


Abstract
Concentrations of unbound drug in the interstitial fluid of the brain are not rapidly measured in vivo. Therefore, measurement of total drug levels, i.e., the amount of drug per gram of brain, has been a common but unheplful practice in drug discovery programs relating to central drug effects. This study was designed to evaluate in vitro techniques for faster estimation of unbound drug concentrations. The parameter that relates the total drug level and the unbound interstitial fluid concentration is the unbound volume of distribution in the brain (Vu,brain). It was measured in vitro for 15 drugs using brain slice uptake and brain homogenate binding methods. The results were validated in vivo by comparison with Vu,brain microdialysis results. The slice method results were within a 3-fold range of the in vivo results for all but one compound, suggesting that this method could be used in combination with total drug levels to estimate unbound interstitial fluid concentrations within reasonable limits. Although successful in 10 of 15 cases, the brain homogenate binding method failed to estimate the Vu,brain of drugs that reside predominantly in the interstitial space or compounds that are accumulated intracellularly. Use of the simple methods described in this article will 1) allow quantification of active transport at the blood-brain barrier in vivo, 2) facilitate the establishment of a relationship between in vitro potency and in vivo activity for compounds acting on central nervous system targets, and 3) provide information on intracellular concentrations of unbound drug.
Determination of drug levels in the brain tissue of experimental animals is routinely undertaken in drug discovery programs for various purposes, including studies on blood-brain barrier (BBB) transport and equilibration. Drug levels are also studied in conjunction with pharmacodynamic experiments to link in vivo effects with in vitro potency, or to elucidate the mechanism and site of action. The practical approach to routinely investigating large numbers of new compounds has been to measure the amount of drug in brain (Abrain), which is given in amount per gram of brain and therefore is commonly referred to as the total brain concentration.
Because assessment of Abrain has historically been the most common method of measuring CNS exposure in drug discovery, medicinal chemistry programs have favored compounds and classes displaying high total CNS-to-plasma concentration ratios. This type of data, expressed as “log BB”, has resulted in the establishment of general criteria for physicochemical properties of compounds with potentially high or low CNS exposure (Kelder et al., 1999). Although this method has the advantage of experimental simplicity, the use of total tissue levels (Abrain) is also associated with limitations. It is generally accepted that it is the unbound drug that exerts the effect on the receptor. Large amounts of drug in the brain do not necessarily mean high concentrations available to the receptor, since the drug may bind to or dissolve in tissue components. Measurements of Abrain alone can thus be very misleading. Similarly, a high brain-to-plasma ratio based on measurements of Abrain may be reflective of extensive binding to brain tissue rather than of unrestrained transport across the BBB.
According to the free drug hypothesis, the unbound drug concentration in tissue is equal to the unbound drug concentration in plasma at equilibrium. This may not be the case for brain tissue since there are active efflux and influx processes at the BBB. Cerebrospinal fluid (CSF) drug concentrations are potentially more closely related to the concentrations of unbound drug in brain interstitial fluid (Cu,brainISF), because of the separation from blood by the blood-CSF barrier. Also, the ependymal lining of the ventricles allows diffusional and convectional exchange with the brain interstitium (Abbott, 2004; Liu et al., 2006). However, the CSF represents a different compartment and the turnover of CSF is different from that of brain ISF (Abbott, 2004). Investigations have demonstrated that drug concentrations in the CSF are not necessarily equal to those in brain ISF (de Lange and Danhof, 2002; Shen et al., 2004). The only method of directly measuring Cu,brainISF is microdialysis. Unfortunately, the utility of this method in drug discovery programs is limited by the time requirements and by specific technical difficulties with lipophilic drugs.
Wang and Welty (1996) introduced the unbound volume of distribution in the brain (Vu,brain) to relate Cu,brainISF to Abrain, where Vblood × Cblood is the amount of drug present in the blood vessels of the brain: 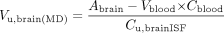
Thus, Vu,brain describes the distribution of drug inside the brain regardless of brain-to-plasma distribution. A low value for Vu,brain, close to the volume of the interstitial space, would thus describe predominantly extracellular distribution, whereas a high value would indicate that the drug enters brain cells and binds to tissue components (Gupta et al., 2006).
If the value of Vu,brain can be reliably obtained for a compound, Cu,brainISF can be calculated from available total drug levels, thus circumventing the need for microdialysis. Methods other than microdialysis that have been used for estimating Vu,brain include the brain slice uptake technique (Kakee et al., 1996) and the brain homogenate binding method (Kalvass and Maurer, 2002; Mano et al., 2002). Recent workers have used the fraction unbound in brain, fu,brain, to describe much the same property (Becker and Liu, 2006; Liu et al., 2006). The fraction unbound in brain, like the fraction unbound in plasma, is an easily understood concept. However, it shares the limitation of the homogenate method from which it originates; there is no distinction made between intra- and extracellular distribution.
The present study aims at evaluating methods for Cu,brainISF estimation to guide industrial drug discovery programs or academic research related to CNS drug exposure. The Vu,brain concept is used as a link between the total brain concentration (Abrain) and the pharmacologically active unbound brain ISF concentration (Cu,brainISF). Along with a characterization of the methods, we present the first comprehensive comparison of in vitro Vu,brain data and in vivo microdialysis measurements. We also discuss how the integrative use of these Vu,brain methods paves the way for estimation of intracellular unbound drug concentrations.
Materials and Methods
Compound Selection. The literature was searched for microdialysis reports containing both unbound and total brain drug concentrations, i.e., the data needed to calculate Vu,brain. Studies that were performed using probe calibration in vivo by retrodialysis were favored. Nearly every compound that fulfilled the criteria was included in the study. The set of 14 compounds listed in Table 1 is pharmacologically diverse, including opioids and their metabolites (morphine, codeine, oxycodone, morphine-3-glucuronide, morphine-6-glucuronide), anti-infectives (alovudine, norfloxacin), antihistamines (R- and S-cetirizine), dopamine agonists (R- and S-apomorphine), an anxiolytic (diazepam), an anticonvulsant (gabapentin), and an anesthetic agent (thiopental). The set is also chemically diverse in terms of ionization state at pH 7.4 and lipophilicity (Table 1). However, it was recognized that most of the included drugs were less lipophilic than the majority of compounds in contemporary drug discovery programs. To balance this, additional microdialysis experiments were performed with a lipophilic base (CP-122721; [cis-n-[[2-methoxy-5-(trifluoromethoxy)phenyl]methyl]-2-phenyl-3-piperidinamine]) and included in the study.
