


Abstract
Prediction of the extent and time course of drug-drug interactions (DDIs) between the mechanism-based inhibitor diltiazem (DTZ) and the CYP3A4 substrate midazolam (MDZ) is confounded by time- and concentration-dependent clearance of the inhibitor. Semiphysiologically based pharmacokinetic (PBPK) models were developed for DTZ and MDZ with the major metabolite of DTZ, N-desmethyldiltiazem (nd-DTZ), incorporated in the DTZ model. Enzyme kinetic parameters (kinact and KI) for DTZ and nd-DTZ were estimated in vitro and used to model the time course of changes in the amount of CYP3A4 in the liver and gut wall, which in turn, determined the nonlinear elimination of MDZ and DTZ, and the corresponding DDI. The robustness of the model prediction was assessed by comparing the results of the prediction to published DTZ pharmacokinetic and DTZ/MDZ interaction data. A clinical study was conducted to further validate the predicted increase of MDZ exposure after DTZ treatment. The model predicted the nonlinear disposition of DTZ after single and multiple oral doses. The clinical study showed that DTZ treatment resulted in 4.1- and 1.6-fold increases in MDZ exposure after oral and intravenous MDZ administration, respectively, suggesting that the DDI in the gut wall plays an important role in the DTZ/MDZ interaction. The semi-PBPK model incorporating the DDI at the gut wall, and the effect of nd-DTZ successfully predicted the nonlinear disposition of DTZ and its interaction with MDZ. Moreover, model simulation suggested that both DTZ and nd-DTZ contributed to the overall inhibitory effect after DTZ administration, and the values of the in vitro estimated inhibition parameters and CYP3A4 turnover rate are critical for the prediction.
Diltiazem (DTZ), a benzothiazepine derivative, is a calcium channel blocker that is widely used in the therapy of angina pectoris and hypertension (Chaffman and Brogden, 1985; Buckley et al., 1990). In humans, the pharmacokinetic properties of DTZ are characterized by intermediate bioavailability with large variation (25–73%), intermediate to high clearance (11.5–21.3 ml/min/kg), extensive plasma protein binding (77–86%), and a large volume of distribution (3–8 l/kg) (Piepho et al., 1982; Hermann et al., 1983; Bianchetti et al., 1991). DTZ undergoes extensive metabolism, with only 0.1 to 4% of the dose excreted unchanged in the urine (Rovei et al., 1980). The major metabolites in human include N-desmethyldiltiazem (nd-DTZ), deacetyldiltiazem, and N-desmethyl, deacetyldiltiazem (Rovei et al., 1980). The N-desmethyl and deacetylated metabolites are present in concentrations of 30 and 10% of the parent, respectively (Rovei et al., 1980).
DTZ displays nonlinear disposition in humans, and metabolism of DTZ has been suggested to be saturable at clinically relevant concentrations despite the fact that saturation is not expected based on the in vitro Km value of DTZ (∼3 μM) (Zelis and Kinney, 1982; Smith et al., 1983; Jones et al., 1999). The area under the plasma concentration-time curve from zero to infinity (AUC0–∞) increases disproportionally after increased single oral doses of DTZ. Moreover, the steady-state plasma concentration is not predictable from that after a single dose using dose-independent models of disposition (Buckley et al., 1990). Multiple-dose administration results in a 60% reduction in oral clearance of DTZ, however, with no significant change in the terminal half-life (Lefebvre et al., 1994).
DTZ is a clinically significant inhibitor of cytochrome P450 3A4 (CYP3A4). Therapeutically important interactions between oral diltiazem and midazolam (MDZ) (Backman et al., 1994), triazolam (Varhe et al., 1996), cyclosporine (Wagner et al., 1988), lovastatin (Azie et al., 1998; Masica et al., 2000), and carbamazepine (Brodie and MacPhee, 1986; Eimer and Carter, 1987) have been documented. A previous study by our group also demonstrated that DTZ (120 mg twice daily for 7 days) caused a 62% decrease in small bowel CYP3A4 activity (Pinto et al., 2005). Thus, irreversible inhibition of CYP3A by DTZ is the most likely explanation for the nonlinear pharmacokinetics of DTZ.
