


Abstract
Some mechanism-based inhibitors cause irreversible inhibition by forming a metabolic intermediate complex (MIC) with cytochrome P450. In the present study, 54 molecules (substrates of CYP3A and amine-containing compounds that are not known substrates of CYP3A) were spectrophotometrically assessed for their propensity to cause MIC formation with recombinant CYP3A4 (+b5). Comparisons of common physicochemical properties showed that mean (±S.D.) mol. wt. of MIC-forming compounds was significantly greater than mean mol. wt. of non-MIC-forming compounds, 472 (±173) versus 307 (±137), respectively. Computational pharmacophores, logistic regression, and recursive partitioning (RP) approaches were applied to predict MIC formation from molecular structure and to generate a quantitative structure activity relationship. A pharmacophore built with SKF-525A (2-diethylaminoethyl 2:2-diphenylvalerate hydrochloride), erythromycin, amprenavir, and norverapamil indicated that four hydrophobic features and a hydrogen bond acceptor were important for these MIC-forming compounds. Two different RP methods using either simple descriptors or 2D augmented atom descriptors indicated that hydro-phobic and hydrogen bond acceptor features were required for MIC formation. Both of these RP methods correctly predicted the MIC formation status with CYP3A4 for 10 of 12 literature molecules in an independent test set. Logistic multiple regression and a third classification tree model predicted 11 of 12 molecules correctly. Both models possessed a hydrogen bond acceptor and represent an approach for predicting CYP3A4 MIC formation that can be improved using more data and molecular descriptors. The preliminary pharmacophores provide structural insights that complement those for CYP3A4 inhibitors and substrates.
The cytochrome P450 (P450) enzymes (EC 1.14.14.1) are membrane-bound proteins that catalyze many oxidations of hydrophobic endobiotics and xenobiotics. The catalytic activity of P450s may be reduced by reversible and irreversible inhibition upon administration of xenobiotics. One type of irreversible inhibition, mechanism-based inhibition, has been the focus of many studies related to P450s recently due to its clinical implications for predicting drug-drug interactions. A mechanism-based inhibitor is one that binds to the active site and then becomes catalytically activated by the enzyme (Silverman, 1988). The activated form of the molecule will irreversibly bind to the enzyme to remove it from the active enzyme pool. Some mechanism-based inhibitors cause this irreversible inhibition by forming a metabolic intermediate complex (MIC) with the heme of the P450 (Franklin, 1977). Primary, secondary, or tertiary amines or methylenedioxy constituents (Supplemental Data Fig. 1) in the molecule are prerequisites for compounds that chelate the heme of the P450 (Franklin, 1977). More recent studies with metabolites of molecules such as indinavir and nelfinavir that lack these functional groups yet still display MIC formation may indicate that other chemical moieties are also involved (Ernest et al., 2005).
The CYP3A family of enzymes is recognized as perhaps the most important for human drug metabolism because they metabolize most commercially available drugs (Wrighton et al., 2000). These P450s are expressed in numerous tissues but affect xenobiotic metabolism and clearance mainly in the liver and small intestine. There are four differentially regulated CYP3A genes in humans, CYP3A4, CYP3A5, CYP3A7, and CYP3A43. Of these genes, CYP3A4 is the most abundant form in the adult liver (Wrighton et al., 2000). The CYP3A forms demonstrate regioselectivity differences for some biotransformations of the same compounds, whereas CYP3A5 generally has lower or comparable metabolic capability than CYP3A4 for common probe substrates (Williams et al., 2002).
In early pharmaceutical drug discovery, the assessment of inhibitory potency with CYP3A4 for new chemical entities is often included as a first tier in vitro screen using fluorescent probes (Crespi and Stresser, 2000). This type of screen is useful for identifying potential inhibition of coadministered drugs and is commonly followed by time-dependent inhibition studies (Wu et al., 2003). These studies may be supplemented with additional in vitro assays with more traditional drug substrate probes using liquid chromatography/mass spectrometry analysis (Ekins et al., 2000b). Data from all of these in vitro screens are increasingly applied to predictive algorithm development (Riley et al., 2001; Gao et al., 2002; Ekins et al., 2003a,b). The interest in computational models based on in vitro data for predicting potential drug interactions via this protein and others (Ekins and Swaan, 2004) represents a possible means to improve productivity of the drug discovery process and remove potential bottlenecks caused by in vitro testing. Due to their highly parallel nature, computational methods are also probably the fastest and most cost-effective method for indicating likely toxic consequences (Ekins et al., 2000a) and suggesting new hypotheses for testing in vitro. A recent review of computational methods for P450s has documented how these approaches have been used over nearly 20 years alongside empirical methods (de Graaf et al., 2005). Others have described and compared the many pharmacophores that have been generated for P450s (Ekins et al., 2001; de Groot and Ekins, 2002), providing insight into the important features for the interaction of ligands and proteins. Computational pharmacophores for CYP3A4 have therefore been derived for substrates and inhibitors using kinetic constants Km, Ki (apparent), and IC50 data. Several studies have shown differential MIC formation for compounds between CYP3A4 and CYP3A5. For example, CYP3A5 did not form a MIC with verapamil (Wang et al., 2004) or saquinavir (Ernest et al., 2005). These differences suggest that the binding sites (and catalytic rate) for both enzymes are subtly different, accommodating some molecules and not others as has been suggested by recent pharmacophores for inhibitors of both enzymes (Ekins et al., 2003b).