Physicochemical description of drugs included in the study
Values of log D7.4 (ACDLogD7.4) and pKa (ACDpKa) were calculated using ACD/Labs databases version 9.03 (Advanced Chemistry Development Inc., Toronto, ON, Canada). Experimentally determined log D7.4 and pKa values were obtained from the literature.
Chemicals. Alovudine (3′-fluorothymidine), R,S-apomorphine, codeine, diazepam, 14C-inulin, norfloxacin, thiopental, and bovine serum albumin (BSA) (initial fractionation by cold alcohol precipitation, lot 40K0896) were obtained from Sigma-Aldrich (St. Louis, MO). Morphine, morphine-3-glucuronide, morphine-6-glucuronide, and oxycodone were obtained from Lipomed (Arlesheim, Switzerland). Tritiated GABA (3H-GABA) was purchased from GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK). Gabapentin was purchased from Toronto Research Chemicals Inc. (Toronto, ON, Canada). Racemic cetirizine, the pure enantiomers S- and R-cetirizine, and an internal standard (ucb20028), [2-[2-(4-benzhydrylidene-piperidin-1-yl)-ethoxy]-ethoxy]-acetic acid chlorhydrate, were supplied by UCB Pharma (Braine l'Alleud, Belgium). CP-122721 was synthesized at AstraZeneca R&D (Mölndal, Sweden) with purity greater than 95%. All other chemicals were of analytical grade. All solvents were of high-performance liquid chromatography grade.
Animals. Male and female Sprague-Dawley rats (Harlan, Horst, the Netherlands) weighing 250 to 350 and 230 to 280 g were used for in vitro experiments and in vivo microdialysis, respectively. Male Dunkin Hartley guinea pigs weighing 450 to 500 g were purchased from Lidköpings Kaninfarm (Lidköping, Sweden). All animals were group-housed at 18-22°C under a 12-h light/dark cycle with free access to food and water for at least 5 days before the experiment. The study was approved by the Animal Ethics Committee of Göteborg (346-2002, 412-2005).
Intracerebral Microdialysis of CP-122721. Adsorption to the FEP (fluorinated ethylene propylene) tubing (CMA Microdialysis, Solna, Sweden) and CMA/12 probe necessitated the inclusion of 0.5% BSA in the perfusion fluid, as described previously (Gupta et al., 2006). Probe recovery and delivery of CP-122721 were found in vitro to be equal, supporting the use of the in vivo retrodialysis calibration method. In vivo experiments in rats were performed as described previously (Bostrom et al., 2006). After retrodialysis and washout with blank perfusate, the drug was administered intravenously as a bolus dose plus a 4-h constant rate infusion to obtain steady state. The size of the bolus dose and the infusion rate were adjusted according to the plasma pharmacokinetics. CP-122721 was dissolved in saline. Microdialysate samples were collected at 20-min intervals from 2 h post-bolus dose until the termination of the experiment at 4 h, when brain tissue was sampled. Vu,brain was calculated using eq. 1, assuming a blood volume of 3% of brain weight (Shockley and LaManna, 1988) and a blood/plasma distribution ratio of unity.
Brain Slice Uptake Experiments. The brain slice uptake experiments were performed as described previously (Kakee et al., 1996), with minor modifications. Drug-naive animals were sacrificed under isoflurane anesthesia, and the brain was removed and immersed in ice-cold oxygenated pH 7.4 buffer (122 mM NaCl, 25 mM NaHCO3, 10 mM glucose, 3 mM KCl; 1.4 mM CaCl2, 1.2 mM MgSO4, 0.4 mM K2HPO4, and 10 mM 4-(2-hydroxyethyl)-N-piperazineethanesulfonic acid). A 6-mm coronal section was cut with a razor and mounted with cyanoacrylate glue onto the tray of a DTK-Zero1 Microslicer (Dosaka, Kyoto, Japan). Eight 300-μm coronal slices of striatal areas were cut. The slices were preincubated at 37°C for 5 min in 10 ml of ECF buffer before the drug, dissolved in ECF buffer, was added. The concentration of drug in the buffer was chosen to match the brain unbound drug concentration observed in the corresponding in vivo microdialysis experiment. The incubations were continuously supplied with a mixture of 5% carbon dioxide in oxygen, keeping the bubbles at some distance from the slice. At prespecified times after addition of drug (15, 30, 60, 120, and 240 min), the slice (∼40 mg) was removed from the solution, dried on filter paper, and weighed. The slices were homogenized in 9 volumes (w/v) of deionized water with an ultrasonic probe (Branson Sonifier 250; Branson Ultrasonics Corporation, Danbury, CT). The ECF buffer and the slice homogenates were stored at -20°C until analysis. Vu,brain was calculated according to eq. 2, where Aslice, Cbuffer, and Vi are the amount of drug in the slice, the concentration of drug in ECF buffer, and the adherent water volume, respectively. 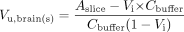
Vi was estimated using 14C-inulin in a separate experiment in which brain slices were incubated for 1, 2, and 4 min. Vi(0.091 ml·g slice-1) was obtained from a plot of Aslice/Cbuffer using zero time back-extrapolation. The possibility of significant drug binding to floating brain debris in the ECF buffer was ruled out for the investigated compounds by quantification of the buffer protein concentration (9-14 μg/ml) with a QuantiPro BCA Assay Kit (Sigma-Aldrich). The ATP content of the slice was determined using an ATP Bioluminescence Assay Kit (CLS II) from Roche Diagnostics (Mannheim, Germany).
The chemical instability of apomorphine enantiomers necessitated the use of 300 μM ascorbic acid in the ECF buffer as an antioxidant. At the end of the incubation, ascorbic acid was added to the buffer at a final concentration of 5 mM. Brain slices from apomorphine incubations were homogenized in 5 mM ascorbic acid. Samples containing apomorphine were stored at -70°C until analysis.