DTZ inhibits CYP3A4 mainly as a mechanism-based inhibitor through metabolic intermediate complex formation as demonstrated by in vitro studies (Jones et al., 1999). Various approaches have been applied for the prediction of in vivo DDIs involving mechanism-based inhibitors (Kanamitsu et al., 2000; Mayhew et al., 2000; Takanaga et al., 2000; Ito et al., 2003; Wang et al., 2004; Venkatakrishnan and Obach, 2005). However, despite several successful predictions, these approaches either do not address the change in inhibitor concentration with time or do not consider the interaction at the gut wall. Furthermore, no effect of active metabolites has ever been incorporated in model development, largely because of the lack of information on metabolite disposition in the literature.
A recent in vitro study suggested that nd-DTZ may play an important role in the interactions between DTZ and other CYP3A4 substrates at a pharmacokinetic level. It has been shown that nd-DTZ is a more potent inhibitor, both reversible and irreversible, of CYP3A4 than the parent drug in vitro (Sutton et al., 1997; Mayhew et al., 2000). However, the steady-state plasma concentration of nd-DTZ is approximately one-third of that of DTZ and its pharmacological effect as a coronary vasodilator is approximately one-fifth of that of DTZ (Hermann and Morselli, 1985). Therefore, given the higher inhibition potency but lower exposure of nd-DTZ compared with DTZ, it is not clear whether nd-DTZ contributes to the overall inhibitory effect observed in vivo after DTZ administration.
Here we present the development and validation of a semiphysiologically based pharmacokinetic (PBPK) model for the prediction of the nonlinear disposition of DTZ and its interaction with MDZ. DDIs in the gut wall and liver, the plasma concentration-time profile of the metabolite, nd-DTZ, and CYP3A4 enzyme pools were incorporated in the model. Furthermore, the only DTZ/MDZ interaction data available in literature are for the immediate-release (IR) preparation of DTZ, for which only changes in the AUC after oral MDZ administration were investigated (Backman et al., 1994). A clinical study was conducted to quantify the extent of the interaction between MDZ and the most commonly used slow-release (SR) preparation of DTZ (Cardizem SR; Biovail Pharmaceuticals, Mississauga, ON, Canada). The robustness of the model prediction was assessed by comparing the results of the prediction to published DTZ PK and DTZ/MDZ data and was further validated with the results of our prospective clinical study of the DTZ/MDZ interaction. Simulation was performed to evaluate the relative contribution of DTZ and nd-DTZ to the overall inhibition of CYP3A4 as probed by MDZ AUC -fold increases. The sensitivity of the model prediction to several key parameters was also evaluated.
