


Abstract
Medetomidine is a chiral imidazole derivate whose dextroenantiomer is pharmacologically active. The major metabolic pathway of dexmedetomidine [(+)-4-(S)-[1-(2,3-dimethylphenyl)ethyl]-1H-imidazole] in humans is N-glucuronidation at the imidazolate nitrogens. We have purified the N3- and N1-glucuronides of dexmedetomidine, termed DG1 and DG2, respectively, according to their elution order in liquid chromatography and determined their structure by 1H nuclear magnetic resonance (NMR). Studying medetomidine glucuronidation by human liver microsomes (HLMs) and recombinant UDP glucuronosyltransferase (UGT) 1A4 indicated that another human UGT plays a major role in these activities. We now demonstrate that this enzyme is UGT2B10. HLMs catalyzed DG1 and DG2 formation, at a ratio of 3:1, with two-enzyme kinetics that contain both a high-affinity component, Km1 values of 6.6 and 8.7 μM, and a low-affinity component, Km2 values > 1 mM. The DG1/DG2 ratio in the case of UGT2B10 was lower, 1.4:1, whereas the substrate affinity for both reactions was high, Km values of 11 and 16 μM. UGT1A4 produced mainly DG1 (DG1/DG2 ratio of 6.6:1) at low substrate affinities, Km values above 0.6 mM, but superior expression-normalized Vmax values. Levomedetomidine [(-)-4-(R)-[1-(2,3-dimethylphenyl)ethyl]-1H-imidazole] glucuronidation by HLMs yielded mostly the N3-glucuronide (LG1, structure determined by NMR), with monophasic kinetics and a Km value of 14 μM. The activity of UGT1A4 toward levomedetomide was low and generated both LG1 and LG2, whereas UGT2B10 exhibited relatively high activity and sharp regioselectivity, yielding only LG1, with a Km value of 7.4 μM. The results highlight the contribution of UGT2B10 to medetomidine glucuronidation and its potential importance for other N-glucuronidation reactions within the human liver.
Medetomidine, an imidazole derivative, is a racemic mixture of two optical enantiomers, dex- and levomedetomidine (Fig. 1). Dexmedetomidine, the pharmacologically active enantiomer of medetomidine, is an agonist of α2-adrenoceptors and is used i.v. as an analgetic, anxiolytic sedative for postsurgical patients (Bhana et al., 2000). Levomedetomidine is practically devoid of pharmacological activity (MacDonald et al., 1991; Savola and Virtanen, 1991). In veterinary use for dogs and cats, medetomidine was originally used as a racemic mixture (Ansah et al., 2000; Kuusela et al., 2000), but it was recently launched as a formulation containing only the pure dextroenantiomer.
Dexmedetomidine undergoes extensive metabolism in the liver, particularly glucuronidation at N3 and N1 of the imidazole ring (Fig. 1; Bhana et al., 2000; Ji et al., 2004). Interestingly, kinetic analyses in human liver microsomes (HLMs) showed that the glucuronidation of the levoenantiomer was considerably more efficient and at much higher affinity than for the dextroform, apparent Km values of 16 versus 370 μM, respectively (Kaivosaari et al., 2002). Dog liver microsomes also formed N-glucuronides from both medetomidine enantiomers but at a lower efficiency than HLMs. In rat liver microsomes, on the other hand, medetomidine N-glucuronidation was barely detectable.
Glucuronidation is catalyzed by UDP-glucuronosyltransferases (UGTs), membrane-bound enzymes of the endoplasmic reticulum that catalyze the glucuronic acid transfer from UDP-glucuronic acid to a large variety of aglycones, both endogenous compounds and xenobiotics (Radominska-Pandya et al., 1999; Tukey and Strassburg, 2000; Wells et al., 2004). In humans, there are some 19 UGTs that are divided into two families, UGT1 (or UGT1A) and UGT2, based on sequence homology and gene structure (Mackenzie et al., 2005). The liver is the major site of glucuronidation in the human body, and most UGTs, but not all, are expressed in liver cells (Tukey and Strassburg, 2000; Mackenzie et al., 2003). The UGTs mostly catalyze the conjugation of a hydroxyl group in the aglycone substrate with glucuronic acid, forming O-glucuronides. However, some UGTs can conjugate different amines, forming N-glucuronides (Green and Tephly, 1998; Hawes, 1998; Zenser et al., 2002; Borlak et al., 2006). Among the human UGTs, UGT1A4 was largely considered as the enzyme “specializing” in N-glucuronidation because it is capable of conjugating different types of amines: primary aromatic amines, secondary and tertiary aliphatic amines, and secondary and tertiary aromatic N-heterocycles (Green and Tephly, 1998; Kaivosaari et al., 2002; Zenser et al., 2002; Kuehl and Murphy, 2003; Rowland et al., 2006). Several other human UGTs, including 1A1, 1A3, 1A7, 1A9, and 2B7, were documented to catalyze different N-glucuronidation reactions (Green and Tephly, 1998; Kaivosaari et al., 2002; Zenser et al., 2002; Staines et al., 2004; Girard et al., 2005; Kaji and Kume, 2005; Borlak et al., 2006; Rowland et al., 2006; Omura et al., 2007). Nevertheless, for all the latter enzymes, N-glucuronidation seems to be a minor reaction, whereas for UGT1A4, it is probably the major type of activity. Until recently, UGT1A4 was assumed to be the only enzyme that specializes in N-glucuronidation. This concept changed last year with the simultaneous discoveries, both in our laboratory and by Chen and coworkers, of the high N-glucuronidation activities of UGT2B10 (Kaivosaari et al., 2007; Chen et al., 2007).