Brain Homogenate Binding. Drug-naive animals were sacrificed under isoflurane anesthesia, the brain was removed, and 3 volumes of a 180 mM phosphate buffer (pH 7.4) were added. The brains were homogenized on ice with an ultrasonic probe and stored at -20°C until required, when the brain homogenate was thawed and the drug added. Equilibrium dialysis of 1 ml of homogenate and buffer was performed in triplicate for 16 h at 37°C in 1 ml of Plexiglas cells mounted with a 5-kDa cutoff Diachema cellulose membrane (Dianorm GmbH, München, Germany). An aliquot of homogenate was sampled before and after coincubation to assess the compound stability. The fraction of unbound drug in diluted brain homogenate, fu,hD, i.e., the buffer-to-homogenate concentration ratio, was used to calculate Vu,brain, while also taking into account the dilution, D, associated with homogenate preparation (eq. 3) (Kalvass and Maurer, 2002). 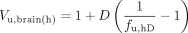
The equation describes a relationship where the lowest possible Vu,brain is 0.8 ml · g brain-1 if there is no binding and the drug occupies only the brain water space. This is an inherent limitation of the technique for Vu,brain predictions of compounds that approach the lowest possible value in vivo, which is the volume of the brain interstitial fluid (0.2 ml · g brain-1).
The chemical instability of apomorphine enantiomers necessitated the use of 50 mM ascorbic acid in the buffer for brain homogenization and equilibrium dialysis. Samples containing apomorphine were stored at -70°C until analysis.
Vu,brain Predictions Based on Log D7.4. A simple Vu,brain prediction model was established using linear regression analysis of in vivo log Vu,brain and log D7.4 of the studied compounds. The in vivo Vu,brain for each compound was predicted from its log D7.4 using the regression line of the other compounds, not including itself. Calculated ACDLogD7.4 values (Table 1) were used for all compounds except morphine glucuronides and cetirizine, for which experimentally obtained values from the literature were considered more reliable.
Analytical Procedures. The amount of drug in the various sample matrices was quantified with reversed phase liquid chromatography and multiple reaction monitoring mass spectrometry (liquid chromatography-tandem mass spectrometry) detection using a Micromass Quattro Ultima instrument (Waters, Manchester, UK) equipped with electrospray run in positive mode for all compounds except thiopental. Gradient elution over 2 min with acetonitrile and 0.2% formic acid with a flow rate of 0.6 ml/min was used. Mass transitions and detailed chromatographic conditions for each compound are given in Table 2.
Conditions for high-performance liquid chromatography-tandem mass spectrometry analysis
Sample preparation was adapted for any compound-specific requirements but followed a general procedure: buffer samples (100 μl) from the brain slice and brain homogenate experiments were added to a 96-deepwell plate (Nalge Nunc International, Rochester, NY) and diluted with a volume of 0.2% formic acid containing an appropriate amount of acetonitrile. Fifty-microliter samples of brain homogenates were protein-precipitated with 150 μl of ice-cold acetonitrile containing 0.2% formic acid. After 1 min of vortexing and 20 min of centrifugation at 4000 rpm (Rotanta/TR; Hettich, Tuttlingen, Germany) at 4°C, the supernatant was transferred to a new plate and appropriately diluted with 0.2% formic acid. Microdialysis samples containing 0.5% BSA were protein-precipitated in a similar manner. External calibration curves with at least five different concentrations were made from a serial dilution in 50% acetonitrile and 0.2% formic acid by standard addition to the blank matrices in a 1:9 volume ratio. Enantioselective analysis of cetirizine was undertaken using a previously reported method (Gupta et al., 2005). The coefficient of correlation, R2, for each calibration curve was 0.990 or higher.

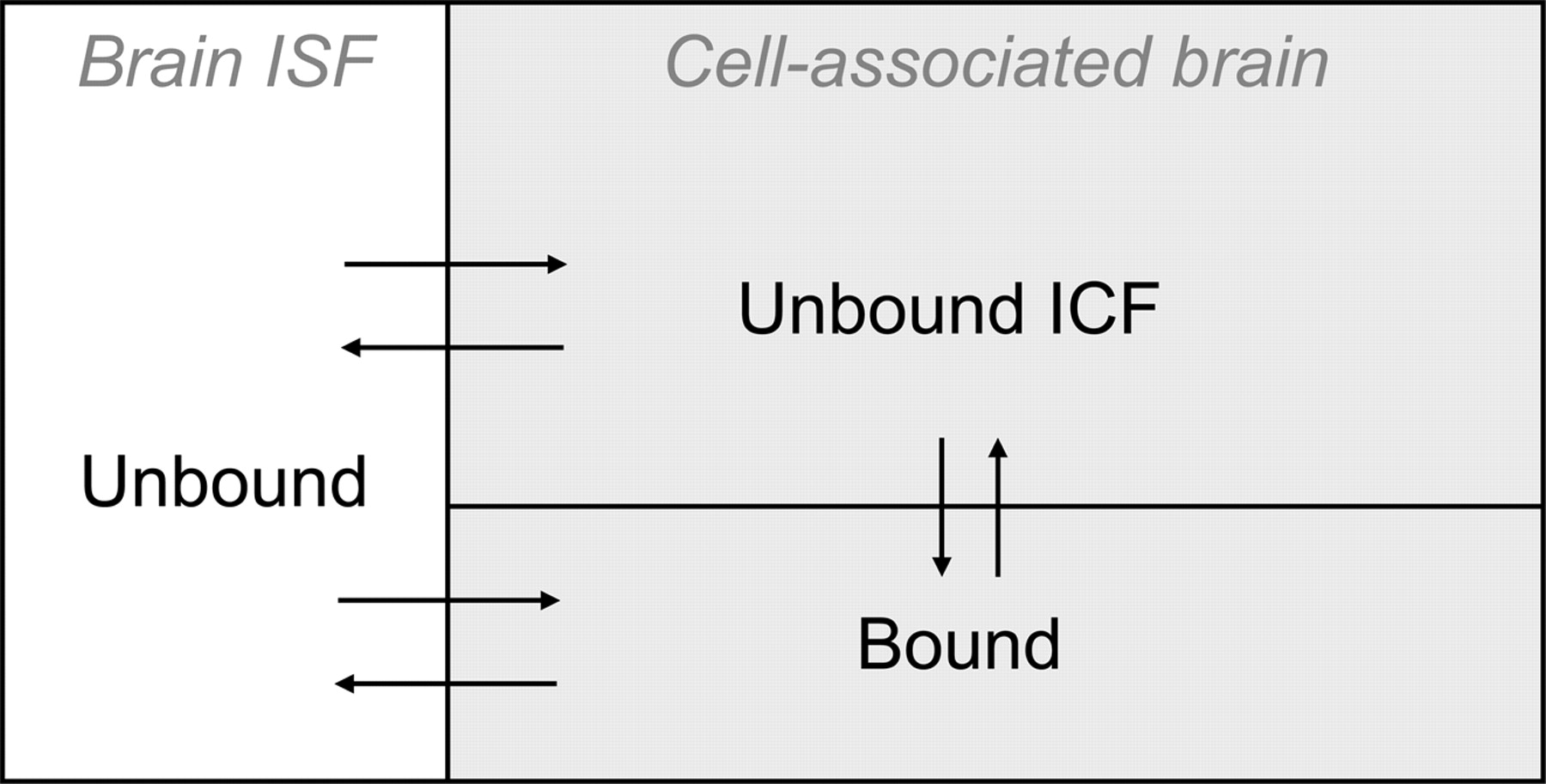
The compartment model describing intrabrain drug distribution. Drug molecules that reside in the brain interstitial fluid compartment (Brain ISF) are unbound by definition. Drug molecules that are associated with the cells are either unbound in the intracellular fluid or bound intra- or extracellularly.