Materials and Methods
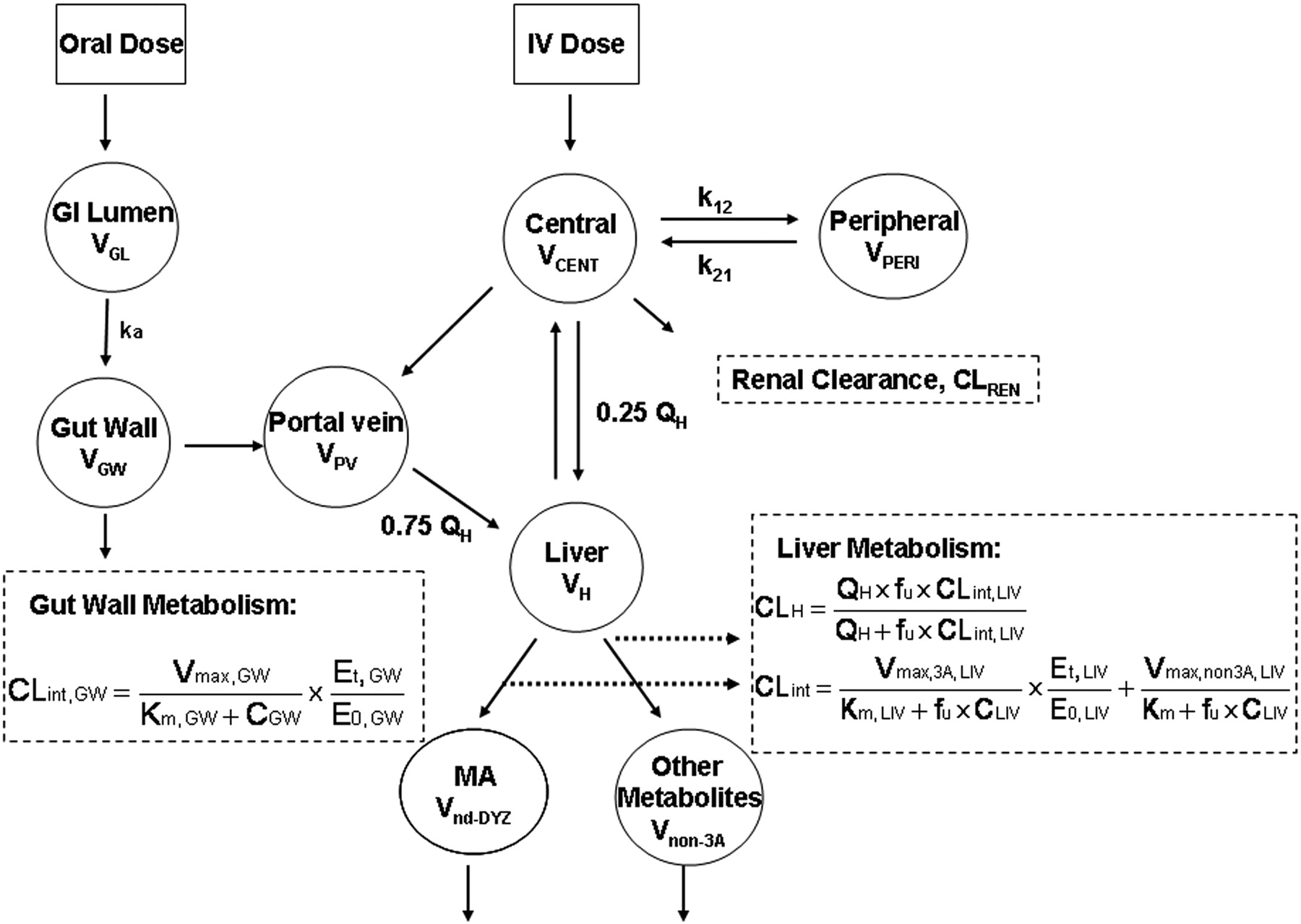
PBPK models were developed for DTZ and MDZ, separately, based on the reported PK and physiological parameters (Tables 1 and 2). CYP3A4 enzyme pools in liver and gut wall, the sites for the interaction between DTZ and MDZ, were modeled as separate compartments (Fig. 1). The gut and hepatic availability and systemic clearance of each drug were expressed as functions of intrinsic clearance, which was related to the amount of active CYP3A4 in the gut wall and liver. Therefore, the time-dependant change in the amount of active CYP3A4 determined the nonlinear disposition of DTZ and the DTZ/MDZ interaction.
Model parameters for MDZ used in the simulations
Model parameters for DTZ and nd-DTZ
PBPK Model Development. The PBPK models for DTZ and MDZ (Fig. 1) were identical except that the DTZ model recognized the generation of an inhibitory metabolite, nd-DTZ. For MDZ, a conventional two-compartment PK model with additional compartments for gut lumen, gut wall, portal vein, and liver were developed. Based on the model, the mass balance equations for drugs transferring between compartments are expressed as eqs. 1 to 5: 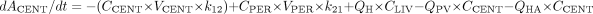
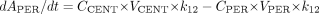
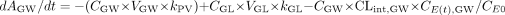
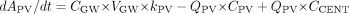
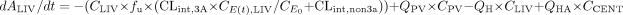 where ACENT, APER, AGW, APV, and ALIV are the amounts of drug in central, peripheral, gut wall, portal vein, and liver compartments, respectively; CCENT, CPER, CGW, CGL, CPV, and CLIV are drug concentrations in central, peripheral, gut wall, portal vein, and liver compartments, respectively; VCENT, VPER, VGL, VGW, VPV, and VLIV are the volumes of central, peripheral, gut lumen, gut wall, portal vein, and liver compartments, respectively; VGW is the same as the volume of water a patient would take to administer the drug, which is usually 250 ml; kGL and kPV are first-order rate constants for drug transferring from gut lumen to gut wall and from gut wall to portal vein, respectively; k12 and k21 are the first-order rate constants for drug transferring from central to peripheral and from peripheral to central compartments, respectively; CE(t), GW and CE(t), LIV are the concentrations of active CYP3A in gut wall and liver at any time and CE0 is the baseline CYP3A concentration at time 0; CLint, GW and CLint, 3A and CLint, non3A are intrinsic clearance in gut wall and the intrinsic clearance for the CYP3A pathway and other metabolism pathway in the liver, respectively; CLREN is renal clearance; fu is fraction unbound in plasma; QH is the liver blood flow; and QPV and QHA are the portal vein blood flow and hepatic artery blood flow and represent 75 and 25% of QH, respectively.