In the present study, 54 molecules were assessed for their propensity to form MIC, whereas 27 molecules possess kinact data with recombinant CYP3A4 in vitro. These data were analyzed along with the generation of simple molecular descriptors to understand any possible relationships between MIC, kinact, and molecule structure. Several computational approaches, e.g., pharmacophores, quantitative structure activity relationships (QSARs) (Ekins and Swaan, 2004), classification trees, and multiple regression methods, were used to generate models to predict these properties separately. These models were applied to the prediction of the probability of a molecule forming an MIC with CYP3A4 using a series of test molecules, whereas the kinact models were internally validated by omitting molecules at random.
Materials and Methods
Chemicals. All of the following classes of study drugs were purchased from Sigma-Aldrich (St. Louis, MO) or United States Pharmacopeia (Rockville, MD) or kindly donated by companies or investigators. NADPH was purchased from Roche Diagnostics (Indianapolis, IN). All other chemicals were at least analytical grade.
Antibiotics: triacetyloleandomycin; erythromycin; N-desmethylerythromycin (United States Pharmacopeia); and clarithromycin, N-desmethylclarithromycin, and 14-hydroxyclarithromycin (donated by Abbott Laboratories, Chicago, IL).
Calcium channel blockers: amlodipine; diltiazem; N-desmethyldiltiazem, desacetyldiltiazem, and desacetyl-N-desmethyldiltiazem (donated by Tanabe Seiyaku Co., Osaka, Japan); R-verapamil; S-verapamil; norverapamil; d-617; and nicardipine.
Central nervous system drugs: amitriptyline; d,l-amphetamine; benzphetamine; brompheniramine; chlorpheniramine; desipramine; diphenhydramine; fenfluramine; fluoxetine; fluvoxamine; imipramine; loperamide; meperidine; methamphetamine; methylenedioxymethamphetamine (MDMA); methylenedioxyethylamphetamine (MDE); 2-methylamino-1-(3,4-methylenedioxyphenyl)butane (MBDB); methylphenidate; mirtazapine; nefazodone; norfluoxetine; nortriptyline; orphenhydramine; paroxetine; phencyclidine; propoxyphene (United States Pharmacopeia); sertraline; and tranylcypromine.
Human immunodeficiency virus protease inhibitors: amprenavir (donated by GlaxoSmithKline, Research Triangle Park, NC); indinavir (donated by Merck, Whitehouse Station, NJ); nelfinavir (donated by Agouron Pharmaceuticals, Inc., New York, NY); ritonavir (donated by Abbott Laboratories); saquinavir (donated by F. Hoffmann-La Roche, Nutley, NJ); and lopinavir (donated by Abbott Laboratories).
Anticancer drugs: tamoxifen; N-desmethyltamoxifen (donated by Zeneca Pharmaceuticals, Wilmington, DE); 4-hydroxytamoxifen; and 3-hydroxytamoxifen.
Miscellaneous drugs: mifepristone and SKF-525A.
Enzyme Preparation. Insect cell membranes containing baculovirus cDNA-expressed CYP3A4 (+b5) were purchased from BD Gentest (Bedford, MA). The P450 content was provided by the manufacturer at the time of purchase.
Estimation of Mechanism-Based Inactivation Parameters. Testosterone 6β-hydroxylation was determined to quantify time- and concentration-dependent loss of CYP3A4 activity in the presence of inactivator (Ernest et al., 2005). The concentration of 6β-hydroxytestosterone was determined by high-performance liquid chromatography with UV detection as described previously (Zhao et al., 2002). The mechanism-based inactivation parameters kinact and KI were obtained from the pseudo first-order decline in the percentage of remaining CYP3A4 activity after preincubation with inactivator by nonlinear regression without weighting using WinNonlin Professional version 4.0 (Pharsight, Mountain View, CA). Details of the data fitting and parameter estimation have been described previously (Ernest et al., 2005).
Metabolic Intermediate Complex Measurements. MIC formation was identified using dual wavelength spectroscopy (UVIKON 933 Double-Beam UV/VIS Spectrophotometer; Research Instruments International, San Diego, CA) by scanning from 380 to 500 nm. The sample cuvette contained protein [200 pmol of CYP3A4 (+b5)], 100 mM sodium phosphate buffer (pH 7.4), inhibitor, and 1 mM NADPH (made with the phosphate buffer), whereas the reference cuvette contained protein, 100 mM phosphate buffer, vehicle, and 1 mM NADPH. All MIC formation experiments were initiated by the addition of NADPH and maintained at 37°C. The absorbance difference spectra for the identification of MIC formation were estimated by subtracting the absorbance at 490 nm of the absorbance scan from the difference of the absorbance scan at 60 min and a background absorbance scan. MIC formation was quantified from absorbance difference spectra using an extinction coefficient of 65 mM-1 cm-1 (Pershing and Franklin, 1982).
Computational Modeling. An initial assessment was undertaken to determine whether MIC and non-MIC-forming compounds could be differentiated with simple calculated molecular properties. Calculated log P (octanol-water partition coefficient) and mol. wt. were determined for each compound with ChemDraw for Excel (CambridgeSoft, Cambridge, MA). Statistical parameters were calculated with SPSS version 12.0 (SPSS Inc., Chicago, IL). In addition, several different computational techniques were evaluated: a pharmacophore method (Catalyst; Accelrys Inc., San Diego, CA), three recursive partitioning (tree) methods, which included ChemTree (Golden Helix Inc., Bozeman, MT), Cerius2 (Accelrys Inc.), and tree function in R software version 2.2.1 (http://www.r-project.org) using different types of descriptors, a linear model (multiple regression and regression tree model) using R software version 2.2.1, and a logistical model using R software version 2.2.1.