Radioactive isotopes were quantified using a Wallac WinSpectral 1414 liquid scintillation counter (GE Healthcare) and an OptiPhase HiSafe 3 scintillation cocktail (Fisher Chemicals, Loughborough, UK). Brain slices were solubilized with 1 ml of Soluene-350 (PerkinElmer Life and Analytical Sciences, Boston, MA) and decolorized with 100 μl of hydrogen peroxide.
Data Presentation and Statistical Analysis. Values of Vu,brain are expressed as means ± standard deviation. Data were log-transformed for the statistical analysis, and the in vivo values of Vu,brain were taken as accurate. Agreement with in vivo Vu,brain data were assessed according to the method of Altman and Bland (Altman and Bland, 1983; Bland and Altman, 1999). For each in vitro method, the significance of the mean bias was tested with Student's t test. The agreement is expressed as the 90% confidence interval ratio (CIR) around the mean, which was calculated using the t distribution. The 90% CIR indicates the likely difference for a future single compound mean across seven slices or three dialysis cells. The 90% confidence interval is the mean difference (bias) divided by the CIR to the mean difference multiplied by the CIR. The in vivo agreement of the Log D7.4-based prediction model was also assessed using 90% CIR.
Definitions and Relationships. The basic assumption of this study was that the intrabrain distribution of a drug can be described by a distributional model in which the drug is unbound in the brain ISF. Distribution occurs by permeation into brain cells and by binding to membranes or proteins located intra- or extracellularly (Fig. 1). Specifically, we assumed that Cu,brainISF measured with a microdialysis probe was representative of the whole brain, i.e., that there would be only limited regional variations in the brain-to-plasma unbound drug concentration ratio. Morphine has been studied in rats and pigs, indicating some spatial differences (Matos et al., 1992; Tunblad et al., 2004), whereas carbamazepine showed no differences in the rat (Van Belle et al., 1995).
The Vu,brain value, in ml · g brain-1 with brain ISF as the reference fluid, reflects the distribution of the drug inside the brain, as distinct from the brain-to-plasma concentration ratio. The amount of drug present in whole brain tissue versus the unbound concentration in brain ISF depends on cell membrane permeability and the affinity of the drug for tissue components. Vu,brain is unrelated to the brain volume of distribution term, VD, which is synonymous with the brain-to-plasma concentration ratio and has blood or plasma as reference. The VD term is commonly used with respect to the intravenous injection technique (Patlak et al., 1983), the in situ brain perfusion method (Dagenais et al., 2000), and positron emission tomography (Koeppe, 2002).
Equations were derived from the definition of Vu,brain (eq. 1) and the distributional model (Fig. 1) to describe how the components influence its numerical value. Accounting for the amount of intravascular drug simplifies the expression for Vu,brain: 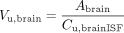
Abrain (μmol · g brain-1) comprises the amount of unbound drug in the ISF plus the amount of drug associated with the cells: 
VbrainISF and Vcell are the physiologic fractional volumes of the brain ISF and brain cells, respectively (ml · g brain-1), and Acell is the amount of drug associated with the cells (μmol · ml cell-1). The distribution volume of unbound drug in the cell, Vu,cell (ml ICF · ml cell-1) is also introduced, as this relates Acell to the intracellular concentration of unbound drug, Cu,cell (μmol · ml ICF-1): 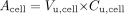
Replacing eq. 6 into eq. 5 and dividing by Cu,brainISF gives: 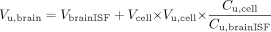
It can be seen from eq. 7 that if no drug enters the brain parenchymal cells, Cu,cell is zero and Vu,brain becomes equal to the volume of ISF, typically a value around 0.2 ml · g brain-1 (Nicholson and Sykova, 1998). This is, from a physiologic perspective, the smallest Vu,brain possible. A value close to the brain water volume (0.8 ml · g brain-1) (Reinoso et al., 1997) may indicate even distribution in the whole brain tissue. Likewise, a Vu,brain larger than 0.8 suggests that the drug has affinity for brain tissue.
It cannot be directly assumed that the concentration of unbound drug in brain ICF is equal to the concentration of unbound drug in brain ISF. Apart from the effects of active transport mechanisms, the lower intracellular pH could cause basic drugs to be trapped intracellularly, as they are not able to permeate the cell membrane in their ionized form. Assuming for acidic and basic drugs that passive diffusion of the un-ionized species dominates permeation of the membrane, the distribution of unbound drug at equilibrium is determined by the drug pKa and the pH in the extra- and intracellular compartments, pHISF and pHcell, respectively (eq. 8 and 9). 