where ACENT, APER, AGW, APV, and ALIV are the amounts of drug in central, peripheral, gut wall, portal vein, and liver compartments, respectively; CCENT, CPER, CGW, CGL, CPV, and CLIV are drug concentrations in central, peripheral, gut wall, portal vein, and liver compartments, respectively; VCENT, VPER, VGL, VGW, VPV, and VLIV are the volumes of central, peripheral, gut lumen, gut wall, portal vein, and liver compartments, respectively; VGW is the same as the volume of water a patient would take to administer the drug, which is usually 250 ml; kGL and kPV are first-order rate constants for drug transferring from gut lumen to gut wall and from gut wall to portal vein, respectively; k12 and k21 are the first-order rate constants for drug transferring from central to peripheral and from peripheral to central compartments, respectively; CE(t), GW and CE(t), LIV are the concentrations of active CYP3A in gut wall and liver at any time and CE0 is the baseline CYP3A concentration at time 0; CLint, GW and CLint, 3A and CLint, non3A are intrinsic clearance in gut wall and the intrinsic clearance for the CYP3A pathway and other metabolism pathway in the liver, respectively; CLREN is renal clearance; fu is fraction unbound in plasma; QH is the liver blood flow; and QPV and QHA are the portal vein blood flow and hepatic artery blood flow and represent 75 and 25% of QH, respectively.

Semi-PBPK model for the description of the disposition of DTZ and MDZ.
Equations 1 to 5 were also applied for the inhibitor DTZ. Furthermore, a metabolite compartment was incorporated for the major metabolite of CYP3A4 pathway, nd-DTZ. Equations 6 and 7 are for nd-DTZ and the metabolites from other pathways (non-CYP3A4 pathway): 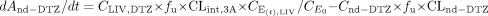
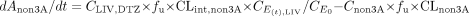 where And-DTZ, and Cnd-DTZ are the amount and concentration of nd-DTZ formed, respectively; Anon3A and Cnon3A are the amount and concentration of metabolites formed from the non-CYP3A4 pathway, respectively; CLIV, DTZ is DTZ concentration in liver; CLint, 3A and CLint, non3A represent the intrinsic clearances of formation of nd-DTZ and other metabolites, respectively; and CLnd-DTZ and CLnon3A are the total clearances (including metabolic and renal clearance) of nd-DTZ and other metabolites, respectively.
where And-DTZ, and Cnd-DTZ are the amount and concentration of nd-DTZ formed, respectively; Anon3A and Cnon3A are the amount and concentration of metabolites formed from the non-CYP3A4 pathway, respectively; CLIV, DTZ is DTZ concentration in liver; CLint, 3A and CLint, non3A represent the intrinsic clearances of formation of nd-DTZ and other metabolites, respectively; and CLnd-DTZ and CLnon3A are the total clearances (including metabolic and renal clearance) of nd-DTZ and other metabolites, respectively.
Without inhibitors, the intrinsic clearance of MDZ in gut wall (CLint, GW, MDZ, assuming only CYP3A in gut wall) and liver (CLint, 3A, MDZ and CLint, non3A, MDZ) can be expressed in terms of Vmax, GW, MDZ, Vmax, 3A, MDZ, Vmax, non3A, MDZ, and Km, MDZ as in eqs. 8 and 9: 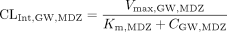
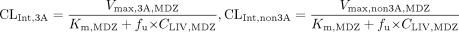 where Vmax, GW, MDZ, Vmax, 3A, MDZ, Vmax, non3A, MDZ, and Km, MDZ are the maximal rate for metabolite formation in gut wall, the maximal rate for metabolite formation through CYP3A4 and non-CYP3A4 pathway in liver, and the Michaelis-Menten constant, respectively, for MDZ. Km values in the gut wall were assumed to be the same as those in the liver.
where Vmax, GW, MDZ, Vmax, 3A, MDZ, Vmax, non3A, MDZ, and Km, MDZ are the maximal rate for metabolite formation in gut wall, the maximal rate for metabolite formation through CYP3A4 and non-CYP3A4 pathway in liver, and the Michaelis-Menten constant, respectively, for MDZ. Km values in the gut wall were assumed to be the same as those in the liver.