Chemical structures of medetomidine enantiomers (*, chiral center) and the regioisomeric N-glucuronides.
Our previous investigation of the glucuronidation activity of recombinant human UGTs 1A3, 1A4, 1A6, and 1A9 toward medetomidine enantiomers revealed that none of these isoforms, not even UGT1A4, which exhibited some activity, account for the high efficiency of medetomidine glucuronidation by HLMs (Kaivosaari et al., 2002). We have now resumed the search for the missing UGT, taking advantage of the availability of a nearly complete set of the human UGTs as recombinant proteins in our laboratory (Kurkela et al., 2007). The renewed effort to understand medetomidine metabolism led to the discovery that UGT2B10 catalyzes the glucuronidation of dexmedetomidine and, particularly, levomedetomidine, exhibiting largely similar affinities and regioselectivity to HLMs. We have also purified the glucuronides and analyzed their structure using 1H NMR. In addition to solving the question about the enzymes involved in medetomidine glucuronidation, the results highlight the importance of UGT2B10 in N-glucuronidation activity within the human liver, alongside UGT1A4.
Materials and Methods
Materials. UDPGA (as triammonium salt) was obtained from Fluka (Buchs, Switzerland), and 14C-UDPGA [glucuronyl-14C(U)] was from PerkinElmer Life and Analytical Sciences (Waltham, MA). Alamethicin (from Trichoderma viride), d-saccharic acid 1,4-lactone, and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO). Dexmedetomidine hydrochloride and levomedetomidine were provided by Orion Pharma (Espoo, Finland).
Pooled HLMs (lot no. 26738, 18 donors), pooled human intestine microsomes (lot no. 36869, five donors), pooled male Wistar Han rat liver microsomes (lot no. 62526), pooled male Cynomolgus monkey liver microsomes (lot no. 1), and pooled male Gottingen minipig liver microsomes (lot no. 26214) were from BD Biosciences (San Jose, CA). Pooled male dog liver microsomes, pooled male mouse liver microsomes (CD-1), guinea pig liver microsomes, and male rabbit liver microsomes were prepared at Orion Pharma. The livers were first homogenized in cold 67 mM Na/K-phosphate buffer, pH 7.4, to obtain a 25% (w/v) homogenate. The liver homogenate was then centrifuged at 1000g for 20 min followed by centrifugation of the first supernatant at 9000g for 20 min. The supernatant of the latter centrifugation was centrifuged at 105,000g for 60 min, after which the microsomal pellet was resuspended by homogenization in the above buffer solution and stored frozen at -70°C until use. Protein concentrations were determined by the method of Bradford (1976), with commercial protein standards.
Recombinant human UGTs were produced in baculovirus-infected insect cells as previously described (Kurkela et al., 2003, 2007) and were used in this study mainly as lysed cell homogenates. The relative expression levels of the different UGT enzymes were determined, side by side, by immunodetection using a tetra-His monoclonal antibody (QIAGEN GmbH, Hilden, Germany) as detailed elsewhere (Kurkela et al., 2007).
Activity Assays. The incubation mixtures for the initial screening assays contained 100 mM phosphate buffer, pH 7.4, 5 mM MgCl2, 0.1 to 0.5 mg protein/ml, 5 mM UDPGA, and 0.5 mM substrate, dex- or levomedetomidine, in a total volume of 100 μl. Incubates of levomedetomidine also contained 1% methanol. The consumption of the substrate was <10% in all the assays. Alamethicin, 37.5 to 100 μg/mg protein, was added to liver and intestine microsomes incubations but not to incubations with recombinant UGTs, except when its contribution to activity was assessed. Saccharolactone, which is often included in glucuronidation reactions, was omitted because in preliminary experiments, it slightly decreased rather than increased the glucuronidation rates of dex- and levomedetomidine by recombinant UGT1A4 and HLMs. The reactions were initiated by the addition of UDPGA, incubated at 37°C for 60 or 120 min, and terminated by the addition of 60 μl of a 5:1 mixture of methanol and 4 M perchloric acid and transferring to an ice-water bath. After subsequent centrifugation, the supernatants were subjected to LC-UV analyses.
Effect of Alamethicin on Dexmedetomidine Glucuronidation. The incubations were performed essentially as described under Activity Assays using 0.5 mM dexmedetomidine, 0.3 mg protein/ml, either HLMs or recombinant UGT1A4, and 12.5, 25, 37.5, 50, or 62.5 μg alamethicin/mg protein. Dexmedetomidine glucuronidation rates were compared with the rates measured in the absence of alamethicin.