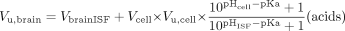
Vu,cell, which describes the affinity of the drug for physical binding inside the cells, was estimated using the brain homogenate binding experiment and taking Vcell into account in the dilution factor: 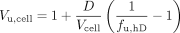
Accordingly, the cells in the homogenate are diluted not only with the added buffer but also in a small volume of brain ISF devoid of plasma proteins. It is assumed in eqs. 5 to 10 that drug binding to the outside of the cell is negligible compared with binding inside the cells. This is a reasonable approximation since for a typical human cell, the outside surface area of the cell membrane represents less than 0.5% of the total membrane surface area (Freitas, 1999). Furthermore, no single type of organelle would have a sufficiently large fractional volume to substantially influence Vu,cell in the case of a moderate concentration difference of unbound drug between the cytosol and the organelle. These approximations are not valid for molecules that are entirely confined to the extracellular domain and significantly bound. Macromolecules with specific protein interactions may possess such a combination of properties. It is, however, most unlikely for low molecular weight molecules for which binding and membrane permeation are largely determined by lipophilicity. Finally, it should be noted that Cu,cell represents the overall concentration of unbound drug in the ICF, although variations may exist among different cell types.
Supplemental Data Appendix I describes suggested procedures for experimental estimation of unbound drug concentrations in brain ISF and ICF as well as the unbound brain-to-plasma and ICF-to-ISF distribution ratios Kp,uu and Kp,uu,cell.
Results
The in vivo Vu,brain of the compounds in this study spanned 3 orders of magnitude, from 0.2 ml · g brain-1 for the morphine glucuronides, indicating exclusive distribution outside brain cells and minimal binding to proteins or membranes, to 210 ml · g brain-1 for CP-122721, revealing extensive tissue binding and distribution to the intracellular space (Table 3). Values for Vu,brain obtained with the investigated methods are also illustrated in Figs. 2 and 3.
Vu,brain values for the model compounds determined by microdialysis, by the slice uptake and homogenate binding methods, and by predictions from log D7.4
The values are given in ml · g brain−1 and are presented as means ± S.D. The microdialysis values are taken from the literature according to the references given. Unless otherwise indicated, continuous intravenous infusions of drug were administered to rats, and sampling of whole brain tissue and striatal microdialysate was undertaken at steady state.
The distribution volume of the extracellular marker 14C-inulin after 4 h of incubation was 0.36 ± 0.037 ml · g brain-1. Because the cell viability from a slice preparation is difficult to assess absolutely or quantitatively, the levels of ATP were monitored during the incubation period. There was little change in ATP concentration from the time of preparation of the brain slice to the end of the 4-h incubation (∼8 nmol · mg protein-1). The Vu,brain of 3 H-GABA, used to demonstrate functionality in terms of cellular transport, reached a maximum (17 ± 4.7 ml · g brain-1) after 60 min and then slowly declined.
The time course of drug uptake in the brain slices was studied by terminating the incubations at various prespecified times (Fig. 2). The extent of uptake of these compounds clearly varied, but they also differed in the time required to reach equilibrium. Since all the compounds had reached equilibrium at 240 min, this time point was used in the calculations. Variability of Vu,brain in slices from different rats was not greater than variability in slices from the same rat (data not shown).
Characterization of the brain homogenate binding method included time course studies using diazepam and gabapentin. Because these studies indicated that 8 h of incubation was necessary to achieve equilibrium between the dialysis cells, overnight incubation for 16 h was assumed sufficient for all compounds.
Agreement between the methods is illustrated in Fig. 3, in which in vitro Vu,brain determined by the slice or homogenate method was plotted against in vivo Vu,brain determined by microdialysis. The brain slice method predicted Vu,brain within a 3-fold range for all but 1 of the 15 compounds, whereas the brain homogenate binding technique predicted Vu,brain within a 3-fold range for 10 of the 15 compounds. There was no statistically significant bias for the in vitro methods in relation to the in vivo data (Table 4). The 90% CIRs expressing the likely (-fold) difference compared with the in vivo data were 3.0 and 6.0 for the slice and homogenate methods, respectively (Table 4).
Statistics of in vitro-in vivo agreement for Vu,brain estimations
The statistical analysis was performed for all 15 compounds. Bias and CIRs indicate the likely range of the difference between the in vitro or in silico estimations of Vu,brain and in vivo values.
There were instances of deviations from agreement between the methods. For example, the homogenate Vu,brain for morphine-3-glucuronide (1.3 ml · g brain-1) was higher than the in vivo value (0.25 ml · g brain-1). The value for this drug using the slice method (0.53 ml · g brain-1) was closer to the in vivo value. The extracellular slice distribution volume of 14C-inulin was 0.36 ml · g brain-1. Furthermore, the gabapentin Vu,brain from the homogenate experiment (1.04 ml · g brain-1) indicated that this drug was not significantly bound to brain tissue. In contrast, in vivo microdialysis and the brain slice method gave values of 5.5 and 4.0 ml · g brain-1, respectively, indicating that the total amount in brain was much higher than ISF concentrations of unbound gabapentin. Conversely, the reverse situation was seen with both cetirizine enantiomers: the brain homogenate method indicated considerable binding to brain tissue (Vu,brain 12 ml · g brain-1) that was indicated to a lesser extent in the slice method (6.5 ml · g brain-1) compared with in vivo microdialysis (2.5 ml · g brain-1).
Linear regression analysis of all data points of a plot of in vivo Vu,brain versus log D7.4 (Fig. 4) indicated a correlation between the lipophilicity of the compound and the in vivo Vu,brain value. The Vu,brain values that were predicted from log D7.4 using the regression line are presented in Table 3. All Vu,brain predictions were made without gabapentin in the model, since the Vu,brain value for gabapentin was known to reflect active transport mechanisms. The Vu,brain predictions from log D7.4 were not as accurate as those using experimental methods. Excluding the Vu,brain prediction of gabapentin gave a 90% CIR of the log D7.4 model of 6.0. Including gabapentin gave a 90% CIR of 9.0. Excluding the in vitro estimates of gabapentin Vu,brain reduced the 90% CIR of the homogenate method to 4.9 but had no effect on the CIR of the slice method (Table 4).