Pharmacophores for MIC Prediction. The Catalyst software was used on a Silicon Graphics Octane workstation (Silicon Graphics, Sunnyvale, CA). After importing the molecular structures, conformers were generated for each compound using the BEST functionality for each molecule and limited to a maximum of 255 conformers with an energy range of 20 kcal/mol. Structural relationships between compounds that form MIC and those that do not form MIC were assessed separately using the common features function (HipHop). The following groups of molecules were selected to test the fit of the hypothesis with unknowns: compounds that form MIC (SKF-525A, erythromycin, amprenavir, and norverapamil); compounds that do not form MIC (mifepristone, sertraline, 4-hydroxytamoxifen, and paroxetine); and compounds that do not form MIC but inactivate CYP3A4 (lopinavir, saquinavir, and nefazodone). These alignments were then used to fit molecules to the hypothesis.

Metabolic intermediate complex formation of SKF-525A (A), phencyclidine (B), and lopinavir (C) with CYP3A4 (+b5). Lines represent the change in absorbance difference for scans at 5 (——), 15 (......), 30 (— — —), and 60 (—..—..—..) minutes.
Recursive Partitioning (ChemTree) for MIC Prediction. The ChemTree recursive partitioning software was run on a Pentium 4 processor. The 54 molecules and experimental data (binary response: 1, compound that forms MIC; 0, compound that does not form detectable MIC) were imported as an .sdf file into ChemTree to generate over 330 path-length descriptors (Young et al., 2002). These descriptors were used to generate either single-tree or 100 random-tree models with the following options: p value threshold for splits, 0.99; maximum segments, 3; parallel threads, 1; and resampling iterations, 10,000.
Recursive Partitioning (Cerius2 CSAR) for MIC Prediction. The Cerius2 4.8 software was used to generate the following default descriptors: sum of atomic polarizabilities, dipole magnitude, radius of gyration, area, mol. wt., molecular volume, density, principal moment of inertia, rotatable bonds, hydrogen bond acceptors, hydrogen bond donors, and AlogP98 (a second method for calculating the octanol-water partition coefficient). The CSAR recursive partitioning method (Hawkins et al., 1997) was used with the 54-molecule training set with the following settings: equally weighted observations, Gini scoring, and scaled prune (0). This model was internally validated using cross-validation: e.g., 10-fold, 5-fold, or 2-fold. A 10-fold cross-validation leaves out 10% test data.
Recursive Partitioning (Tree Function in R) for MIC Prediction. The R version 2.2.1 software was used to predict MIC formation based on the 12 prescribed descriptors. The recursive partitioning method (Breiman et al., 1984) was used with the 54-molecule training set (with equally weighted observations and Gini scoring). This model was internally validated using 5-fold cross-validation, and the tree is pruned based on misclassification.
Logistic Regression for MIC Prediction. The logistic regression model was implemented in an R function, generalized linearized model (glm) (R version 2.2.1). The model was generated with the 12 Cerius2 descriptors to predict MIC formation. Fifty-four compounds served as the training set. The glm() algorithm is based on the maximizing likelihood approach. A forward stepwise forward variable selection strategy was used to select descriptors. The optimal model was internally validated based on a 5-fold cross-validation.
Pharmacophore, tree models, and the logistic regression model were tested with molecules outside of the training set using recently published data for 12 compounds (Yamazaki and Shimada, 1998; Kasahara et al., 2000; Kim et al., 2001; Kajita et al., 2002; Chatterjee and Franklin, 2003; Wu et al., 2003) with MIC formation for CYP3A4.
Linear Multiple Regression and Regression Tree Models for kinact, KI, and kinact/KI Prediction. The kinact and KI values were estimated from in vitro studies with 27 compounds as described previously. Linear multiple regression and regression tree models were fit with the 12 descriptors listed under “Recursive Partitioning (Tree Function in R) for MIC Prediction” to predict kinact, KI, kinact/KI. Stratified randomization was used to divide samples into three strata based on 33 percentile and 67 percentile of kinact and/or KI values. Within each stratum, 2/3 of the samples (n = 18) were randomly selected into the training set, and the remaining 1/3 (n = 9) were selected into the external validation set. All variables were log-transformed.
Two R functions, linear model (lm), and tree() (R version 2.2.1) were implemented for regression tree model construction. These two predictive models were based on 5-fold cross-validation on training set samples.
Results
Formation of Metabolic Intermediate Complex. MIC formation with each compound and CYP3A4 (+b5) was determined spectro-photometrically. Figure 1 illustrates absorbance difference changes by SKF-525A, a compound that inactivates CYP3A4 and forms MIC, phencyclidine, a compound that is not metabolized by CYP3A4 and does not form MIC, and lopinavir, a compound that inactivates CYP3A4 but does not form MIC. The minimum detectable absorbance difference is 0.002, which corresponds to 31 pmol of complex or 16% of total CYP3A4. The percentage of maximum MIC was estimated by comparing the amount of MIC that was formed with the amount of CYP3A4 (+b5) that was used in the experiment. If a compound formed less than 16% of maximum MIC, it was considered to be a compound that does not form MIC. The MIC formation results are shown in Table 1.
Data on MIC formation with recombinant expressed CYP3A4 (+b5) and used for training sets
ClogP and mol. wt. were calculated with ChemDraw for Excel (CambridgeSoft). All data were produced in this study except where explicitly referenced. For predicted MIC formation, a value of >0.5 was used for MIC formation; a value of <0.5 was used for no MIC formation.