Glucuronide Analyses by LC-UV and Radioactivity Detection. The samples were analyzed using an Agilent Series 1100 liquid chromatograph (Agilent Technologies, Palo Alto, CA) with a thermostated column compartment at 30°C. A Waters Symmetry 150- × 4.6-mm C18 column (Waters, Milford, MA) and a guard column of the same material were used. The isocratic mobile phase consisted of methanol (38%) and 50 mM sodium phosphate buffer, pH 3.0 (62%), at a flow rate of 1 ml/min. The glucuronides were monitored by an Agilent series 1100 UV detector at 215 nm. The retention times of the glucuronides were as follows: dexmedetomidine glucuronides, DG1 and DG2, 7.0 and 9.2 min, respectively; and levomedetomidine glucuronides, LG1 and LG2, 6.5 and 9.1 min, respectively. The glucuronide conjugates were quantified by radioactivity detection as described earlier (Kaivosaari et al., 2001). Briefly, the substrate was incubated as described under Activity Assays, with HLMs as the enzyme source, but in the presence of both 14C-UDPGA (3.7–7.4 kBq) and nonradiolabeled UDPGA (0.02–0.5 mM). The formed glucuronides were monitored using both the UV detector and a Packard 500TR series radioactivity detector (PerkinElmer Life and Analytical Sciences) with Flo-One version 3.61 software. A 0.5-ml flow cell was used, and the flow rate of the scintillation liquid (Ultima-Flo; PerkinElmer Life and Analytical Sciences) was 3 ml/min. A calibration curve was constructed for the UV detector [glucuronide (picomoles) versus UV peak area (mAU per second)], and the UV detector was then used to quantify the glucuronides formed in the actual samples incubated only with nonradiolabeled UDPGA.
Characterization of Glucuronides by LC-MS. Dex- and levomedetomidine were incubated as described under Activity Assays, with either HLMs or recombinant UGT1A4. Control samples were incubated similarly, but in the absence of UDPGA. The samples were first subjected to LC-UV analysis, during which the glucuronide fractions DG1, DG2, LG1, and LG2 were manually collected. The separated glucuronide fractions were concentrated by solid phase extraction and injected into LC-MS using a Waters Symmetry C18 20- × 3.0-mm column. The isocratic mobile phase consisted of methanol (20%) and 0.1% formic acid in water (80%) at a flow rate of 0.4 ml/min. The glucuronides were monitored by an Agilent 1100 series single quadrupole mass spectrometer (model G-1946A) with atmospheric pressure electrospray ionization using the following settings: N2 as drying gas (13.0 l/min, 350°C); nebulizer pressure, 50 pounds per square inch gauge; Vcap (positive/negative), ±4 kV; and fragmentor voltage (positive/negative), ±150 V. The glucuronides were monitored as [M+H]+ ions at m/z 377 or as [M-H]- ions at m/z 375.
Glucuronide Biosynthesis and Purification. The incubation mixtures contained 100 mM phosphate buffer, pH 7.4, 5 mM MgCl2, 0.4 mg HLM protein/ml, 2 mM UDPGA, 0.1 mg/ml Triton X-100 (0.25 mg/mg microsomal protein), and either 1 mM dexmedetomidine or 0.5 mM levomedetomidine (in 1% methanol) in a total volume of 50 ml. To avoid substrate inhibition, levomedetomide was added in several portions during the first 6 hours of the incubation. Altogether, 11.8 mg of dexmedetomidine and 5.0 mg levomedetomidine were added. The biosyntheses were carried out in a shaking dry bath at 37°C for 30 h and terminated by protein precipitation with 4 M perchloric acid (5 ml) and tubes transfer to an ice-water bath, followed by protein removal from the mixture by centrifugation. The pH of the supernatants was adjusted to between 1 and 2 with 1 M hydrochloric acid, and the glucuronides were purified by SPE (Oasis MCX, 3 ml, 60 mg; Waters). The cartridge was first conditioned with 1 ml of methanol and 1 ml of 1 M hydrochloric acid, the sample was then loaded, and the glucuronides were eluted with a 15:85 mixture of methanol and 5% ammonium hydroxide in water. DG1 and DG2 were subsequently separated using an automated fraction collector (Agilent Analyt-FC) coupled to the LC system, as described under LC-UV and Radioactivity Detection. In the case of levomedetomidine, 95% of the formed glucuronide consisted of LG1, and further purification by LC-UV was not required. After SPE and fractionation, the methanol was evaporated from the samples under nitrogen stream, and the glucuronides were further purified by SPE (Oasis HLB, 3 ml, 60 mg; Waters). The cartridge was preconditioned with 1 ml of methanol and 1 ml of water, the sample was loaded, and the cartridge was washed with 3 ml of water. The glucuronides were eluted with methanol that was subsequently evaporated. The precipitates were dissolved in 0.5 ml of water, and the glucuronides were thereafter lyophilized.
NMR Spectroscopy. NMR spectra of dexmedetomidine, DG1, DG2, and LG1 samples dissolved in 280 μl of D2O in Shigemi NMR tubes (Shigemi, Inc., Allison Park, PA) were recorded. In one-dimensional 1H spectra, presaturation was used for water suppression. For two-dimensional NOESY spectra, matrices of 2048 × 256 points were collected. A cosine-bell weighting function was employed in both dimensions. A mixing time of 700 ms was used. All spectra were measured at 23°C except for DG1, which was measured at 35°C due to overlap of HDO and GH1 signals at 23°C. The 1H chemical shifts were referenced to HDO, 4.80 and 4.65 ppm, for measurements at 23 and 35°C, respectively. All the experiments were carried out on a Varian Unity INOVA 600 MHz spectrometer, equipped with a 15N/13C/1H triple-resonance cold probe and an actively shielded z-axis gradient system (Varian, Inc., Palo Alto, CA).