Time course of slice Vu,brain estimations of the 15 model compounds. Error bars represent the standard deviations for five to seven slices.
Kp,uu,cell, the ratio of intracellular to extracellular unbound drug concentrations was calculated for each compound using Vu,brain from the slice method and Vu,cell from the homogenate method (Supplemental Data Appendix I, eq. A2). Five of the six basic compounds had a Kp,uu,cell greater than 1. Neutral compounds had ratios close to or slightly below 1. The hydrophilic morphine-glucuronides had the lowest ratios, followed by the zwitterionic cetirizine enantiomers. Gabapentin had a Kp,uu,cell of 4.5 (Fig. 5).
Discussion
Since only the unbound drug is available to occupy extracellular receptors, estimation of Cu,brainISF could explain why some compounds fail to demonstrate in vivo activity despite in vitro potency and reasonable amounts of drug in brain (Abrain). Estimation of Cu,brainISF also allows quantification of the extent of BBB drug transport and investigation of the function of active transporters in vivo without confounding by nonspecific brain tissue binding. Since methods for routine measurement of unbound drug concentrations are lacking or have not yet been sufficiently evaluated in vivo, much research still relies on the easily measured Abrain. As an attractive alternative to microdialysis, which directly measures Cu,brainISF in vivo, we propose the combined use of in vivo Abrain and in vitro estimates of Vu,brain to calculate Cu,brainISF. This approach is less labor-intensive than microdialysis and likely to be more successful with lipophilic drugs.
The slice method estimated Vu,brain within a 3-fold range of in vivo results for 14 of the 15 compounds investigated; Vu,brain for morphine-6-glucuronide was slightly more than 3 times greater than the in vivo result. This indicates that the slice method has potential for accurately estimating the brain distribution of compounds with diverse properties. The brain homogenate binding method did not provide the same level of agreement with in vivo results; Vu,brain fell within the 3-fold range for only 10 of the 15 compounds. For example, Vu,brain for the morphine-glucuronides, which are known to reside in the interstitial space in vivo (Xie et al., 2000; Bouw et al., 2001), was around 1 ml · g brain-1 in the homogenate method. This discrepancy can be explained by the inherent inability of the homogenate method to differentiate between intra- and extracellular distribution due to disruption of cell membranes in the homogenate. Thus, the homogenate method measures the physical binding to brain constituents, which does not determine Vu,brain alone. This was clearly demonstrated for gabapentin, which is actively transported into brain cells by the system L α-amino acid transporter (Su et al., 1995). Because this process cannot be captured in the homogenate method, the Vu,brain value of close to 1 contrasted with the higher in vivo value of 5.5 and the slice Vu,brain value of 4 ml · g brain-1.
