtained by subtracting the uptake at 0.5 min from that at 2 min, and the uptake clearance at 1 μM valsartan is defined as 100%. Circles, squares, and triangles represent the uptake in lots OCF, 03-013, and 094, respectively. Each point represents the mean ± S.E. (n = 3).
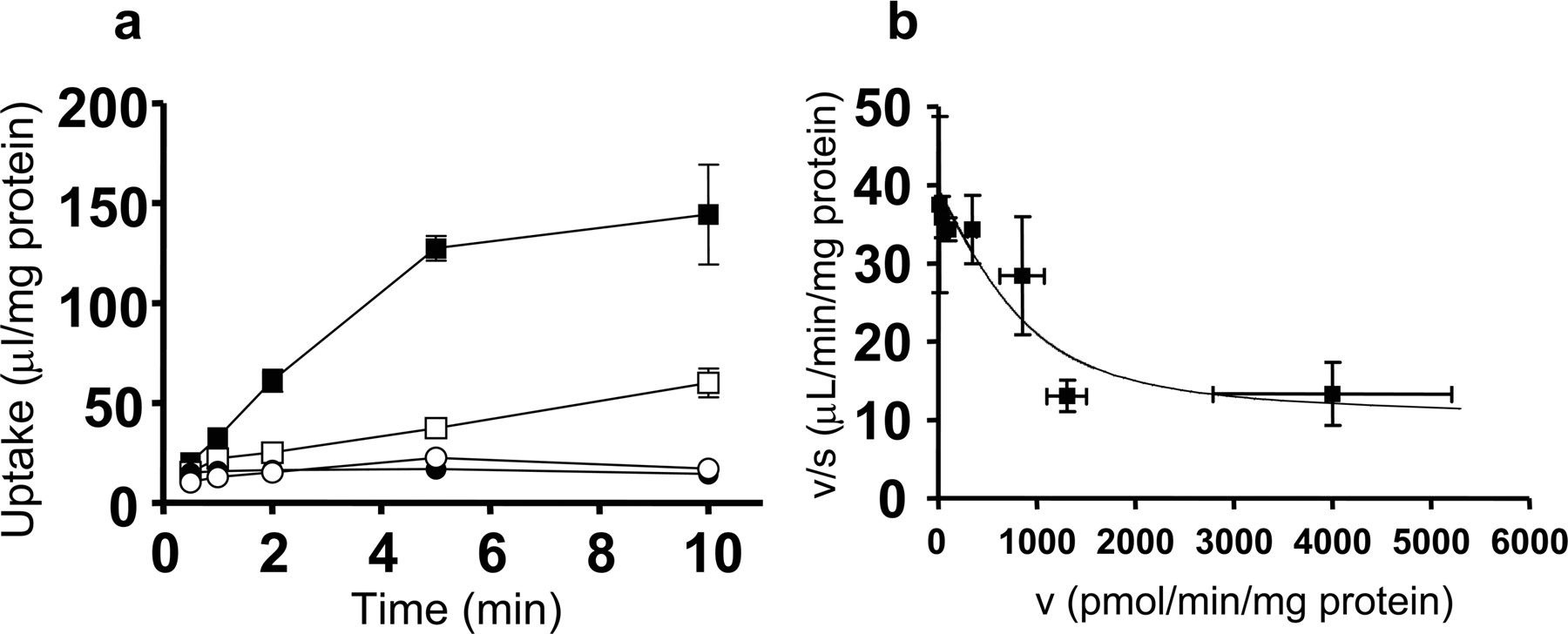
ATP-Dependent Uptake of Valsartan in Human MRP2-Expressing Membrane Vesicles. To confirm that valsartan is a substrate of human MRP2, the time-dependent uptake of [3H]valsartan by membrane vesicles prepared from MRP2-expressing LLC-PK1 cells was examined. [3H]Valsartan was significantly taken up into the membrane vesicles expressing MRP2 in an ATP-dependent manner (Fig. 7a), and this uptake could be saturated with Km, Vmax, and Pdif values of 30.4 ± 17.7 μM, 895 ± 578 pmol/min/mg protein, and 8.88 ± 3.05 μl/min/mg protein, respectively.