Kinetic Analyses. The incubations for kinetic analyses of HLMs, UGT1A4, and UGT2B10 were performed essentially as described under Activity Assays, typically using 10 concentrations (2.5–2000 μM) of either dex- or levomedetomidine. To increase substrate solubility, incubates of levomedetomidine contained up to 4% methanol. This methanol concentration inhibited the reactions by less than 10% (results not shown). Under the assay conditions, metabolite formation was linear with respect to both protein concentration (1–2 mg protein/ml UGT1A4 or UGT2B10 or 0.2 mg protein/ml HLMs) and time (60–180 min). The kinetic constants were estimated by nonlinear regression analysis using SigmaPlot Enzyme Kinetics Module version 1.1 (SPSS Inc., Chicago, IL). The initial reaction velocity data were fitted to the following equations (Cornish-Bowden, 1995; Houston and Kenworthy, 2000):
The Michaelis-Menten equation: 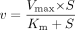 where v is the reaction velocity, Vmax is the maximum velocity, Km is the Michaelis constant, and S is the substrate concentration.
where v is the reaction velocity, Vmax is the maximum velocity, Km is the Michaelis constant, and S is the substrate concentration.
The Hill equation describing sigmoidal kinetics:  where n is the Hill coefficient.
where n is the Hill coefficient.
The substrate inhibition equation: 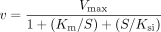 where Ksi is the constant describing the substrate inhibition interaction.
where Ksi is the constant describing the substrate inhibition interaction.
The two-enzyme equation:  where the subscripts 1 and 2 denote the high- and low-affinity enzymes of the reaction, respectively. Uniform or 1/v weighting was used. Goodness of fit to the equations was evaluated by visual inspection of the Michaelis-Menten, Eadie-Hofstee, and residual plots, the S.E. values of the parameters, and R2 values.
where the subscripts 1 and 2 denote the high- and low-affinity enzymes of the reaction, respectively. Uniform or 1/v weighting was used. Goodness of fit to the equations was evaluated by visual inspection of the Michaelis-Menten, Eadie-Hofstee, and residual plots, the S.E. values of the parameters, and R2 values.
Results
Medetomidine Glucuronidation by Human and Animal Microsomes. We have measured the glucuronidation of medetomidine enantiomers in HLMs and several animal species. HLMs exhibited a much higher glucuronidation rate than any other liver microsomes sample for all the four metabolites, DG1, DG2, LG1, and LG2 (Fig. 2). Besides HLMs, only dog liver microsomes glucuronidated medetomidine at considerable rates.
HLMs displayed high preference for the G1 regioisomer formation from both dex- (80% DG1) and levomedetomidine (95% LG1) (Fig. 2). Essentially similar high preference for G1 formation was seen in dog liver microsomes, whereas the regioselectivity of Cynomolgus monkey liver microsomes was different; the formation of the G1 metabolite of either medetomidine enantiomer was very low, and relatively much more of each G2 glucuronide was formed.
The glucuronidation activity of human intestine microsomes toward medetomidine enantiomers was also assayed and turned out to be very low (Fig. 2). The sharp difference in activity between human liver to intestine microsomes is noteworthy, particularly because UGT1A4 that until now was assumed to be the major, if not the sole, human enzyme catalyzing the N-glucuronidation of imidazole-containing drugs (Kaivosaari et al., 2002), is expressed to some extent in the human intestine (Kaivosaari et al., 2007).
Recombinant Human UGT Activities. We have tested 16 different recombinant human UGTs for their ability to glucuronidate dex- and levomedetomidine. The UGTs were originally used as isolated membrane preparations, but after finding out that the activity of UGT2B10 toward nicotine sharply declined upon the isolation of microsomal membranes from baculovirus-infected insect cells (Kaivosaari et al., 2007), we have switched to using cell homogenates rather than isolated membranes. Prior to that, we have observed that the levomedetomidine glucuronidation activity of UGT2B10 also decreases upon the membrane isolation procedure (data not shown).