Mechanism-Based Inactivation Parameters. The kinact and KI values for 27 compounds were estimated from in vitro preincubations with CYP3A4 (+b5). The estimates for each compound are listed in Table 2. The kinact values ranged from 0.03 to 1.12 min-1, and KI values ranged from 0.1 to 12.5 μM.
Estimated kinact, KI, and kinact/KI generated with recombinant expressed CYP3A4 (+b5)
All data were produced in this study except where explicitly referenced.
Computational Methods. An initial comparison of the distributions of the molecular weights of 27 molecules that formed MIC to the 27 molecules that do not form MIC (Table 1; Supplemental Data Fig. 2) indicated mean values of 472 (standard deviation = 174; range, 263.4-798) and 308 (standard deviation = 137; range, 133.2-670.9), respectively, which were statistically significant applying a t test with p < 0.05. The mean calculated log P for MIC-forming compounds, 3.92 (standard deviation = 1.44; range, 1.44-6.81), was not significantly different when compared with the mean calculated log P for non-MIC-forming compounds, 3.86 (standard deviation = 1.53; range, 1.24-7.05), applying the same statistical tests.
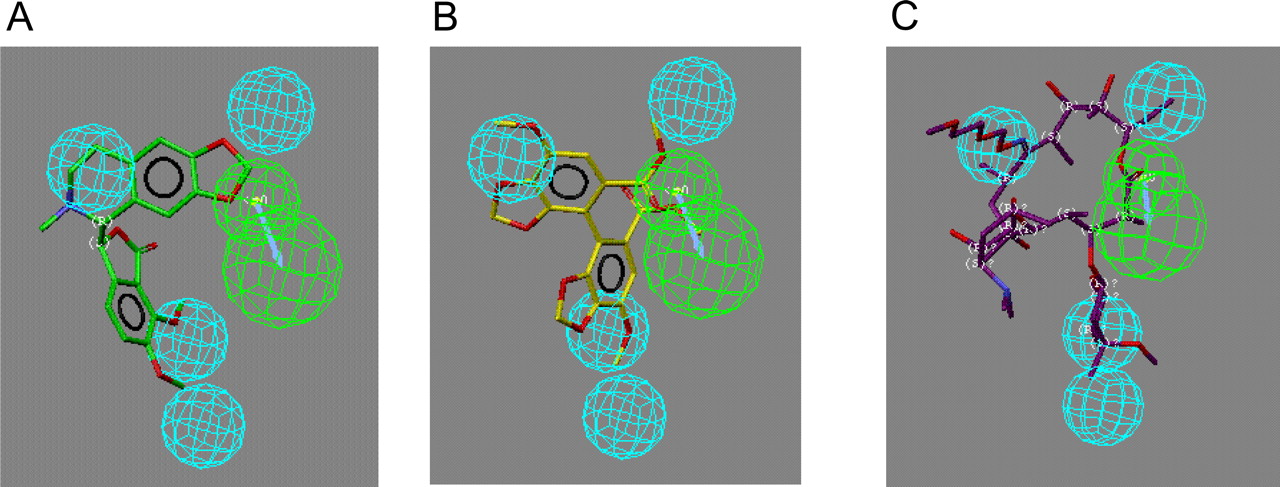
Pharmacophores for MIC Prediction. Catalyst was used to generate common-feature alignment (HipHop) pharmacophores for compounds that formed MIC and those that did not. These preliminary pharmacophores represent the disposition of common chemical features such as hydrogen bond acceptor, hydrogen bond donor, and hydrophobe in 3D space. The pharmacophore based on the four MIC-forming compounds (SKF-525A, erythromycin, amprenavir, and norverapamil) indicated that four hydrophobic features and a hydrogen bond acceptor were common (Fig. 2A). The pharmacophore for non-MIC-forming compounds was smaller and possessed two hydrophobes and one hydrogen bond donor (Fig. 2B). The pharmacophore for compounds that were CYP3A4 inactivators but did not form MIC was considerably larger and richer in features (five hydrophobes and two hydrogen bond acceptor features) than the MIC pharmacophore (Fig. 2C). These pharmacophores represent a qualitative approach, and the MIC-forming compound pharmacophore was used to evaluate the mapping of several other literature compounds, including (+)-β-hydrastine, dimethyl-4,4′-dimethoxy-5,6,5′,6′-dimethylenedioxybiphenyl-2,2′decarboxylate (DDB), and roxithromycin, which show a good agreement with most of the features (Fig. 3).

Common-feature pharmacophores for (A) MIC-forming compounds (SKF-525A, erythromycin, amprenavir, and norverapamil), (B) non-MIC-forming compounds (mifepristone, sertraline, 4-hydroxytamoxifen, and paroxetine), and (C) non-MIC-forming compounds that inactivate CYP3A4 (lopinavir, saquinavir, and nefazodone). Blue spheres, hydrophobic; green feature, hydrogen bond acceptor; purple spheres, hydrogen bond donor.