Pharmacokinetics of Valsartan in SD Rats and EHBRs. After i.v. administration of 1 mg of valsartan per kg of body weight, we compared the plasma concentration and the biliary excretion of valsartan in SD rats and EHBRs. The plasma concentration was significantly higher in EHBRs compared with SD rats (Fig. 8a). After 2 h, 70% of the total radioactivity injected was excreted into bile in SD rats, whereas in EHBRs, only 15% was excreted (Fig. 8b). The pharmacokinetic parameters of valsartan after i.v. administration in SD rats and EHBRs are summarized in Table. 5. The plasma AUC0-∞ of EHBRs was 17 times higher than that of SD rats, and the total body plasma clearance and biliary clearance were markedly decreased in EHBRs to 6% and 2% of control SD rats, respectively.
Summary for the pharmacokinetic parameters of valsartan in SD rats and EHBRs
Pharmacokinetic parameters are expressed as mean ± S.E. (n = 3).

Time profiles of the transcellular transport of valsartan across MDCKII monolayers expressing transporters. Transcellular transport of valsartan (0.1 μM) across MDCKII monolayers expressing OATP1B1/MDR1 (a), OATP1B1/MRP2 (b), and OATP1B1/BCRP (c) was observed. Open circles and closed circles represent the transcellular transport in the apical-to-basal and basal-to-apical directions, respectively. Each point represents the mean ± S.E. (n = 3).

Concentration dependence of the transcellular clearance (a) and intrinsic clearance across the apical membrane (b) in OATP1B1/MRP2-expressing MDCKII cells. The transcellular transport of valsartan across OATP1B1/MRP2 MDCKII monolayer was observed for 2 h at 37°C (a). The intrinsic clearance for the transport of valsartan across the apical membrane (PSapical) was determined by dividing the transcellular transport velocity of valsartan determined over 2 h by the cellular concentration determined at the end of the experiments (2 h) (b). Triangles and squares represent the transcellular transport across OATP1B1/MRP2 double transfectants and vector-transfected control cells, respectively. Each point represents the mean ± S.E. (n = 3).
Discussion
In humans, valsartan is mainly excreted into bile, mostly in the unchanged form without any significant metabolism (Waldmeier et al., 1997). Considering this physicochemical property, we hypothesized that a series of transporters of organic anions could be involved in the hepatic transport of valsartan. Therefore, in this study, we identified transporters that mediate the hepatic uptake and biliary excretion of valsartan in humans.
First, we performed uptake experiments using OATP1B1 and OATP1B3 expression systems. These transporters are thought to be important for the transport of organic anions in human liver because they are selectively expressed in the liver and can accept a wide variety of compounds. We found that valsartan can be taken up via both OATP1B1 and OATP1B3. In contrast, no significant uptake of valsartan was observed in OATP2B1-expressing cells compared with vector-transfected control cells, suggesting that the contribution of OATP2B1 is negligible as far as hepatic uptake is concerned. To understand the relative importance of OATP1B1 and OATP1B3 in the hepatic uptake of valsartan, we cannot easily compare the uptake clearance of OATP1B1 and OATP1B3 in transporter expression systems because the relative expression level of OATP1B1 and OATP1B3 is different in human hepatocytes and expression systems. To overcome this problem, Hirano et al. (2004) established a methodology for estimating the contribution of OATP1B1 and OATP1B3 to the hepatic uptake of compounds. We used the approach called the relative activity factor (RAF) method (Hirano et al., 2004), in which we use transporter-specific ligands (estrone-3-sulfate for OATP1B1, CCK-8 for OATP1B3). To determine the contribution of transporters, we used three different batches of human hepatocytes prepared from independent donors because Shitara et al. (2003b) reported that there were large interbatch differences in uptake activity in human cryopreserved hepatocytes. Valsartan was taken up by all batches of human hepatocytes in a saturable manner. In the presence of 100 μM valsartan, the time course of the uptake of valsartan was almost flat for 5 min, whereas the uptake of 1 μM valsartan was clearly observed by 2 min, indicating that 100 μM valsartan is enough to saturate the transporter-mediated uptake. This is consistent with our results showing that the Km values of OATP1B1 and OATP1B3 were much lower than 100 μM. We observed the self-saturation of the uptake of valsartan in human hepatocytes at three concentrations (1, 10, and 100 μM), and the uptake clearance at 10 μM was decreased to half of that at 1 μM (Fig. 4).