After the administration of DTZ, the inactivator will produce a corresponding reduction in intrinsic clearance to CLint′, as expressed in eqs. 10, considering that DTZ and nd-DTZ act simultaneously as inhibitory species: 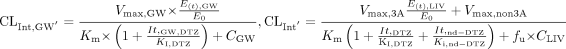 where Ki, DTZ and Ki, nd-DTZ are the competitive inhibition constant for DTZ and nd-DTZ, respectively. Note that nd-DTZ was considered to have no effect on gut wall CYP3A4 because of its minimal concentration in gut wall. Equations 8 to 10 also apply to the inhibitor, DTZ, because a change in the amount of active enzyme by the inhibitor, in turn, causes a change in the intrinsic clearance and determines the nonlinear disposition of the inhibitor itself. We assume that MDZ does nothing to DTZ or nd-DTZ.
where Ki, DTZ and Ki, nd-DTZ are the competitive inhibition constant for DTZ and nd-DTZ, respectively. Note that nd-DTZ was considered to have no effect on gut wall CYP3A4 because of its minimal concentration in gut wall. Equations 8 to 10 also apply to the inhibitor, DTZ, because a change in the amount of active enzyme by the inhibitor, in turn, causes a change in the intrinsic clearance and determines the nonlinear disposition of the inhibitor itself. We assume that MDZ does nothing to DTZ or nd-DTZ.
The PK parameters for the development of the MDZ PK model were estimated using a population approach, in which the concentration-time data from 112 healthy subjects were combined from five clinical studies, as reported in a previous study by our group (Chien et al., 2006). The PK parameters are listed in Table 1 and were used for the simulation in the current study. The PK parameters for DTZ and nd-DTZ were obtained from data in the literature and are listed in Table 2. Because the IR preparation of DTZ is seldom used today, the disposition of the SR preparation of DTZ and its interaction with MDZ were also simulated. The SR formulation of DTZ was modeled by modifying the kGL from 0.9 (for the IR formulation) to 0.2 and applying a lag time of 4 h.

The determinants of the amount of CYP3A at any time (E(t)) without inhibitors (A), with only DTZ as inhibitor (B), and with DTZ and nd-DTZ as inhibitors (C).
Enzyme Model Development. The amount of CYP3A4 in liver and gut wall was modeled as illustrated in Scheme 1. Scheme 1A shows that at steady state without inactivator in vivo, the amount of active CYP3A4 enzyme (E0) is determined by R0, the rate of enzyme synthesis (zero order), and the rate of enzyme degradation, which is governed by the first-order degradation rate constant, kdeg. In general, the rate of change of active enzyme [E(t)] is given by eq. 11, 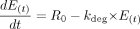 at baseline steady state (eq. 12),
at baseline steady state (eq. 12), 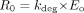 where E0 is the amount of active enzyme at the time 0.
where E0 is the amount of active enzyme at the time 0.
After the treatment with DTZ, assuming that either DTZ or nd-DTZ acts as the inactivator, another pathway for enzyme inactivation is present, as depicted in Scheme 1B. The rate of inactivation is determined by the pseudo-first-order rate constant, kobs as shown in eq. 13: 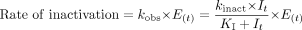 where kinact is the rate constant that defines the maximal rate of inactive enzyme formation, It is the inhibitor concentration at time t, and KI is the inhibitor concentration when kobs = kinact/2. The differential equation for the amount of active CYP3A4 can then be expressed as in eq. 14:
where kinact is the rate constant that defines the maximal rate of inactive enzyme formation, It is the inhibitor concentration at time t, and KI is the inhibitor concentration when kobs = kinact/2. The differential equation for the amount of active CYP3A4 can then be expressed as in eq. 14: 
Scheme 1C depicts the situation in which both DTZ and nd-DTZ act as CYP3A4 inactivators. An additive model was used for the simultaneous exposure to the two inactivators. The effect of competitive inhibition between the two inhibitors is also taken into account by incorporating a (1 + It/Ki) term into the model, as shown in eq. 15: 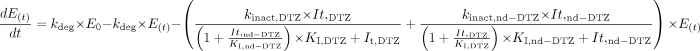
The reversible (Ki) and irreversible (kinact and KI) inhibition parameters were estimated for DTZ and nd-DTZ with human liver microsome (HLMs) using a method as described previously (Jones et al., 1999). The values of the parameters were presented in a previous article by our group and are listed in Table 2 (Zhang et al., 2009). In vivo approaches estimated the half-life for CYP3A to be between 1 and 6 days (Lai et al., 1978; Fromm et al., 1996; Hsu et al., 1997; Greenblatt et al., 2003; Zhang et al., 2008). We have examined the recovery of intestinal and hepatic CYP3A activity after a week-long course of clarithromycin (Gorski et al., 2002; Wang et al., 2004). By fitting the inactivation rate equation to the data, a half-life of CYP3A was determined to be approximately 28 h (kdeg ≈ 0.03 h–1). Therefore, a kdeg value of 0.03 h–1 was used for the simulation in the current study (Table 2). Moreover, because the unbound fraction of DTZ in HLMs is approximately 0.78 (Austin et al., 2002), a correction of KI of DTZ by the unbound fraction in HLMs was not made. The same was assumed for the metabolites of DTZ.