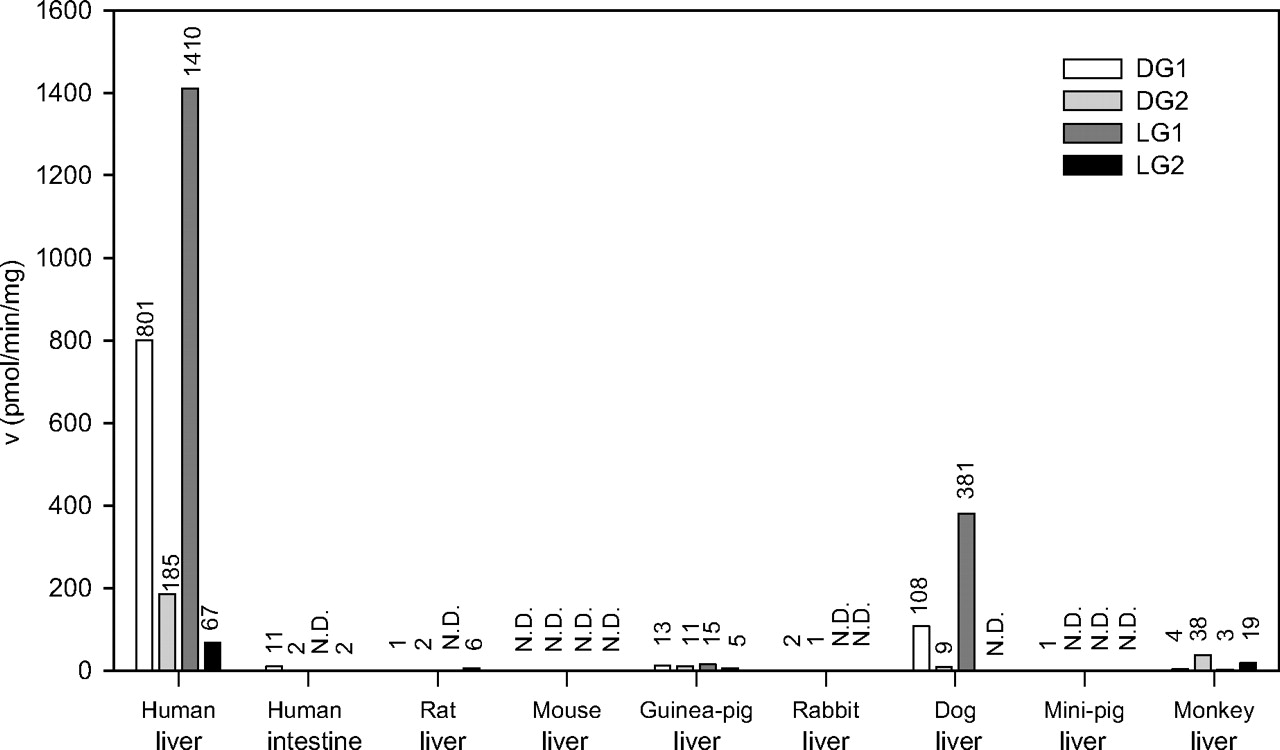
Medetomidine glucuronidation activity of human liver and intestine microsomes and liver microsomes of different animals. The activities were measured at 0.5 mM substrate concentration, 0.2 to 0.5 mg/ml protein, 2-h incubation, in duplicate or triplicate (<15% difference), and mean data are presented. N.D., activity below quantification limit (0.2 pmol/min/mg protein).
The relative expression level of each recombinant UGT in the cell homogenate was measured by immunodetection to calculate the normalized activities and obtain a better comparison of their turnover rates. Four human UGTs, namely 1A3, 1A4, 2B7, and 2B10, catalyzed, to different degree, the glucuronidation of medetomidine enantiomers when present at 0.5 mM substrate concentration (Table 1). Under these conditions, UGT1A4 exhibited high normalized activity toward dexmedetomidine, with a preference for DG1 formation (87%) (Table 1). UGT2B10 glucuronidated dexmedetomidine rather poorly and without a significant regioselectivity. On the other hand, when levomedetomidine glucuronidation was assayed, the normalized activity of UGT2B10 was higher than that of UGT1A4 (Table 1). In addition, levomedetomidine glucuronidation by UGT2B10 was highly regioselective, producing only the LG1 metabolite. Conversely, the regioselectivity of UGT1A4 in levomedetomidine glucuronidation was significantly lower than in the case of dexmedetomidine. The normalized activities of UGT1A3 and UGT2B7 toward medetomidine enantiomers were very low, and no G1 metabolites were detected in the case of UGT2B7 (Table 1).
Formation of the four different medetomidine glucuronides by recombinant UGTs Most human UGTs were assayed, and those exhibiting activity above the quantification limit (0.2 pmol/min/mg protein) are shown in the table. The other tested UGTs were 1A1, 1A5, 1A6–1A10, 2B4, 2B11, 2B15, 2B17, and 2B28. The substrate concentration in the assays, either dex- or levomedetomidine, was 0.5 mM, the protein concentration was 2.4 to 3.5 mg/ml, and the reaction time was 120 min. Activities were measured in duplicate (<15% difference), and mean data are presented.
Effect of Alamethicin on Dexmedetomidine Glucuronidation. Treatment of HLMs with the pore-forming peptide alamethicin activated DG1 and DG2 formations by about 4-fold (Fig. 3A), and maximal activation was observed when the alamethicin concentration was ≥25 μg/mg protein. On the contrary, no activation by alamethicin was observed with the recombinant UGT1A4 (Fig. 3B). Likewise, alamethicin activated HLMs, but not UGT1A4, in the formation of LG1 and LG2 (data not shown). In light of these observations, alamethicin was only used in assays on the glucuronidation activity of HLMs, but not with recombinant UGTs.