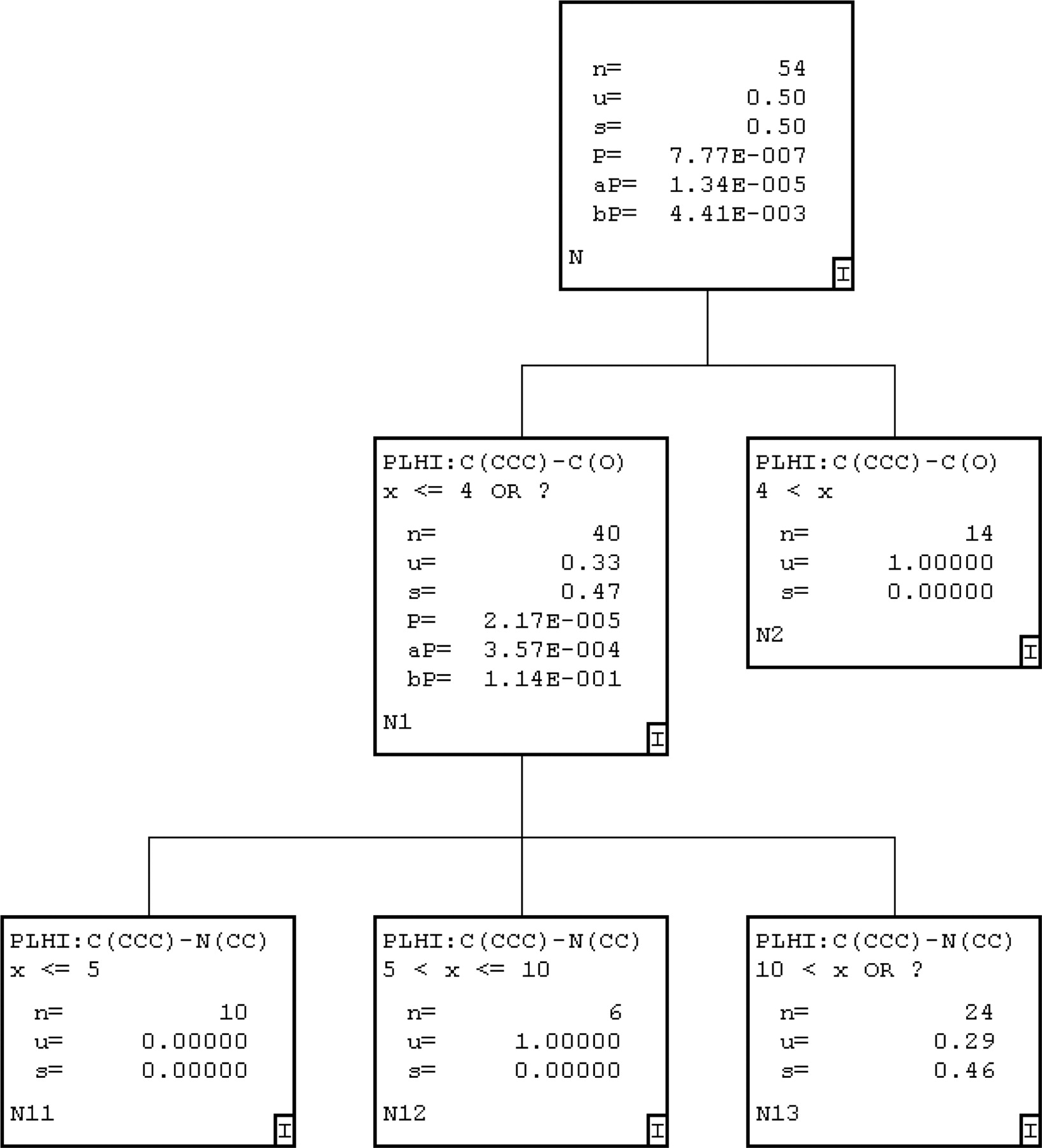
Recursive Partitioning (ChemTree) for MIC Prediction. Two different recursive partitioning methods were used to produce models for MIC prediction. The first method was used to produce a single-tree model, and the second model produced a 100 random-tree model by applying ChemTree to all 54 molecules in the training set. The single-tree model (Fig. 4) was produced that used two path-length descriptors (Young et al., 2002), C(CCC)-C(O) (e.g., this descriptor represents a carbon atom attached to three other carbon atoms at a distance from a carbon-oxygen bond) and C(CCC)-N(CC), of 330 general descriptors that were initially generated. This model misclassified seven molecules in the training set based on a score less than 0.5 for compounds that do not form MIC and a score greater than 0.5 for compounds that do form MIC, such that 87% were correctly predicted (Table 1). The 100 random-tree model generated with the same training set used 21 of the 330 available descriptors. The most frequently used descriptors were C(CCC)-N(CC), C(CCC)-C(CCC), C(CCC)-C(CN), C(N)-C(O), and C(CC)-N(CC) (representing connectivity between the atoms listed). When the observed versus predicted values for the 100-tree model were analyzed, 5 of the 54 compounds (91% accuracy) were misclassified as non-MIC-forming compounds (Table 1).
The single-tree and 100-tree models both incorrectly assigned 2 of the 12 compounds in the validation test set (Supplemental Data Fig. 3), representative of 83% accuracy (Table 3). These compounds were olopatadine M1 and miocamycin.
Observed and predicted data for molecules in the validation test set
Recursive Partitioning (Cerius2 CSAR) for MIC Prediction. The second recursive partitioning tree model was generated with 12 descriptors in Cerius2 (Fig. 5; Supplemental Data Table 1). This CSAR model correctly classified all of the molecules in the training set using the same 54 compounds that were used in the ChemTree models. This model was internally validated using 10-fold, 5-fold, and 2-fold cross-validation, resulting in 74, 80, and 76% correct predictions, respectively. This stability in the predictions for the molecules that were left out perhaps also suggests the stable nature of this model for external testing. This model incorrectly assigned roxithromycin M2 and M3 as non-MIC-forming and miocamycin as MIC-forming, i.e., 3 of 12 compounds in the validation test set, which represents 75% accuracy (Table 3).