Model Validation and Prediction. The model was applied to simulate data of four sets of published data describing DTZ disposition (both IR and SR formulations) and DTZ IR/MDZ interaction studies (Table 3). Using the PK and enzyme inhibition parameters, eqs. 1 to 15 were numerically solved to simulate the time course of the concentration of DTZ, nd-DTZ, MDZ, and the amount of active CYP3A4 in liver and gut wall, using the Pharsight Trial Simulator 2.2 (Pharsight, Mountain View, CA). The values of three key parameters (kinact, KI, and kdeg) were varied 10-fold within the simulation environment to quantify the sensitivity of the prediction of the DTZ/MDZ interaction.
Simulation scenarios used in the study
Clinical Study on DTZ/MDZ Interaction. Three healthy subjects (two males and one female) aged between 20 and 40 years were enrolled in a two-phase, open-label, fixed-order study. All subjects gave written informed consent, and the study protocol was approved by the institutional review board at Indiana University–Purdue University Indianapolis.
Subjects were instructed to avoid taking any nonprescription or prescription medications and to abstain from alcohol and grapefruit juice-containing products for 1 week before the start of the study and throughout the study. Subjects were excluded if they were allergic to DTZ or MDZ or had clinically significant abnormalities in medical history, physical examination, routine serum chemistry, and urinalysis. Pregnant women, as determined by a pregnancy test, were excluded. On phase 1 day 1, after an overnight fast, the subjects reported to the General Clinical Research Center. Intravenous catheters were placed in each forearm for the administration of drug and withdrawal of blood samples. Just before subjects received the dose of MDZ, a baseline blood sample (5 ml) and a urine sample were obtained. MDZ (0.05 mg/kg, midazolam hydrochloride injection; American Pharmaceutical Partners, Inc., Schaumburg, IL) was infused intravenously at a constant rate over 30 min. Nine blood samples (6 ml each) were collected at baseline, 30 and 45 min, and 1, 1.5, 2, 4, 8, and 12 h after MDZ administration. On the morning of day 2, a 4-mg oral dose of MDZ hydrochloride syrup (Roxane Laboratories, Inc., Columbus, OH) was administered with 240 ml of tap water, and nine blood samples (6 ml each) were collected at 30 and 45 min and at 1, 1.5, 2, 4, 8, 12, and 24 h after oral MDZ administration. The subjects began taking 120 mg of DTZ twice daily for 6 days, and on the morning of day 6 after the initiation of DTZ treatment, the subjects returned to the General Clinical Research Center and phase 2 began. Intravenous and oral MDZ were given exactly the same as on study day 1 and day 2 of phase 1.
Serum concentrations of MDZ, DTZ, and nd-DTZ were measured using a liquid chromatography/mass spectrophotometry method as described previously (Pinto et al., 2005). PK parameters (Cmax, Tmax, AUC0–∞, and t1/2) of MDZ before and after DTZ treatment were estimated by noncompartmental analysis using WinNonlin (version 4.0; Pharsight). A two-tailed Student's t test or Wilcoxon rank-sum test, where appropriate, was performed to compare the pharmacokinetic parameter estimates before and after DTZ treatment. Differences were considered statistically significant at p < 0.05.
Results
Plasma Profiles of DTZ and nd-DTZ. Plasma concentration-time profiles for DTZ IR and SR and nd-DTZ were simulated using the interaction model with the pharmacokinetic, physiological, and enzyme kinetic parameters listed in Tables 1 and 2 and were compared with the reported data in literature. Figure 2A shows the concentration-time profiles of plasma DTZ after DTZ IR administration at four different single oral doses (60, 120, 180, and 210 mg). Overall, the simulated and reported profiles were comparable (Rovei et al., 1980). Moreover, the dose-normalized AUC0–∞ (both observed and predicted) increased with the increase in dose, indicating nonlinear disposition, as shown in Fig. 2B.