Glucuronide Characterization by LC-MS. To verify that both G1 and G2 are the expected monoglucuronide metabolites, dex- and levomedetomidine were subjected to glucuronidation catalyzed by HLMs and recombinant UGT1A4, the enzyme previously shown to catalyze such activity (Kaivosaari et al., 2002), and the regioisomeric glucuronide peaks were separated and collected by LC-UV and analyzed by MS. The electrospray ionization-MS spectra of all the four metabolites, DG1, DG2, LG1, and LG2, regardless of whether they were formed by HLMs or UGT1A4, showed an ion m/z 377 at positive ion mode, corresponding to protonated glucuronide. In negative ion mode, ion m/z 375 was observed, corresponding to deprotonated monoglucuronide. These ions were not detected in control samples incubated similarly, except for the absence of UDPGA. All the four glucuronides, DG1, DG2, LG1, and LG2, were also detected by radioactivity detector when incubated with HLMs and radiolabeled 14C-UDPGA (data not shown).

The effect of alamethicin on dexmedetomidine N-glucuronidation by HLMs (A) and recombinant human UGT1A4 (B) at 0.5 mM substrate concentration.
Biosynthesis and NMR Spectra. HLMs treated with Triton were used for the biosynthesis of dex- and levomedetomidine glucuronides. During the incubation, 10% of dexmedetomidine and 37% of levomedetomidine were converted into glucuronide conjugates. DG1 and DG2 were separated by LC connected to fraction collector. After subsequent purification and lyophilization, the samples for NMR analyses contained 0.25 mg of DG1 and 0.13 mg of DG2. In the case of levomedetomidine, LG1 comprised 95% of the end product, whereas the amount of LG2 was insufficient for NMR analyses. Therefore, LG1 and LG2 were not separated, and the purified, lyophilized NMR sample contained 1.6 mg of LG1 with about 5% LG2 as impurity.
The 1H signals of dexmedetomidine, DG1, DG2, and LG1 were assigned from their one-dimensional 1H and NOESY spectra (Table 2). The signals from H11 could be distinguished from the H9 signals through their NOE correlation to H15. Similarly, from the imidazole ring protons whose signals are singlets, only H5 has NOE correlations to H6 or H8. The G1 protons were identified by their characteristic chemical shifts near 5 ppm, whereas H6 signals resonate near the G1 signals, but they are quartets due to coupling to the H8 methyl protons. Interglycosidic NOE correlations indicated that DG1 and DG2 are the N3- and N1-glucuronides of dexmedetomidine, respectively, and LG1 is the N3-glucuronide of levomedetomidine, as shown in Figs. 1 and 4. Further analysis of the NOESY spectra is discussed below.
NMR data, 1H chemical shifts of dexmedetomidine, its N3- and N1-glucuronides, DG1 and DG2, respectively, and N3-glucuronide of levomedetomidine, LG1
Medetomidine Glucuronidation Kinetics. Kinetic analysis of dexmedetomidine glucuronidation by HLMs exhibited two-enzyme kinetics, strongly suggesting the involvement of two different UGTs (Fig. 5). The apparent Km1 values (the high-affinity components) for DG1 and DG2 formations by HLMs were both below 10 μM, whereas the Km2 values (the low-affinity components) were much higher, >1 mM (Table 3). The apparent Vmax values indicated a preference for DG1 formation. When recombinant UGTs were assayed for dexmedetomidine glucuronidation, large differences between UGT1A4 and UGT2B10 were observed. The affinity of UGT1A4 was low, Km of 0.6 to 0.7 mM for both DG1 and DG2 formation, but the Vmax values (normalized for expression level), particularly in the case of DG1, were relatively high (Table 4; Fig. 5). UGT2B10, on the other hand, exhibited much higher affinity for dexmedetomidine, Km of 10 to 20 μM, but its turnover rate with this substrate was low (Table 4; Fig. 5). It may be noted, however, that UGT2B10 was previously shown to be a sensitive enzyme that loses activity upon membrane isolation (Kaivosaari et al., 2007); thus, it is possible that it also loses activity upon storage. In any case, the Vmax values of recombinant UGTs and HLMs are not directly comparable because we do not have suitable antibodies to estimate the level of UGT1A4 and UGT2B10 in the HLM sample. On the other hand, the Km values are not dependent on expression level; therefore, we find it highly meaningful that the Km values for dexmedetomidine glucuronidation by UGT2B10 were very close to the high-affinity Km1 values observed in HLMs, whereas the Km values of UGT1A4 resembled the low-affinity Km2 values in HLMs (Tables 3 and 4).
Kinetics of dex- and levomedetomidine glucuronidation by human liver microsomes The data are presented as mean ± S.E. of parameter fit. The assay conditions are detailed in the legend to Fig. 5.
Kinetic parameters of dex- and levomedetomidine glucuronidation by recombinant human UGT1A4 and UGT2B10 Data are presented as mean ± S.E. The assay conditions are detailed in the legend to Fig. 5.

Proposed structures for 3 medetomidine glucuronides based on experimental one- and two-dimensional NMR data. A, DG1 (N3-glucuronide of dexmedetomidine. B, DG2 (N1-glucuronide of dexmedetomidine. C, LG1 (N3-glucuronide of levomedetomidine). Key NOE correlations used in the identification of the different glucuronides are indicated by dashed lines. All distances between correlating protons are smaller than 5Å in the depicted structures.

