


Abstract
There are documented clinical drug-drug interactions between bupropion and the CYP2D6-metabolized drug desipramine resulting in marked (5-fold) increases in desipramine exposure. This finding was unexpected as CYP2D6 does not play a significant role in bupropion clearance, and bupropion and its major active metabolite, hydroxybupropion, are not strong CYP2D6 inhibitors in vitro. The aims of this study were to investigate whether bupropion's reductive metabolites, threohydrobupropion and erythrohydrobupropion, contribute to the drug interaction with desipramine. In human liver microsomes using the CYP2D6 probe substrate bufuralol, erythrohydrobupropion and threohydrobupropion were more potent inhibitors of CYP2D6 activity (Ki = 1.7 and 5.4 μM, respectively) than hydroxybupropion (Ki = 13 μM) or bupropion (Ki = 21 μM). Furthermore, neither bupropion nor its metabolites were metabolism-dependent CYP2D6 inhibitors. Using the in vitro kinetic constants and estimated liver concentrations of bupropion and its metabolites, modeling was able to predict within 2-fold the increase in desipramine exposure observed when coadministered with bupropion. This work indicates that the reductive metabolites of bupropion are potent competitive CYP2D6 inhibitors in vivo and provides a mechanistic explanation for the clinical drug-drug interaction between bupropion and desipramine.
Bupropion is a norepinephrine/dopamine reuptake inhibitor currently indicated for the treatment of major depressive disorder (Wellbutrin; GlaxoSmithKline, Research Triangle Park, NC) and smoking cessation (Zyban; GlaxoSmithKline). Clinical interactions involving bupropion and coadministered CYP2D6 substrates desipramine (Shad and Preskorn 1997; Jefferson et al., 2005), dextromethorphan (Güzey et al., 2002; Kotlyar et al., 2005), and venlafaxine (Kennedy et al., 2002) are well documented. However, CYP2D6 plays an insignificant role in bupropion clearance. In humans, bupropion is extensively metabolized to three active metabolites: hydroxybupropion, which is formed primarily via CYP2B6, and the amino-alcohol isomers threohydrobupropion and erythrohydrobupropion, which are formed via reduction of the carbonyl group (Schroeder, 1983; Golden et al., 1988; Faucette et al., 2000; Hesse et al., 2000). Presumably, the mechanism for the clinical interaction with desipramine, which is primarily eliminated via CYP2D6 (Gram, 1974; Distlerath and Guengerich, 1984; Brøsen et al., 1986), is the result of CYP2D6 inhibition by bupropion and/or its metabolite(s). However, published data show that bupropion and hydroxybupropion are weak CYP2D6 inhibitors in vitro (IC50 = 58 and 74 μM, respectively) (Hesse et al., 2000). Because unbound human plasma concentrations of the active metabolites are 2.3- to 12-fold higher than bupropion levels, it is possible that the CYP2D6 inhibition observed in the clinic is the result of more potent CYP2D6 inhibition by threohydrobupropion or erythrohydrobupropion compared with bupropion and hydroxybupropion. In addition, another hypothesis is that bupropion and/or its metabolites are metabolism-dependent CYP2D6 inhibitors. Therefore, the objective of this study was to investigate reversible and metabolism-dependent CYP2D6 inhibition by bupropion, hydroxybupropion, threohydrobupropion, and erythrohydrobupropion in vitro in human liver microsomes using the CYP2D6 probe substrate bufuralol. The calculated in vitro kinetic constants and estimated in vivo liver concentrations of bupropion and its metabolites were then used retrospectively to predict the change in desipramine exposure following coadministration with bupropion.
Materials and Methods
Materials. Bupropion hydrochloride, threohydrobupropion, erythrohydrobupropion, (S)-hydroxybupropion, paroxetine, and [13C,2H3]1-hydroxybufuralol were synthesized at GlaxoSmithKline (Research Triangle Park, NC). Quinidine, glucose 6-phosphate, glucose-6-phosphate dehydrogenase, and NADP+ were purchased from Sigma-Aldrich (St. Louis, MO). Bufuralol hydrochloride and hydroxybufuralol maleate were purchased from Ultrafine Chemicals (Manchester, UK). Pooled human liver microsomes (prepared from at least 15 donors) were obtained from Xenotech LLC (Lenexa, KS). Microsomal protein and cytochrome P450 (P450) content were provided by the manufacturer.