The ATP-dependent transport of valsartan in MRP2-expressing LLC-PK1 cells. Time profiles for the uptake of valsartan were measured in isolated membrane vesicles prepared from LLC-PK1 cells expressing MRP2 (a). Membrane vesicles were incubated at 37°C with valsartan (0.1 μM) in the medium in the presence of ATP (closed symbols) or AMP (open symbols) for designated periods (0.5, 1, 2, 5, or 10 min). Squares and circles represent the uptake of membrane vesicles expressing MRP2 and control vesicles infected only with adenovirus containing tetracycline-responsive transcriptional activator, respectively. The concentration dependence of MRP2-mediated uptake of valsartan is shown as Eadie-Hofstee plots (b). The uptake of valsartan for 2 min was determined at various concentrations (0.3–300 μM). Each point represents the mean ± S.E. (n = 3).

Biliary elimination of valsartan in male SD rats and EHBRs. Rats were injected with valsartan (1 mg/kg body weight dissolved in phosphate-buffered saline) into a femoral vein after cannulation of the bile duct. The time profiles of the plasma concentration (a) and cumulative biliary excretion (b) of valsartan in SD rats (open circles) and EHBRs (closed circles) are shown. Each point represents the mean ± S.E. (n = 3).
According to the manufacturer's interview form, the maximum plasma concentration (Cmax) and AUC are directly proportional to the dose in Japanese healthy male subjects. After oral administration of 160 mg of valsartan, the Cmax is 12 μM and the plasma unbound fraction is 5 to 7% (Colussi et al., 1997), indicating that the unbound plasma concentration of valsartan is 0.60 to 0.84 μM. This value is lower than the Km values obtained from OATP-expressing systems and human hepatocytes. This is consistent with the fact that valsartan exhibits linear pharmacokinetics over the clinical dose range. The absolute value of the uptake clearance of reference compounds in each batch of hepatocytes was different from the reported values (Hirano et al., 2004). We have sometimes experienced that the viability of the hepatocytes and the uptake clearance of compounds are different between the individual tubes even when they are prepared from the same human liver. To overcome this problem, we always check the uptake clearance of reference compounds such as E217βG in parallel with the test compounds and the relative values of the uptake clearance of reference compounds and test compounds can be discussed.
Regarding the contribution of OATP1B1 and OATP1B3 to the hepatic uptake of valsartan, our results indicate that both OATP1B1 and OATP1B3 are involved in the uptake of valsartan in human hepatocytes, although the estimated contribution of each batch of hepatocytes was different (Table. 3). Previous reports suggest that pitavastatin, a novel 3-hydroxy-3-methylglutaryl CoA reductase inhibitor, and estradiol-17β-glucuronide, a typical substrate of both OATP1B1 and OATP1B3, are taken up into hepatocytes predominantly via OATP1B1 (Hirano et al., 2004), whereas the hepatic uptake of fexofenadine, an H2-receptor antagonist, is thought to be mainly via OATP1B3 rather than OATP1B1 (Shimizu et al., 2005). Therefore, although OATP1B1 is generally believed to be responsible for the hepatic uptake of several kinds of drugs, we think that the relative importance of OATP1B1 and OATP1B3 depends on the drugs themselves, and this estimation method is useful for predicting their contribution to hepatic clearance. Very recently, we found that telmisartan is recognized exclusively by OATP1B3, and not OATP1B1 (Ishiguro et al., 2006). So, even within the same category of drugs, the relative contribution of OATP1B1 and OATP1B3 is different. This might result in differences in the pharmacokinetics and subsequent pharmacological effects when the function of certain transporters is changed by a variety of conditions, such as genetic polymorphism (e.g., OATP1B1*15) (Nishizato et al., 2003). Therefore, we think that it is important to evaluate the contribution of each transporter to the hepatic elimination of drugs because we can then estimate the change in the overall hepatic clearance quantitatively when the expression level and/or transport function of certain transporters is changed by pathophysiological conditions, single-nucleotide polymorphisms, and transporter-mediated drug-drug interactions. This kind of information may make it possible to predict the pharmacokinetics and subsequent pharmacological effects and side effects of drugs under certain conditions.