The predicted DTZ and nd-DTZ concentration profiles after the administration of a single dose of 90 mg of DTZ IR are presented in Fig. 3A and demonstrated good agreement with the observed data (Yeung et al., 1993). Shown in Fig. 3B are the predicted and observed concentration-time profiles of DTZ SR after the administration of 120 mg of DTZ SR. The profile of DTZ SR was compared with that of DTZ IR at the same dose and was predicted to have a longer Tmax (∼7 h), lower Cmax (∼80 mg/l), and slower terminal declining phase than those for the IR preparation. These simulated characteristics of DTZ SR were consistent with the observed data (Lefebvre et al., 1994). The above findings indicate the validity of the structure of the DTZ model and the parameters used in the present simulation for DTZ and nd-DTZ. The MDZ model was validated in a similar manner, and results have been presented elsewhere (Chien et al., 2006).
Effects of DTZ and nd-DTZ on Liver and Gut Wall CYP3A and MDZ AUC. The simulated DTZ and nd-DTZ concentrations and their effects on the active CYP3A4 enzyme activity in the liver and gut wall after 60 mg of DTZ twice daily for five doses are shown in Fig. 4, A and B. The observed DTZ concentration after the fourth dose agreed with the predicted DTZ profile (Backman et al., 1994). In response to DTZ administration, a maximum of 55 and 90% inactivation of liver and gut wall CYP3A4 was predicted, respectively (Fig. 4B). Furthermore, the decrease in the amount of CYP3A4 led to an increase in the AUC of MDZ by 3-fold when 15 mg of MDZ was administered after DTZ treatment (Fig. 4C), which is consistent with the reported 3.75-fold increase (Backman et al., 1994).
After administration of DTZ SR (120 mg twice daily for 7 days), the plasma DTZ concentration was predicted to increase gradually and reach steady state in approximately 2 days with a maximal concentration at steady state of approximately 120 mg/l as shown in Fig. 5A. The observed plasma DTZ concentrations on day 1 and day 6 of DTZ administration showed excellent agreement with the predicted data (Lefebvre et al., 1994). On the other hand, the nd-DTZ concentration was predicted to decrease after repeated doses of DTZ and reached a plateau of 20 mg/l. This decrease is probably due to the inactivation of CYP3A4 in liver, leading to a decreased formation rate of nd-DTZ. No observed data are available for nd-DTZ under this scenario. Furthermore, after DTZ administration, the active CYP3A4 in liver was predicted to fall gradually and reach a plateau of approximately 40% of remaining activity. Upon the discontinuation of DTZ treatment, the activity recovered gradually, returning to the original level in approximately 5 days. In contrast, gut wall CYP3A4 was predicted to decline almost immediately after the first dose of DTZ and reach a plateau of approximately 10% (Fig. 5B). We previously determined that a 38 ± 10% of the CYP3A4 activity remains in the gut wall under this dosing scenario (Fig. 5B). In our previous study, intestinal biopsies were obtained in 10 healthy subjects after receiving DTZ (Cardizem SR) 120 mg twice daily for 7 days and 10 healthy control subjects, and intestinal CYP3A4 activity was determined by incubating the small bowel tissue homogenate with MDZ in vitro. The predicted profile of gut wall CYP3A4 activity using the PBPK model is consistent with the findings of the prior clinical study.

Predicted and observed concentration-time profile (A) and dose-normalized AUC0–∞ (B) for DTZ after increasing single oral doses. In A, the lines are predicted concentration profiles using the semi-PBPK model. The symbols are observed concentration profiles after a DTZ dose of 60 mg (•), 120 mg (▴), 180 mg (▪), and 210 mg (♦) (Rovei et al., 1980).