P450 Inhibition Assays. Inhibition of bufuralol 1′-hydroxylase (CYP2D6) activity was investigated in human liver microsomes in the presence and absence of bupropion and its metabolites. Bupropion, hydroxybupropion, threohydrobupropion, erythrohydrobupropion, and a positive control inhibitor (quinidine) were preincubated for 5 min with microsomes (0.1 mg/ml) and bufuralol (5 μM, approximately = Km) in 50 mM potassium phosphate buffer, pH 7.4, at 37°C. Final concentrations of inhibitor were 0.003 to 30 μM (bupropion), 0.02 to 200 μM (hydroxybupropion), and 0.1 to 100 μM (threohydrobupropion, erythrohydrobupropion). Reactions were initiated by the addition of an NADPH-regenerating system and terminated after 10 min for threohydrobupropion and erythrohydrobupropion and 30 min for bupropion and hydroxybupropion by the addition of an equal volume of acetonitrile. Incubations absent of cofactor were performed as controls. All the incubations were performed in duplicate. A solution containing internal standard ([13C,2H3]1-hydroxybufuralol) was added to the threohydrobupropion and erythrohydrobupropion samples and centrifuged before liquid chromatography/tandem mass spectrometry analysis. Ki determinations for threohydrobupropion and erythrohydrobupropion were conducted at concentrations corresponding to approximately 0.33 × IC50, IC50, 3 × IC50, and 10 × IC50 and at final bufuralol concentrations of 1.25, 2.5, 5, 10, and 25 μM. Ki determinations for bupropion and hydroxybupropion were conducted using final bufuralol concentrations of 1, 3, 10, and 30 μM, bupropion concentrations of 0.003, 0.03, 0.3, 3, and 30 μM, and hydroxybupropion concentrations of 0.02, 0.2, 2, 20, and 50 μM. Ki values were also determined for the CYP2D6 positive control inhibitor quinidine. To evaluate metabolism-dependent CYP2D6 inhibition, bupropion, threohydrobupropion, erythrohydrobupropion, hydroxybupropion, or positive control paroxetine were preincubated for 20 min with cofactor, buffer, and microsomes before addition of bufuralol (5 μM). The reactions were then processed as described above. Metabolism-dependent inhibition was investigated by measuring the shift in IC50 rather than determining the metabolism-dependent inhibitor parameters, kinact and KI.
Data Analysis. 1′-Hydroxybufuralol formation was quantified in the erythrohydrobupropion and threohydrobupropion incubations using a liquid chromatography/tandem mass spectrometry system that comprised a PE Sciex API 4000 or API300 (Applied Biosystems, Foster City, CA) with Ionics EP10+ mass spectrometer, CTC HTS PAL autosampler, and Shimadzu (Kyoto, Japan) pump. For the bupropion and hydroxybupropion incubations, 1′-hydroxybufuralol formation was quantified with high-performance liquid chromatography and fluorescence detection (excitation/emission, 252/302 nm) using calibration standards of the authentic reference standard 1′-hydroxybufuralol. For all the inhibitors, rates of 1′-hydroxybufuralol production at each concentration of inhibitor were expressed as a percentage of the mean uninhibited control rate. Graphical analysis of the data and IC50 calculations were performed using GraFit version 5.0.8 (Erithacus Software Ltd., Staines, UK). For Ki determinations, the data were fitted to the Michaelis-Menten equations for competitive, mixed (competitive and noncompetitive), and noncompetitive inhibition using nonlinear regression, and graphical analysis was performed according to Eadie-Hofstee plots. Metabolism-dependent inhibition of CYP2D6 activity was inferred from a decrease (>2-fold) in IC50 obtained following a 20-min NADPH preincubation relative to that obtained with the control preincubation.