Representative kinetic plots (main figures) and Eadie-Hofstee plots (insets) for dex- and levomedetomidine N-glucuronidation by human liver microsomes (A and B), recombinant human UGT1A4 (C and D), and UGT2B10 (E and F). The formation of the N3-glucuronide, DG1 or LG1, is presented by • and solid line. The formation of N1-glucuronide, DG2 or LG2, is indicated by □ and dashed line. The incubation conditions in dexmedetomidine were either 0.2 mg/ml protein HLM for 60 min, 1 mg/ml UGT1A4 for 120 min, or 1 mg/ml UGT2B10 for 240 min. In levomedetomidine assays, the conditions were 0.2 mg/ml HLM for 30 min, 2 mg/ml UGT1A4 for 120 min, or 1 mg/ml UGT2B10 for 120 min. The glucuronidation rates represent the mean (±S.D.) of two or three independent determinations. The rates were normalized according to the relative expression levels. The respective kinetic constants are presented in Tables 3 and 4.
Levomedetomidine glucuronidation by HLMs was highly regioselective, and more than 95% of the product was LG1 (Fig. 5). In addition, the kinetics of LG1 formation differed significantly from LG2 formation by HLMs. LG1 formation by HLMs exhibited monophasic kinetics with a high affinity, Km of 14 μM, and some substrate inhibition (Table 3; Fig. 5). On the other hand, LG2 was formed by HLMs at a low rate and at a low affinity. Interestingly, metabolism of levomedetomidine by recombinant human UGT2B10 yielded only LG1, whereas UGT1A4 produced both LG1 and LG2 at comparable rates (Table 4; Fig. 5). The kinetics of LG1 formation by UGT2B10 highly resembled HLMs with respect to both high affinity, Km of 7.4 μM, and mild substrate inhibition (Fig. 5). UGT1A4, on the other hand, exhibited much lower affinity than either HLMs or UGT2B10 for levomedetomidine, with a somewhat lower Km value for the LG1 formation (0.3 mM) than for LG2 (1.6 mM) (Table 4; Fig. 5). In addition and in contrast to the situation with dexmedetomidine, the normalized turnover rates of UGT1A4 with levomedetomidine were low, significantly lower than those of UGT2B10 in the formation of LG1 (Table 4).
Discussion
The present study emerged from the drive to clarify open questions left after our previous work on medetomidine metabolism (Kaivosaari et al., 2002). Among other things, it was concluded then that the activity and, particularly, the high affinity exhibited by HLMs for levomedetomidine could not be solely explained by the contribution of UGT1A4. Finding and characterizing another human UGT, or UGTs, that glucuronidate medetomidine may explain the observed enantioselectivity and the kinetics.
Analyses of liver microsomes from different animals and human intestine microsomes indicated that N-glucuronidation of medetomidine is largely restricted to the human liver (Fig. 2). Conjugation of dex- and levomedetomidine by HLMs yields two glucuronides from each enantiomer (Fig. 2). LC-MS analyses indicated that all the four regioisomers are monoglucuronides. We have biosynthesized and purified the main three glucuronides, DG1, DG2, and LG1, and determined their structure by NMR (Table 2; Fig. 4). A comparison of the 1H chemical shifts of dexmedetomidine and DG2 revealed that the aromatic ring chemical shifts did not change significantly upon glucuronidation (Table 2). In addition, no NOEs were observed between the glucuronic acid moiety and the aromatic ring, indicating that these units are not close to each other in DG2. The only interglycosidic NOEs detected in the case of DG2 were between the imidazole ring protons H2/H5 and GH1 and GH2 (Fig. 4). Because the intensity of the NOE correlations between GH1 and H2 was approximately as strong as the correlation between GH1 and H5, the average distance between these pairs of protons should be similar. This is possible only if the glycosidic linkage is formed at the imidazole nitrogen N1, supporting the assignment of DG2 (Fig. 4B).
The NOESY spectra of LG1 and DG1 revealed a strong NOE between GH2 and H2, a much weaker NOE between GH1 and H2, and no interglycosidic NOEs to H5 (Fig. 4, A and C). This NOE pattern strongly suggests that in these metabolites the glucuronic acid is attached to nitrogen N3. It can be seen from the structures (Fig. 4) that the H5s in both DG1 and LG1 are further apart from the glucuronic acid than H2. Several NOEs were visible between the aromatic ring and the glucuronic acid in both DG1 and LG1, demonstrating that the aromatic ring stacks against the H1/H3/H5 face of the glucuronic acid. This further verified that LG1 and DG1 were N3-glucuronides as shown in Fig. 4 because such stacking would not be possible in the G2 configuration.
Four of the 16 human UGTs of subfamilies 1A and 2B catalyzed medetomidine glucuronidation (Table 1). Under the assay conditions, namely the presence of high substrate concentration, the highest (normalized) activity toward dexmedetomidine was exhibited by UGT1A4, whereas lower activities were also observed with three other enzymes, in the order UGT2B10 >> UGT1A3 ≈ UGT2B7 (Table 1). In the case of levomedetomidine, the picture was changed, and the highest glucuronidation rate was measured with UGT2B10, whereas the activities of the other UGTs were considerably lower. Hence, it seemed that although the activities of UGTs 1A3 and 2B7 do not contribute significantly to medetomidine glucuronidation in HLMs, the contributions of UGT1A4 and, particularly, UGT2B10 are fundamental. The importance of UGT2B10 was also in good agreement with the activity difference between HLMs and human intestine microsomes (Fig. 2) because UGT2B10 is well expressed in the human liver, but not in the intestine (Kaivosaari et al., 2007).