As suggested by the gabapentin result, the Vu,brain value for any transporter substrate at the level of brain parenchymal cells will vary according to which of these methods is used. Multidrug resistance-associated proteins have been located beyond the BBB in microglia, astrocytes, neurons, and oligodendrocytes (Dallas et al., 2006). Whenever Vu,brain is influenced by active transport mechanisms in the brain parenchyma, the slice method can be expected to provide more accurate estimates. Furthermore, intracellular accumulation of basic drugs will also occur as the ionized species is trapped by the lower pH of the intracellular fluid. Cu,cell could potentially be 2-fold higher than Cu,brainISF, depending on the pKa of the drug and assuming a difference of 0.3 pH unit between intra- and extracellular compartments (Davson and Segal, 1996). This phenomenon was observed for the basic model compounds in our study.

Relationship between in vivo Vu,brain values and in vitro slice values (A), in vitro brain homogenate values (B), and Vu,brain values (C) predicted from log D7.4. The solid line represents perfect agreement. The dashed lines represent a 3-fold over- or underestimation compared with in vivo Vu,brain values. Symbols for drugs are defined in Table 3.
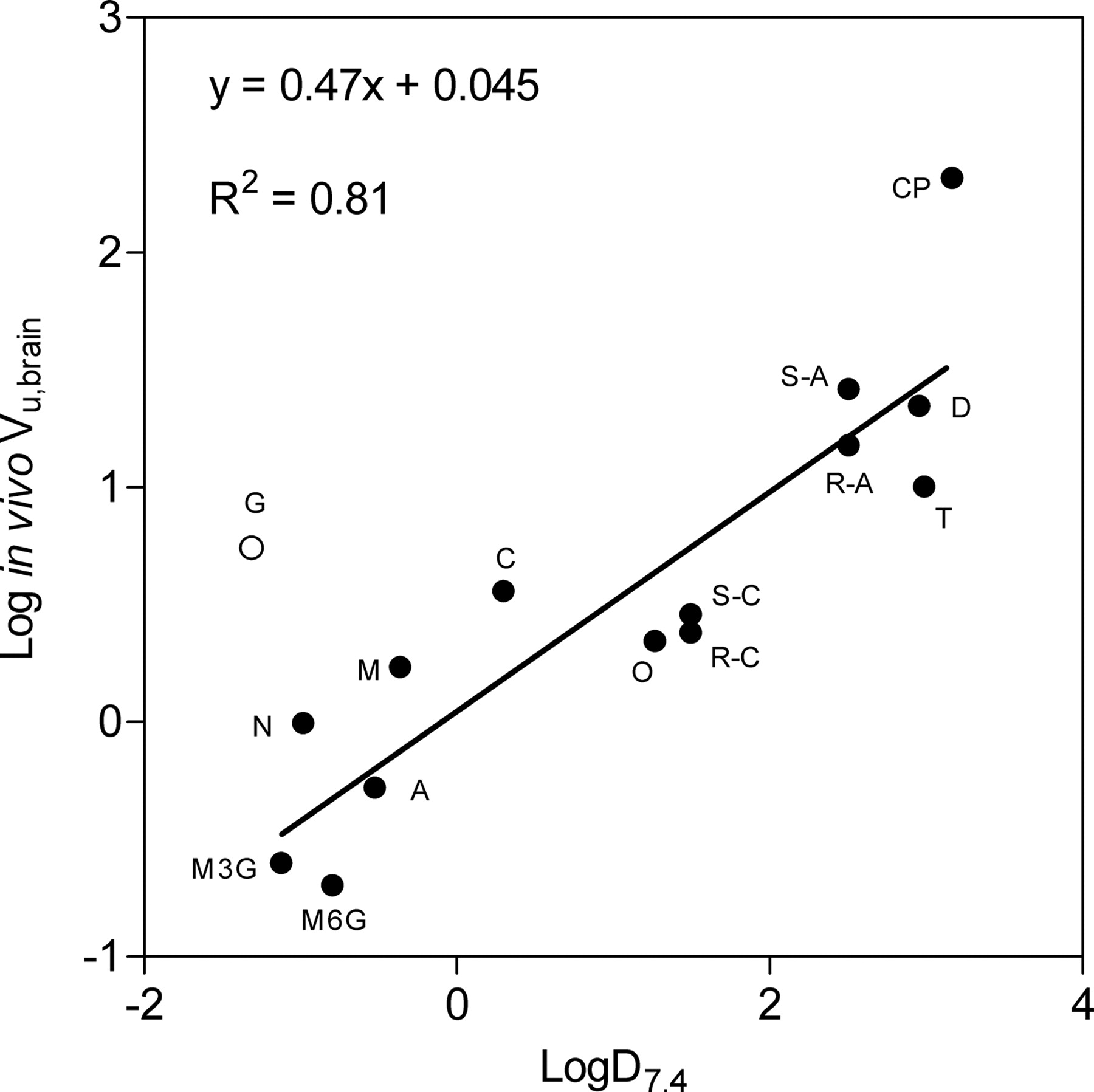
Relationship between in vivo Vu,brain values and lipophilicity estimated as log D7.4. The equation and solid line show the best fit of the linear regression analysis. Gabapentin (open circle) was excluded from the analysis based on information of active uptake.
Whereas the cells are entirely disrupted in the brain homogenate method, the cellular integrity of incubated brain slices could also be compromised near the cut surfaces, thus affecting discrimination between intra- and extracellular compartments. In fact, the measured slice distribution volume of the extracellular marker 14C-inulin (0.36 ml · g brain-1) was higher than in vivo values (Nicholson and Sykova, 1998). Slice viability, measured as the ATP levels, was stable; ATP levels were similar to those in previous reports (12-14 nmol · mg protein-1) (Lipton and Whittingham, 1984). Factors potentially affecting the in vivo characteristics of the brain slice include the choice of slicing technique, thickness of slice, oxygen supply, composition and pH of the medium, incubation time, and degree of medium convection at the surface of the slice (Lipton and Whittingham, 1984).
The attainment of equilibrium is essential for any method that measures Vu,brain, including in vivo microdialysis. In vivo experiments have established that equilibration of gabapentin concentrations between the intra- and extracellular compartments is rapid compared with BBB transport (Wang and Welty, 1996), but this finding cannot be generally extrapolated. In the in vitro systems, the equilibration time is dependent on the permeability of the brain slice or the dialysis membrane in the homogenate method. The initial distance to reach equilibrium should also be considered. At the start of the slice incubations, all the drug is in the medium, potentially far from equilibrium. Although this is not a problem if sufficient incubation time is allowed, equilibrium time could theoretically reach impractical levels as Vu,brain values increase. It was concluded that 4 h of incubation was sufficient for compounds with a Vu,brain not exceeding 200 ml · g brain-1.
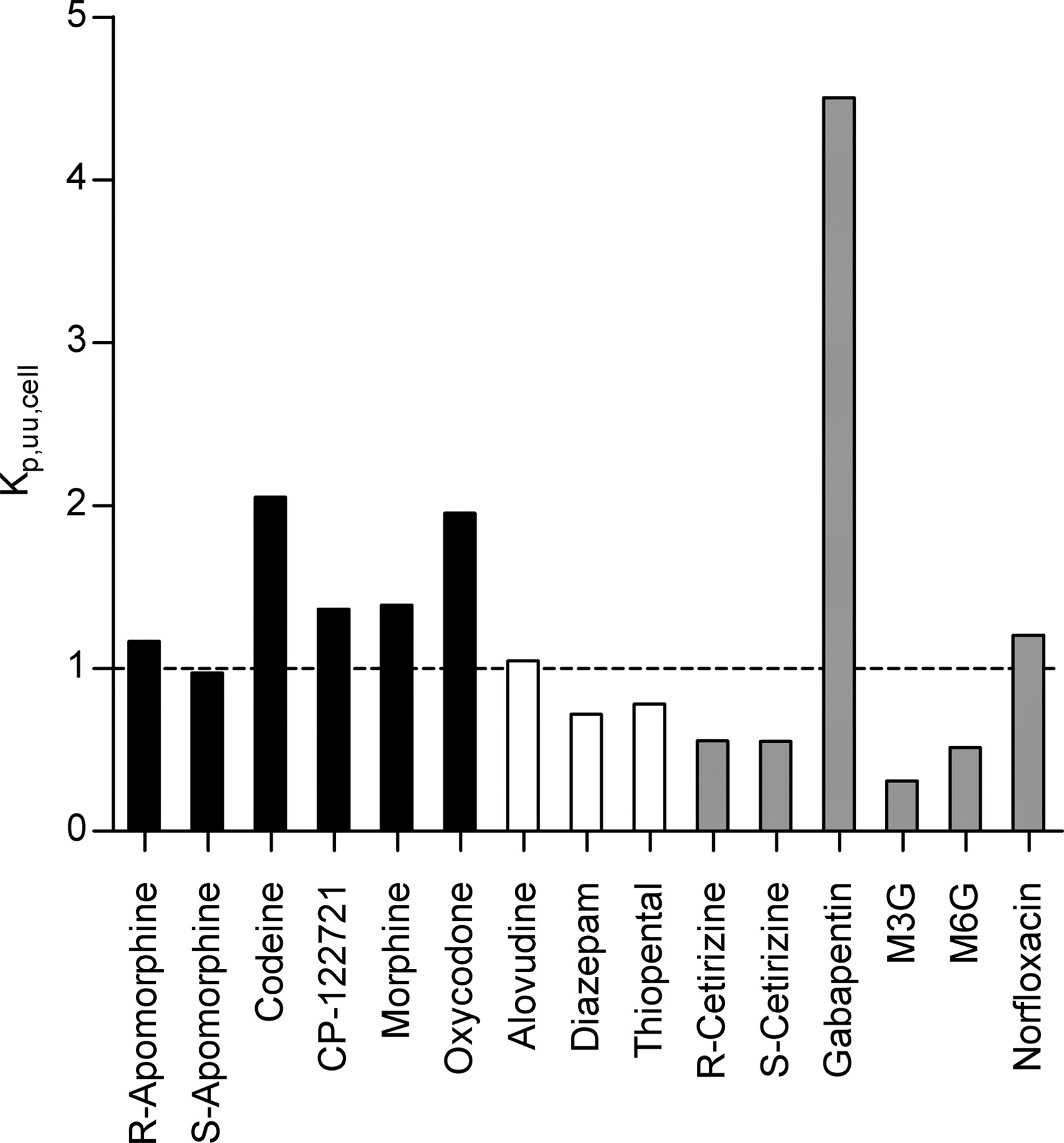
The Kp,uu,cell of the model compounds represents the ratio of concentrations of unbound drug in the intracellular and interstitial brain fluids (Cu,cell/Cu,brainISF). Basic compounds (black bars) generally had higher ratios than neutral compounds (open bars). The zwitterionic compounds, gabapentin and morphine-3-glucuronide (gray bars), had the highest and lowest Cu,cell/Cu,brainISF ratios, respectively.