Prediction of Clinical Area under the Drug Concentration-Time Curve Change. The area under the drug concentration-time curve (AUC) ratios of p.o. administered CYP2D6 substrates in the presence and absence of inhibitor(s) have been successfully predicted using the following equation, which assumes that all the inhibitors act via the same inhibition mechanism (Rostami-Hodjegan and Tucker, 2004; Ito et al., 2005; Hinton et al., 2008): 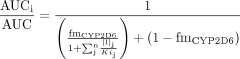 where AUCi/AUC is the area under the plasma concentration-time curve of substrate in the presence and absence of inhibitor(s) (after repeat p.o. dosing), fmCYP2D6 is the fraction of a substrate drug cleared by the inhibited pathway via a particular P450 enzyme, [Ij] is the jth inhibitor concentration, 1-fmCYP2D6 is the clearance via other P450 enzymes and/or renal clearance, and Kij is the jth in vitro inhibition constant. Substituting the clinical unbound plasma Cmax of bupropion and hydroxybupropion for [I], the calculated Ki parameters for these entities (Table 1), and desipramine fmCYP2D6 (0.9) (Brøsen et al., 1993; Ito et al., 2005) into eq. 1 (reflecting the previously published data from Hesse et al., 2000), the apparent AUCi/AUC ratio was predicted. The calculations were repeated using estimated liver concentrations of these two entities shown in Table 1. Liver concentrations were estimated by multiplying the clinical unbound plasma Cmax by the liver/plasma ratio of 5.5:9.4, determined from rat quantitative whole-body autoradiography (qWBA) studies (internal data; not published). Adding the clinical unbound plasma concentrations for threohydrobupropion and erythrohydrobupropion and the calculated Ki parameters for these entities (Table 1), the AUCi/AUC ratio was predicted using eq. 1. The predicted AUC change was recalculated using the estimated liver concentrations of all four entities shown in Table 1.
where AUCi/AUC is the area under the plasma concentration-time curve of substrate in the presence and absence of inhibitor(s) (after repeat p.o. dosing), fmCYP2D6 is the fraction of a substrate drug cleared by the inhibited pathway via a particular P450 enzyme, [Ij] is the jth inhibitor concentration, 1-fmCYP2D6 is the clearance via other P450 enzymes and/or renal clearance, and Kij is the jth in vitro inhibition constant. Substituting the clinical unbound plasma Cmax of bupropion and hydroxybupropion for [I], the calculated Ki parameters for these entities (Table 1), and desipramine fmCYP2D6 (0.9) (Brøsen et al., 1993; Ito et al., 2005) into eq. 1 (reflecting the previously published data from Hesse et al., 2000), the apparent AUCi/AUC ratio was predicted. The calculations were repeated using estimated liver concentrations of these two entities shown in Table 1. Liver concentrations were estimated by multiplying the clinical unbound plasma Cmax by the liver/plasma ratio of 5.5:9.4, determined from rat quantitative whole-body autoradiography (qWBA) studies (internal data; not published). Adding the clinical unbound plasma concentrations for threohydrobupropion and erythrohydrobupropion and the calculated Ki parameters for these entities (Table 1), the AUCi/AUC ratio was predicted using eq. 1. The predicted AUC change was recalculated using the estimated liver concentrations of all four entities shown in Table 1.
Estimated human clinical concentrations and Ki values for CYP2D6 inhibition by bupropion and metabolites
Clinical Study Design. An open-label study investigating the effects of multiple doses of bupropion on the pharmacokinetics of a single dose of desipramine was performed in 15 male subjects genotyped as CYP2D6 extensive metabolizers. Subjects received a single 50-mg p.o. dose of desipramine on day 1, bupropion (150 mg/day p.o.) on days 8 to 10 and 150 mg twice daily on days 11 to 21. On day 22, subjects received a single 150-mg p.o. dose of bupropion and a 50-mg dose of desipramine. The effect of bupropion dosing on desipramine exposure was assessed based on pharmacokinetic parameters calculated from serum concentrations of desipramine before and after treatment with bupropion.

Representative Eadie-Hofstee plots for CYP2D6 inhibition by threohydrobupropion (A) and erythrohydrobupropion (B) in human liver microsomes. Bufuralol concentrations were 1.25, 2.5, 5, 10, and 25 μM.