Test set MIC-forming compounds fitted to the MIC common-features pharmacophore. A, (+)-β-hydrastine; B, dimethyl-4,4′-dimethoxy-5,6,5′,6′-dimethylene dioxybiphenyl-2,2′decarboxylate; and C, roxithromycin. Blue spheres, hydrophobic; green feature, hydrogen bond acceptor.

An example of a single automatically generated recursive partitioning tree for CYP3A4 (+b5) MIC data. The parent population has 54 binary observations with a mean of 0.5 because half are MIC-forming and half are non-MIC-forming. The path-length descriptor C(CCC)-C(O) (Young et al., 2002) is an integer distance range within the molecule and can be split significantly (p = 7.77 e-7) two ways. From this point, the remaining 40 compounds are split into 3 subsets using C(CCC)-N(CC).
The combination of Cerius2 molecular descriptors with ChemTree descriptors enabled another 100-tree model to be built with ChemTree. This used 37 of 342 available descriptors. The most frequently used descriptors in addition to those path-length descriptors previously mentioned included the radius of gyration from the Cerius2 descriptors. This 100-tree ChemTree recursive partitioning model was used to generate predictions for molecules that were not in the training set, and the mean score determined as described above. This model was used to score the validation set of literature molecules for MIC formation and incorrectly assigned olopatadine M1 and miocamycin as MIC-forming, 2 of 12, which represents 83% accuracy (Table 3).
Recursive Partitioning (Tree Function in R) for MIC Prediction. Another recursive partitioning tree model (logistic model tree function in R) was generated using the 12 descriptors from Cerius2 (Fig. 6; Supplemental Data Table 1). This tree model was formed by optimizing the Gini function. It was then pruned by minimizing the misclassification rate in a 5-fold cross-validation. The final tree model for the 54-molecule training set correctly classified 96% of the molecules in the training set and contained hydrogen bond acceptor, radius of gyration, sum of atomic polarization, and dipole magnitude descriptors. This model was also used to predict the tendency of the literature test set molecules to form an MIC. Among 12 compounds in the external validation set, the model incorrectly assigned miocamycin as MIC-forming, which relates to 92% accuracy (Table 3).

Cerius2 CSAR tree model for CYP3A4 (+b5) MIC data. In this decision tree, a branch downward indicates that the criteria were satisfied, i.e., true. A branch upward indicates that the criteria were not satisfied at that particular decision point.
Logistic Regression Model for MIC Prediction. Logistic regression was generated with an R function glm and the 12 Cerius2 molecular descriptors (Supplemental Data Table 1) to predict MIC formation. Logistic regression uses a logit transformation of descriptors in linear combinations to predict the probability of a binary outcome variable. Usually, maximum likelihood estimation is used for estimating regression parameters. Fifty-four drug compounds served as the training set (Table 1), and 12 compounds represented the external validation set (Table 3). The stepwise feed-forward procedure was chosen to select and optimize prediction model. The internal validation was based on a 5-fold cross-validation.
The logistic regression training data produced a predictive model in which the hydrogen bond acceptor descriptor was the most important predictor (p = 0.002). The following equation was used to predict MIC formation applying the hydrogen bond acceptor descriptor: 
The logistic regression model had 80% prediction accuracy for the 54 compounds in the training set. Among 12 compounds in the external validation set, it incorrectly assigned miocamycin as MIC-forming, which represents 92% prediction accuracy (Table 3).
Regression Tree Models for kinact, KI, and kinact/KI Prediction. Recursive partitioning tree models (tree function in R) were generated with 12 descriptors in Cerius2 (Table 2; Supplemental Data Table 1) for kinact, KI, and kinact/KI prediction. The tree models were formed by optimizing the Gini function. It was then pruned by minimizing the sum of square of error in a 5-fold cross-validation. The final tree models exhibited coefficients of determination (r2) of 0.41, 0.39, 0.64 for kinact, KI, and kinact/KI prediction, respectively, in the 18 training samples and r2 values of 0.12, 0.15, 0.05 in the nine validation samples (Table 4). The kinact/KI model contained sum of atomic polarizabilities and hydrogen bond donor descriptors; the kinact model contained AlogP98 and hydrogen bond donor, and the KI possessed dipole magnitude and density descriptors.
Coefficient of determination values for regression tree models and linear multiple regression used in the prediction of kinact, KI, and kinact/KI

The R version 2.2.1 tree model for CYP3A4 (+b5) MIC formation.
Linear Regression Models for kinact, KI, and kinact/KI Prediction. Linear regression was implemented with an R function lm. A stepwise feed-forward procedure and a 5-fold cross-validation were used to select optimal prediction models. The final regression models provided r2 values of 0.16, 0.26, and 0.40 in the 18 training samples for kinact, KI, and kinact/KI prediction, respectively, and r2 values of 0.23, 0.26, and 0.11 in the nine validation samples (Table 4). The kinact/KI model contained the rotatable bond descriptor; the kinact model contained the radius of gyration descriptor, and the KI model possessed the sum of atomic polarization descriptor.