The level of uncertainty in in vivo estimates of Vu,brain should also be considered. Even the most careful probe implantation causes a transient loss of BBB integrity. If leakage of drug occurs, it can produce artifactually low values of Vu,brain, since elevated concentrations around the site of the probe are no longer representative of the overall Cu,brainISF, and the Abrain is measured in whole brain. The invasiveness of microdialysis has been much discussed, but the large number of studies showing very low unbound drug brain-to-plasma ratios indicates that microdialysis measures the Cu,brainISF reasonably accurately (de Lange et al., 1994; Xie et al., 2000; Gupta et al., 2006).
In the context of the methodological issues discussed above, and considering that the microdialysis experiments were performed at different laboratories, our findings demonstrated remarkable in vitro-in vivo agreement for Vu,brain measurements. Whether the 3-fold range in agreement of this study provides enough accuracy for determination of intrabrain distribution patterns remains open for discussion. It is, however, our opinion that this would be acceptable for most situations in drug discovery programs. Our recommendation is to use the slice method when estimating the Cu,brainISF of compounds that have not been previously characterized in this respect. The slightly easier homogenate method could be used for certain series of compounds after demonstrating agreement with slice or microdialysis Vu,brain values. Using similar methods, Becker et al. (2006) concluded that the brain slice method was equal to or better than the homogenate method for predicting total brain-to-plasma ratios in P-glycoprotein-deficient mice. In silico predictions of Vu,brain based on physicochemical properties may prove more useful than indicated in our study; incorporation of additional molecular descriptors and a larger training dataset of in vivo or slice Vu,brain values would, however, be required.
Counterintuitively, brain tissue binding, as reflected by Vu,brain, does not affect exposure of the brain to unbound drug: the steady-state Cu,brainISF is specifically determined by systemic exposure to unbound drug and the unbound drug brain-to-plasma concentration ratio (Hammarlund-Udenaes et al., 1997; Liu and Chen, 2005; Syvanen et al., 2006). Thus, the interest in estimating Vu,brain is associated with the ability to convert Abrain to Cu,brainISF. Suggested applications of Vu,brain measurements are outlined in Supplemental Data Appendix I.
It is doubtful that there will ever be a direct way of measuring Cu,cell. The difficulty is in knowing whether the cell-associated amount of drug reflects cellular binding or the uptake and efflux processes that determine Cu,cell. In this article, we have provided a theoretical framework and methodology for making that discrimination; the slice method gives the amount of drug associated with the cells (Acell) which is, in turn, converted to Cu,cell with the homogenate estimate of intracellular binding (Vu,cell). This integrative use of the slice and homogenate methods allowed us to estimate the slice Cu,cell/Cu,brainISF ratio (Kp,uu,cell) for the 15 study compounds. In effect, the slice and homogenate methods may be used in parallel to provide insight into whether active transport systems are operating beyond the BBB. This was clearly observed for gabapentin in comparison with the other drugs. The framework of Cu,cell estimations could also be adapted to tissues other than brain and used in a variety of research areas.
In conclusion, there is a recognized need in drug discovery programs for methods of estimating unbound drug concentrations in the brain in an efficient, reliable manner. Estimations of Vu,brain using the slice method agreed well with in vivo microdialysis measurements. Deviation from in vivo results was greater with the homogenate method. It is therefore suggested that total brain concentrations from in vivo experiments be combined with results from brain slice studies. This will 1) allow quantification of active transport at the BBB in vivo, 2) provide a better understanding of the relationship between in vitro potency and in vivo activity for compounds acting on CNS targets, and 3) in combination with the homogenate method, provide additional information on intracellular concentrations of unbound drug.
Acknowledgments
We thank Professor Tetsuya Terasaki for kindly introducing the brain slice technique and AstraZeneca R&D Mölndal for generous sharing of expertise.
Footnotes
-
This work was supported by AstraZeneca R&D Mölndal.
-
Nomenclature list and Appendix I are available as supplemental data.
-
doi:10.1124/dmd.107.015222.
-
ABBREVIATIONS: BBB, blood-brain barrier; BSA, bovine serum albumin; CIR, confidence interval ratio; CNS, central nervous system; CSF, cerebrospinal fluid; ICF, intracellular fluid; ISF, interstitial fluid; CP-122721, [cis-n-[[2-methoxy-5-(trifluoromethoxy)phenyl]methyl]-2-phenyl-3-piperidinamine].
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Received February 19, 2007.
- Accepted June 18, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}