Results and Discussion
Clinical interactions resulting in increased exposure of CYP2D6-metabolized drugs following coadministration with bupropion have been observed despite published in vitro data showing that bupropion and a major active metabolite, hydroxybupropion, are relatively weak CYP2D6 inhibitors (IC50 = 58 and 74 μM, respectively) (Hesse et al., 2000). The purpose of this study was to evaluate whether bupropion's other active metabolites, threohydrobupropion and erythrohydrobupropion, are potent CYP2D6 inhibitors in vitro and whether bupropion and its metabolites are metabolism-dependent CYP2D6 inhibitors in vitro.
In human liver microsomes, erythrohydrobupropion and threohydrobupropion were more potent inhibitors of CYP2D6-mediated bufuralol 1′-hydroxylation (Ki = 1.7 and 5.4 μM, respectively) (Fig. 1) than hydroxybupropion (Ki = 13 μM) or bupropion (Ki = 21 μM) (Table 1). The mechanism for CYP2D6 inhibition best fit a competitive model. It has been suggested that the clinical drug-drug interaction with bupropion and CYP2D6 substrates may be the result of mechanism-based CYP2D6 inhibition by bupropion (Kotlyar et al., 2005). Our investigations showed that neither bupropion nor its metabolites were metabolism-dependent CYP2D6 inhibitors (data not shown). In addition, neither bupropion nor its metabolites were reversible or metabolism-dependent inhibitors of the major P450 enzymes investigated in pooled human liver microsomes.
The in vitro kinetic constants and unbound human plasma concentrations (Cmax) of bupropion and its metabolites were then used retrospectively to predict the increase in desipramine exposure in vivo. Accurate prediction of the magnitude of clinical drug interactions is dependent on the in vivo inhibitor concentration available to the enzyme in the liver. Because the concentration at the enzyme site cannot be measured directly, the use of surrogate inhibitor in vivo concentrations, particularly unbound hepatic inlet concentrations, has been successful in predicting clinical drug interactions (Obach et al., 2006). Hepatic inlet concentration estimations, however, require knowledge of inhibitor dose and absorption rate, and fraction of inhibitor passing through the intestine unchanged. In