Discussion
Considerable research has focused on computational algorithms that can be used in drug discovery for predicting molecular properties. These include rule-based models for predicting the likelihood of absorption (Lipinski et al., 1997), methods that have more graphical outputs for predicting binding to P450s (Ekins et al., 2001), or abstract QSAR methods applied to many proteins and properties relevant to absorption, distribution, metabolism, and excretion/toxicity (Ekins and Swaan, 2004). Such calculations can be performed with very large numbers of molecules to act as a molecule selection filter. Comparative molecular fields analysis and pharmacophore approaches have been used to model P450 enzymes involved in drug metabolism and which have a role in important drug-drug interactions (de Groot et al., 1996, 1999a,b; Jones et al., 1996; Ekins et al., 2001). Recursive partitioning methods have been used extensively with large sets of molecules and either continuous (Chen et al., 1998, 1999) or binary data for therapeutic target endpoints and P450 inhibition (Ekins et al., 2003a) and toxicity properties such as Ames mutagenicity status (Young et al., 2002). Linear regression is perhaps one of the earliest approaches used for QSAR of P450 (Hansch and Zhang, 1993). The goal of a logistic regression analysis is to find the best-fitting, most parsimonious probabilistic model to describe the relationship between an outcome and a set of predictors. What distinguishes the logistic regression model from the linear regression model is that the outcome variable in logistic regression is categorical. In fitting the logistic regression model, a maximum likelihood-based approach is used to estimate the regression parameters (Hosmer and Lemeshow, 1989). Logistic regression analysis has been used for a QSAR-modeling antibacterial study (Cronin et al., 2002) as well as in other areas. In the present study, we have applied pharmacophore, recursive partitioning, and logistic regression methods to aid in discriminating between MIC-forming compounds and those that do not form an MIC.
A preliminary comparison between CYP3A4 (+b5) MIC and non-MIC-forming compounds based on the mean molecular weight showed a significant difference between the two groups of compounds (p < 0.05), indicating that larger molecules are more likely to form MIC in vitro with CYP3A4 (+b5). Another comparison was done on compounds that were CYP3A4 substrates compared with non-CYP3A4 substrates, which resulted in a similar outcome; i.e., there was a significant difference in the molecular weights between the two groups. This difference may be due to the specific set of data used in our study (the macrolide antibiotics and anti-human immunodeficiency virus compounds tend to have higher molecular weights) and may not be a universal characteristic of substrates/inhibitors of CYP3A4. However, there was no significant difference between the ClogP between the two groups of compounds. The qualitative approach of common-feature pharmacophore alignments (Catalyst, 2003) for selected compounds suggests that CYP3A4 (+b5) MIC formation requires at least multiple (four) hydrophobic interactions and a hydrogen bond acceptor interaction. Conversely, non-MIC-forming compounds seem to possess fewer hydrophobic pharmacophore features with hydrogen bond donor rather than acceptor interactions (Fig. 2). Molecules that are inactivators but apparently do not form an MIC also seem to be generally larger with more hydrophobic and hydrogen bond acceptor features that MIC and non-MIC-forming compounds.
The recursive partitioning approach confirmed these findings to some extent, as a key descriptor in the ChemTree lowest root mean square scoring tree for the first split was C(N)-O(CC). This highlights a hydrophobic interaction, whereas the second split C(CCC)-C(CN) probably represents a hydrogen bond acceptor interaction some distance from a hydrophobic feature. The key molecular descriptors used in all models as well as the summary of the training and test set correlations are described in Table 5. Similarly, in the single-tree ChemTree model (Fig. 4) selected for comparison with this multiple-tree model, the descriptor C(CCC)-C(O) represents the hydrogen bond acceptor, whereas the descriptor C(CCC)-N(CC) represents the hydrophobic interaction. When the different molecular descriptors generated in Cerius2 CSAR were used with the ChemTree descriptors, different recursive partitioning models can be produced that suggest the radius of gyration is an important descriptor as well. This is interesting because the initial Cerius2 CSAR tree model does not include any splits based on this descriptor. The radius of gyration is calculated using the following equation: 
Summary of molecular descriptors selected and training and test set correlations for MIC predictions
where N is the number of atoms and x, y, and z are the atomic coordinates relative to the center of mass (Cerius2, 2003). When this descriptor is used, all 12 molecules with a radius of gyration less than 3.75 are not MIC-forming, whereas a value between 3.75 and 3.95 suggests MIC formation for five compounds. This descriptor is, however, less discriminating for the 23 molecules with radius of gyration values above 3.95. Compared with the hydrogen bond and hydrophobic descriptors, radius of gyration is probably less important. Although the ChemTree and Cerius2 tree models classify most (10 of 12) of the literature test set molecules correctly, it should also be noted that the addition of larger numbers of trees did not improve the prediction accuracy in this case. The logistic regression model only used a hydrogen bond acceptor descriptor, and it possessed the highest prediction accuracy (91.6%) for the test set.
The linear and regression tree models for kinact, KI, and kinact/KI prediction produced poor statistics for training and testing. In addition, there was no consistency on the selected molecular descriptors among the optimal prediction models. This potentially indicates either limitations in the feature set or the complexity of modeling such parameters with these methods alone.
The presence of an amine function does not always imply that a molecule will form an MIC (Kajita et al., 2002). For example, 3-hydroxytamoxifen and 4-hydroxytamoxifen do not form MIC, whereas tamoxifen and n-desmethyltamoxifen form MIC in vitro (Zhao et al., 2002). Some macrolide antibiotics such as clarithromycin form MIC (Mayhew et al., 2000), whereas others such as miocamycin do not (Kasahara et al., 2000), which suggests the amine may be sterically hindered in the latter case such that it cannot orientate correctly in the CYP3A4 (+b5) binding site. This type of interaction may not be picked up with the 2D-QSAR tree methods and may result in the misclassifications observed.