Next, to identify which transporters are involved in the biliary elimination of valsartan, we investigated the transcellular transport of valsartan in MDCKII cells coexpressing OATP1B1/MDR1, OATP1B1/MRP2, and OATP1B1/BCRP. As a result, the basal-to-apical transcellular transport of valsartan in OATP1B1/MRP2 double transfectant was the largest among three kinds of double transfected cells, whereas that in OATP1B1/MDR1 transfectant was slightly observed, and any significant transport could not be observed in OATP1B1/BCRP transfectant (Fig. 5). In parallel, we observed significant vectorial transport of estradiol-17β-glucuronide in OATP1B1/MDR1, OATP1B1/MRP2, and OATP1B1/BCRP double transfected cells at the same level as in a previous report (Matsushima et al., 2005). The ratio of the basal-to-apical transport to that in the opposite direction in OATP1B1/MRP2 cells was the highest among these double transfectants in the case of estradiol-17β-glucuronide and pravastatin (Matsushima et al., 2005), which were reported to be excreted mainly via MRP2, judged from the impairment of the biliary excretion in EHBR, an Mrp2-deficient rat (Yamazaki et al., 1997; Morikawa et al., 2000). These results suggest that valsartan is also mainly excreted by MRP2. We observed the saturation of transcellular transport in OATP1B1/MRP2 double transfectants with a Km value of 27.5 μM (Fig. 6a; Table. 4). This Km value is smaller than that of the efflux transport across the apical membrane (PSapical) (99.0 μM) (Fig. 6b; Table. 4). From a kinetic viewpoint, if the intrinsic efflux clearance across the apical membrane, on which efflux transporters are over-expressed in MDCKII cells, is larger than that across the basal membrane, the transcellular clearance is almost equal to the uptake clearance. Therefore, the Km value of transcellular transport represents that of OATP1B1-mediated uptake clearance. From our results, the Km value of transcellular transport was greater than that of uptake by OATP1B1 in HEK293 cells. This discrepancy may be due to the fact that the host cells are different or the uptake process is not a rate-limiting step for the transcellular transport of valsartan. We also checked the ATP-dependent uptake of valsartan in MRP2-expressing membrane vesicles and found time-dependent saturable uptake (Fig. 7). The Km value obtained from membrane vesicles is smaller than that of PSapical in OATP1B1/MRP2 double transfectants. This is reasonable because we did not estimate the unbound intracellular concentration of valsartan in double transfectants, and the Km value normalized by the unbound concentration should be lower than the current value.
In addition, we examined the biliary excretion of intravenously administered valsartan in SD rats and EHBRs, in which the MRP2 expression is hereditarily defective. The elimination of valsartan from blood was drastically delayed in EHBRs compared with SD rats, and the biliary excretion clearance in EHBRs was 44 times lower than that in SD rats (Fig. 8), indicating that MRP2 is responsible for the biliary excretion of valsartan, at least in rats. The contribution of transporters might show a species difference. However, taking the results of the transcellular transport in double transfected cells and the ATP-dependent uptake in MRP2-expressing membrane vesicles into consideration, it appears that MRP2 also plays an important role in the biliary excretion in humans.
Further investigations will be required to determine the contribution of efflux transporters to the overall biliary excretion in humans by quantitative comparison of the relative expression levels of each transporter in double transfectants and human liver samples. Information about the contribution of each transporter to hepatic drug transport will be provided by clinical studies investigating the effect of genetic polymorphisms in certain transporters on the pharmacokinetics of different drugs.
In conclusion, we have demonstrated that both OATP1B1 and OATP1B3 are responsible transporters for the hepatic uptake of valsartan, and the efflux clearance of valsartan is mainly via MRP2.
Acknowledgments
We are grateful to Masaru Hirano (The University of Tokyo, Tokyo, Japan) for helpful advice and to Satoshi Kitamura (The University of Tokyo, Tokyo, Japan) for supporting our experiments. We also express our great appreciation to Novartis Pharma AG (Basel, Switzerland) for providing us 3H-labeled and unlabeled valsartan and to Dr. Yuko Tsukamoto and Dr. Ryosei Kawai (Novartis Pharma K.K., Tokyo, Japan) for fruitful discussion.
Footnotes
-
This work was partly supported by Health and Labor Sciences Research Grants from the Ministry of Health, Labor, and Welfare for the Research on Advanced Medical Technology and by Grant-in Aid for Young Scientists B (17790113) from the Ministry of Education, Culture, Sports, Science, and Technology.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.008938.
-
ABBREVIATIONS: OATP, organic anion-transporting polypeptide; AUC, area under the plasma concentration-time curve; BCRP, breast cancer resistance protein; CCK-8, cholecystokinin octapeptide; E1S, estrone-3-sulfate; E217βG, estradiol-17β-glucuronide; EHBR, Eisai hyperbilirubinemic rat; MRP2, multidrug resistance-associated protein 2; MDR1, multidrug resistance 1; PS, permeability surface; SD, Sprague-Dawley; CL, clearance.
- Received December 13, 2005.
- Accepted April 18, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}