the case of bupropion, all three metabolites are formed in the liver, which confounds the use of hepatic inlet concentrations for the metabolites. Thus, the most meaningful in vivo concentration to use to predict the increase in desipramine exposure (AUCi/AUC) is unbound clinical plasma concentrations (Cmax) of bupropion and its metabolites, along with estimated liver/plasma ratios from preclinical rat qWBA studies. Bupropion qWBA data were not available, and thus qWBA data from rats dosed with 14C-hydroxybupropion were used as estimates. Therefore, liver/plasma ratios for bupropion and its metabolites could be different if each compound had been individually dosed in separate qWBA studies. The purpose of this study, however, was to provide via in vitro experiments a mechanistic explanation for the clinical bupropion-CYP2D6 interaction, rather than to provide an absolute quantitative prediction of a clinical interaction.
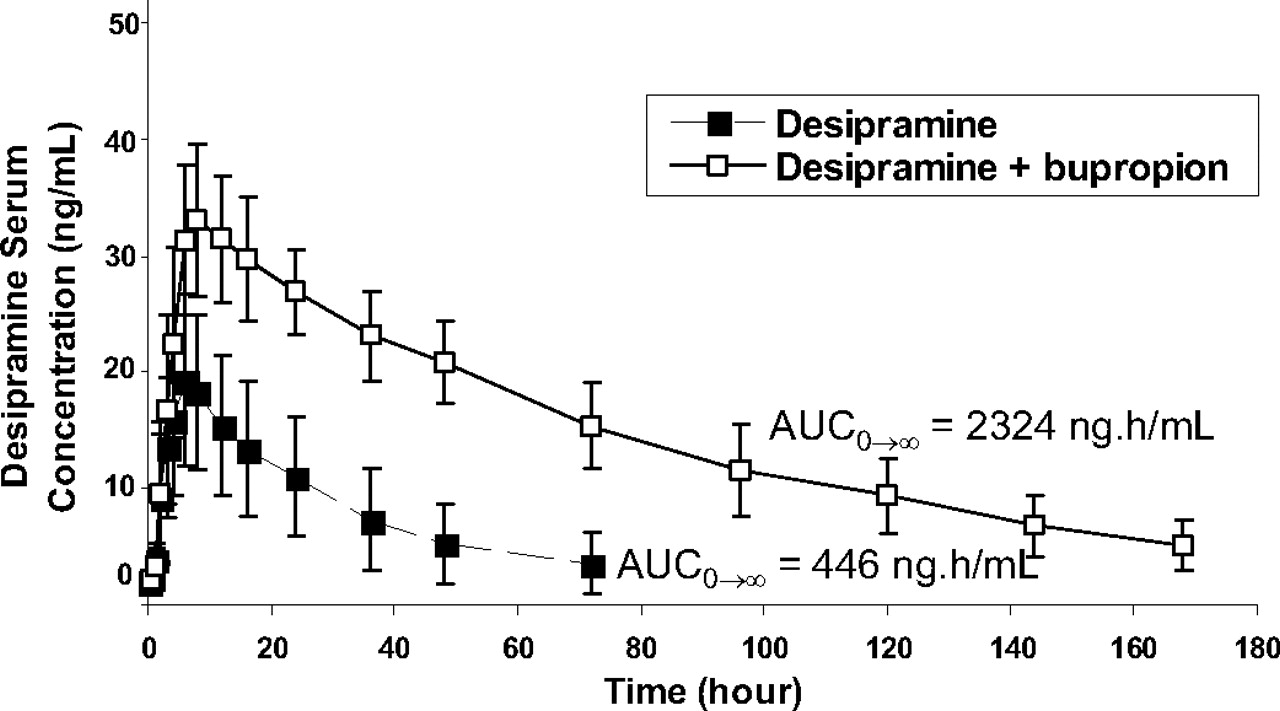
Average serum concentrations of desipramine in extensive CYP2D6 metabolizers given a single p.o. dose of 50 mg of desipramine with (□) and without (▪) 300 mg of bupropion codosing for 11 days.

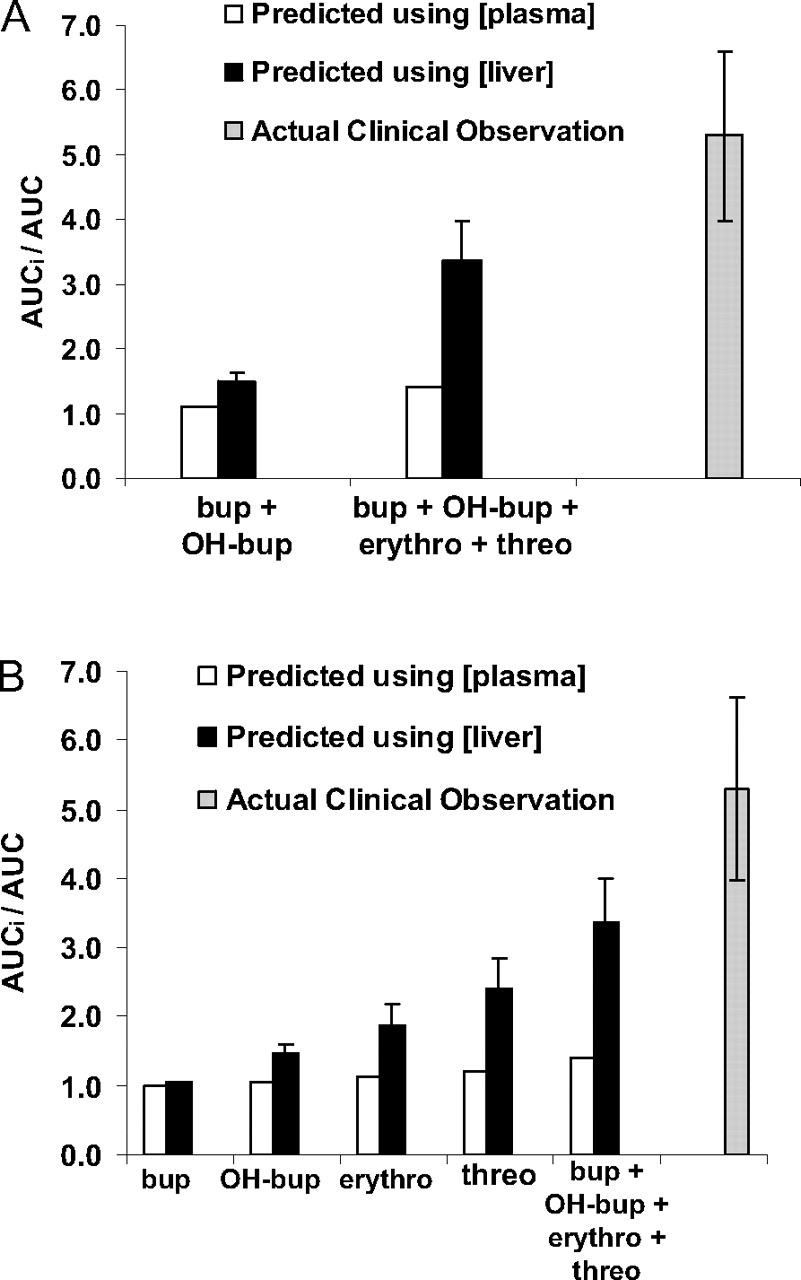
A, desipramine AUCi/AUC predicted in vitro using inhibition constants and plasma and estimated liver concentrations of bupropion and hydroxybupropion only, using bupropion, hydroxybupropion, threohydrobupropion, and erythrohydrobupropion parameters, and AUCi/AUC observed in clinic. B, the predicted desipramine AUC change when only bupropion's (or its individual metabolites) Ki, plasma, and estimated liver concentrations were used in the prediction. See Table 1 for bupropion and metabolite Ki values and plasma and liver concentrations. Bup, bupropion; OH-bup, hydroxybupropion; threo, threohydrobupropion; erythro, erythrohydrobupropion.
The predicted AUC change was compared with the AUC ratio observed in CYP2D6-extensive metabolizers administered 300 mg of bupropion for 10 days and a single 50-mg dose of desipramine on day 11. In this study, a 5.2- and 1.9-fold increase in the AUC0→∞ and Cmax of desipramine, respectively, was observed when desipramine was codosed with bupropion (product label) (Fig. 2). Using the in vitro