Prior to kinetic analyses, we have tested the effect of alamethicin on the activity of HLMs and recombinant UGTs. In agreement with previous results (e.g., Fisher et al., 2000), alamethicin increased turnover rate in HLMs, but it had no significant effect on the activity of recombinant UGTs (Fig. 3). It may also be noted that we have used the recombinant UGTs as cell homogenates because we have recently found that, for a yet-unknown reason, the activity of recombinant UGT2B10 decreases sharply upon membrane isolation (Kaivosaari et al., 2007). In the case of UGT1A4, the normalized activities in cell homogenate and isolated membranes were very similar, so the use of cell homogenate was clearly advantageous in this study. Cell homogenates might also be a more suitable test material in other studies with recombinant human UGTs, particularly when the activity of UGT2B10 is examined.
Detailed kinetic analyses of medetomidine glucuronidation by HLMs, UGT1A4, and UGT2B10 were carried out. In HLMs, levomedetomidine glucuronidation yielded specifically (≥95%) the LG1 metabolite, i.e., N3-glucuronide, with a high affinity obeying classical Michaelis-Menten kinetics (Fig. 5). The formation of LG2 was low (<5%) and indicated low affinity. DG1, the N3-glucuronide, was the preferred regioisomer also in the case of dexmedetomidine glucuronidation, but the regioselectivity, 70 to 80% DG1, was not as striking as with levomedetomidine. The conjugation of dexmedetomidine to DG1 and DG2 by HLMs clearly obeyed two-enzyme kinetics (Fig. 5), and the estimated high-affinity Km values (6.6 and 8.7 μM, respectively) were very close to the 14 μM Km value for LG1 formation (Table 3). The detailed kinetic study we have now performed gives new insight into the results of a previous study (Kaivosaari et al., 2002) in which we overlooked the high-affinity component of dexmedetomidine glucuronidation by HLMs. Based on the present results, it is concluded that one UGT with high affinity, UGT2B10, is mainly responsible for levomedetomidine glucuronidation (i.e., LG1 formation), whereas two enzymes, UGT2B10 and UGT1A4, are involved in dexmedetomidine glucuronidation (DG1 and DG2 formation).
The kinetic profiles of HLMs and UGT2B10 in levomedetomidine glucuronidation were very similar, including LG1 formation as the main or only metabolite, high affinity in LG1 formation, and mild substrate inhibition (Fig. 5). In the case of dexmedetomidine, however, both UGT1A4 and UGT2B10 contributed to the activity observed in HLMs. UGT2B10 conjugated dexmedetomidine to both DG1 and DG2 with a high affinity, but low capacity (Fig. 5; Table 4), corresponding to the high-affinity component in the activity of HLMs. UGT1A4, on the other hand, produced both DG1 and DG2, with a preference for DG1, at higher turnover rates, but at a much lower affinity than UGT2B10. It may be noted that although levomedetomidine appeared to be an almost pure UGT2B10 substrate (Fig. 5; Table 4), the small contribution of UGT1A4 to levomedetomidine glucuronidation may still be significant, at least for the low level of LG2 formation by HLMs at a low affinity.
We have shown recently that UGT2B10 is mainly responsible for the N-glucuronidation of nicotine in human liver (Kaivosaari et al., 2007). Based on these and the present findings, it is tempting to suggest that UGT2B10 is also responsible for the N-glucuronidation of few other xenobiotics, including drugs for which the high-affinity glucuronidation by HLMs could not be explained earlier (Breyer-Pfaff et al., 2000; Nakajima et al., 2002).
In summary, N-glucuronidation of medetomidine enantiomers is largely restricted to humans and shows marked regio- and stereospecificity. Although both UGT1A4 and UGT2B10 contribute to the glucuronidation of dexmedetomidine, the analgesic sedative drug, the other enantiomer, levomedetomidine, is practically a specific substrate for UGT2B10. The results further highlight the importance of UGT2B10 in N-glucuronidation of xenobiotics in the human liver.
Acknowledgments
We thank Eija Puukangas, Satu Teliö, and Johanna Mosorin for skillful technical assistance.
Footnotes
-
This study was partly supported by the Academy of Finland (Project 210933) and by the Sigrid Juselius Foundation.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.021709.
-
ABBREVIATIONS: dexmedetomidine, (+)-4-(S)-[1-(2,3-dimethylphenyl)ethyl]-1H-imidazole monohydrochloride; levomedetomidine, (-)-4-(R)-[1-(2,3-dimethylphenyl)ethyl]-1H-imidazole; HLM, human liver microsome; UGT, UDP glucuronosyltransferase; NMR, nuclear magnetic resonance; UDPGA, UDP-glucuronic acid; LC, liquid chromatography; DG1, DG2, LG1, and LG2, N3- and N1-glucuronides of dex- and levomedetomidine, respectively; MS, mass spectrometry; SPE, solid phase extraction; NOESY, nuclear Overhauser enhancement spectroscopy; NOE, nuclear Overhauser enhancement.
- Received March 31, 2008.
- Accepted May 7, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}