It would also seem that requirements for CYP3A4 (+b5)-mediated MIC formation requires other molecular properties in addition to primary, secondary, and tertiary amines or methylenedioxyphenyl features. From this study, differentiating between MIC-forming and non-MIC-forming compounds is apparently complex, probably requiring numerous hydrophobic and hydrogen bond acceptor features for MIC formation. The pharmacophore for MIC-forming compounds (Fig. 2A) is similar to previously published pharmacophores for CYP3A4 inhibitors (Ekins et al., 1999a, 2003b) and autoactivators (Ekins et al., 1999b), indicating that these binding sites may overlap to some extent. Compounds that inactivate but do not form MIC with CYP3A4 may possess all of these features but be too large overall. This would suggest some important size and molecular feature constraints on binding CYP3A4 (+b5), which lopinavir, saquinavir, and nefazodone seem to exceed because they possess many more molecular features than MIC- and non-MIC-forming compounds. The non-MIC-forming compounds indicate that there may be a minimal set of molecular features for MIC formation to occur. It is interesting to speculate that these features may affect the orientation of a molecule in the CYP3A4 binding site and the observation of MIC formation. For example, the number of rotatable bonds correlates with the molecular weight (r2 = 0.68, using the data for 54 molecules from Supplemental Data Table 1), and the number of hydrogen bond acceptors also correlates with the molecular weight (r2 = 0.75, using the data for 54 molecules from Supplemental Data Table 1); these larger compounds in turn tend to be MIC-forming, at least up to a point. In contrast, the correlation between molecular weight and the number of hydrogen bond donors is much lower (r2 = 0.42, using the data for 54 molecules from Supplemental Data Table 1). This may indicate that molecules with greater conformational flexibility and size can position themselves optimally to form an MIC, whereas smaller, more rigid molecules are less likely to be able to fulfill the key pharmacophore features. However, with the increasing conformational flexibility of molecules could come the increased likelihood that fewer of the MIC formation events become observable with our current detection method, as many of the population of conformations do not result in MIC formation. A recent X-ray structure of the MIC-forming compound erythromycin in CYP3A4 did not suggest that the molecule was in a productive binding mode, as the site of metabolism was too far from the heme (Ekroos and Sjogren, 2006), yet the authors did not indicate whether the orientation could result in the MIC.
It is important to note that this study used molecular descriptors calculated on the parent molecule to predict MIC formation, when in fact a metabolite is actually binding to the heme. Our results indicate that the descriptors selected from the parent molecules are transferable in capturing the important features in the metabolites. In and of itself, this may be valuable, as our approach therefore does not require the a priori knowledge or prediction of the metabolite/s involved. However, once the metabolite(s) is known, it may be possible to use combined descriptors for the parent and metabolites as demonstrated previously for predicting N-dealkylation (Balakin et al., 2004). Future work may assess the chemical reactivity of the molecules that form MIC and attempt to determine the binding interactions with heme and elsewhere in the protein using site-directed mutagenesis. In addition, the models could be further tested with much larger diverse test sets, as those molecules selected in this study from the literature represent six core structures.
An in vitro model was proposed that can predict the clinically observed inhibition of CYP3A4 after administration of clarithromycin, fluoxetine, and diltiazem (Mayhew et al., 2000). In summary, the in vitro rates for the loss of CYP3A4 activity were used to predict the in vivo rates. Overall, the in vitro-in vivo predictions agreed with the clinical data. The clinical significance of being able to predict compounds that form MIC in humans is the constant polypharmacy that many humans subject themselves to on a daily basis. If one could predict a priori that a compound will form an MIC in humans and what the kinact value is, then it is possible to remove these types of structures from combinatorial libraries and screening collections. In this study, models were developed and tested for predicting compounds that form MIC with CYP3A4 (+b5). Ultimately, based on the results of this study, the different QSAR models generated complement the in vitro testing, representing a preliminary approach to predicting CYP3A4 (+b5) MIC formation that may be improved with the addition of further molecules to the training set, different descriptors, or other algorithms. These approaches may be equally applicable to naturally occurring compounds as well as to drugs. The modeling methods could also be applied to other P450s that are known to form MIC (Chatterjee and Franklin, 2003), such as CYP3A5 and CYP2D6, enabling a comparison with the CYP3A4 data presented herein, providing insight into the features important for MIC formation, and representing a more complete picture for pharmaceutical research.
Acknowledgments
We gratefully acknowledge Dr. Raymond Galinsky (Purdue University School of Pharmacy and Pharmaceutical Sciences, West Lafayette, IN), who provided small amounts of MDMA, MDE, and MBDB for MIC assessment, and Dr. Shikha Varma-O'Brien (Accelrys Inc.) for assistance with Cerius2 model descriptors.
Footnotes
-
This work was supported by grants from the National Institutes of Health (Grants AG13718 to S.D.H. and GM74217 to S.D.H.) and the U.S. Food and Drug Administration (Grant FD-T-001756-01 to S.D.H.).
-
doi:10.1124/dmd.106.014613.
-
ABBREVIATIONS: P450, cytochrome P450; MIC, metabolic intermediate complex; QSAR, quantitative structure activity relationship; SKF-525A, 2-diethylaminoethyl 2:2-diphenylvalerate hydrochloride; MDMA, methylenedioxymethamphetamine; MDE, methylenedioxyethylamphetamine; MBDB, 2-methylamino-1-(3,4-methylenedioxyphenyl)butane; CSAR, classification structure activity relationship; glm, generalized linearized model; DDB, dimethyl-4,4′-dimethoxy-5,6,5′,6′-dimethylenedioxybiphenyl-2,2′decarboxylate.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Received December 28, 2006.
- Accepted May 24, 2007.
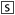
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}