kinetic constants and the unbound plasma concentrations (Cmax) of bupropion and hydroxybupropion only (Table 1; eq. 1), the change in desipramine exposure (AUCi/AUC) in vivo was underpredicted (AUC ratio = 1.07). Because tissue correction factors have been previously shown to improve DDI predictions (Hinton et al., 2008), the liver concentrations of bupropion and hydroxybupropion were estimated using a liver/plasma ratio of 5.5:1–9.4:1 (from rat whole-body autoradiography studies). Incorporating these estimated liver concentrations into eq. 1, the predicted AUC ratio increased marginally to 1.4 to 1.6. When erythrohydrobupropion and threohydrobupropion were included in the calculation using only unbound plasma concentrations (Cmax) for all four entities, the predicted AUC ratio remained marginal at 1.4. In contrast, adjusting the estimated liver concentrations of bupropion, hydroxybupropion, erythrohydrobupropion, and threohydrobupropion yielded a predicted AUC change increase of 2.9- to 3.8-fold, which is within 2-fold of the in vivo AUCi/AUC of desipramine following coadministration with bupropion (AUC ratio = 5.2) (Fig. 3). This predicted AUC ratio is considerably greater than if only bupropion or any of its metabolites were used for the prediction (Fig. 3B). Thus, incorporation of bupropion metabolites' in vitro kinetic constants and estimated liver concentrations in addition to those of bupropion improves the in vivo drug-interaction prediction.
In conclusion, the bupropion metabolites erythrohydrobupropion and threohydrobupropion are more potent CYP2D6 inhibitors in vitro than bupropion or hydroxybupropion. These data offer a mechanistic explanation for the observed clinical drug interaction between bupropion and drugs metabolized by CYP2D6, which cannot be explained by the in vitro inhibitory potency of bupropion alone.
Acknowledgments
We thank Joseph Polli for helpful discussions and review of this manuscript.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.020198.
-
ABBREVIATIONS: P450, cytochrome P450; AUC, area under the drug concentration-time curve; qWBA, quantitative whole-body autoradiography.
- Received January 9, 2008.
- Accepted April 15, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}