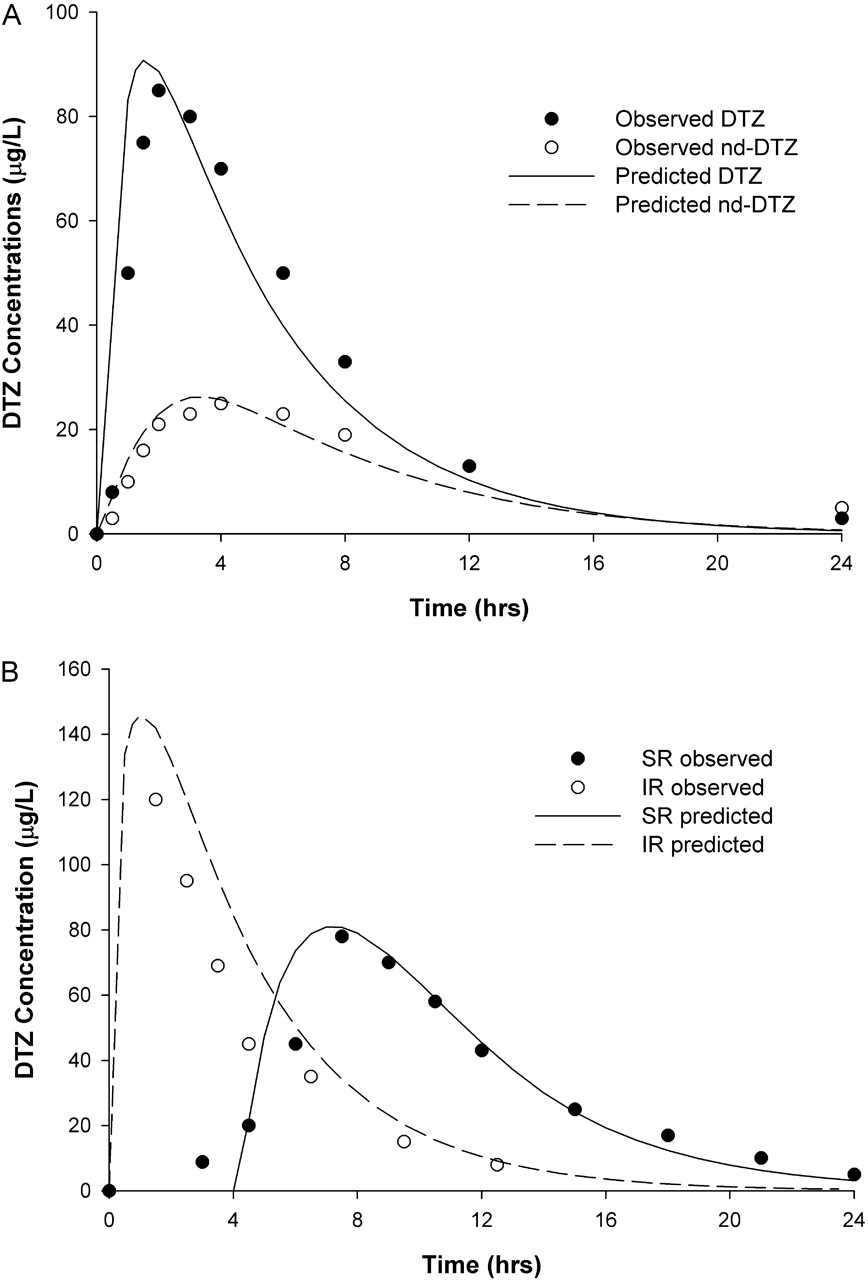
Predicted and observed concentration-time profiles for DTZ and nd-DTZ after a single oral dose of 60 mg of DTZ immediate-release formulation (A), and for DTZ after a single oral dose of 120 mg of DTZ immediate-release formulation and DTZ sustained-release formulation (B). • and ○, corresponding observed data (Yeung et al., 1993; Lefebvre et al., 1994).
Clinical Study on DTZ SR/MDZ Interaction. Pharmacokinetic parameters of MDZ (mean ± S.D.) after administration of a 4-mg p.o. dose or a 0.05-mg/kg i.v. dose of MDZ before and after pretreatment with DTZ (Cardizem SR, 120 mg twice daily) for 6 days to three volunteers are listed in Table 4. DTZ significantly increased the maximum serum concentration (p < 0.05) and the AUC (p < 0.05) of MDZ, without affecting the terminal half-life or time to reach the maximal serum concentration for both oral and intravenous MDZ. The mean -fold increases of MDZ AUC were 4.1- and 1.6-fold for oral and intravenous MDZ, respectively.
Pharmacokinetic parameters of MDZ after administration of a 4-mg p.o. dose or a 0.05-mg/kg i.v. dose of MDZ before and after pretreatment with DTZ SR (Cardizem SR, 120 mg twice daily) for 7 days to three volunteers
Data are mean ± S.D.
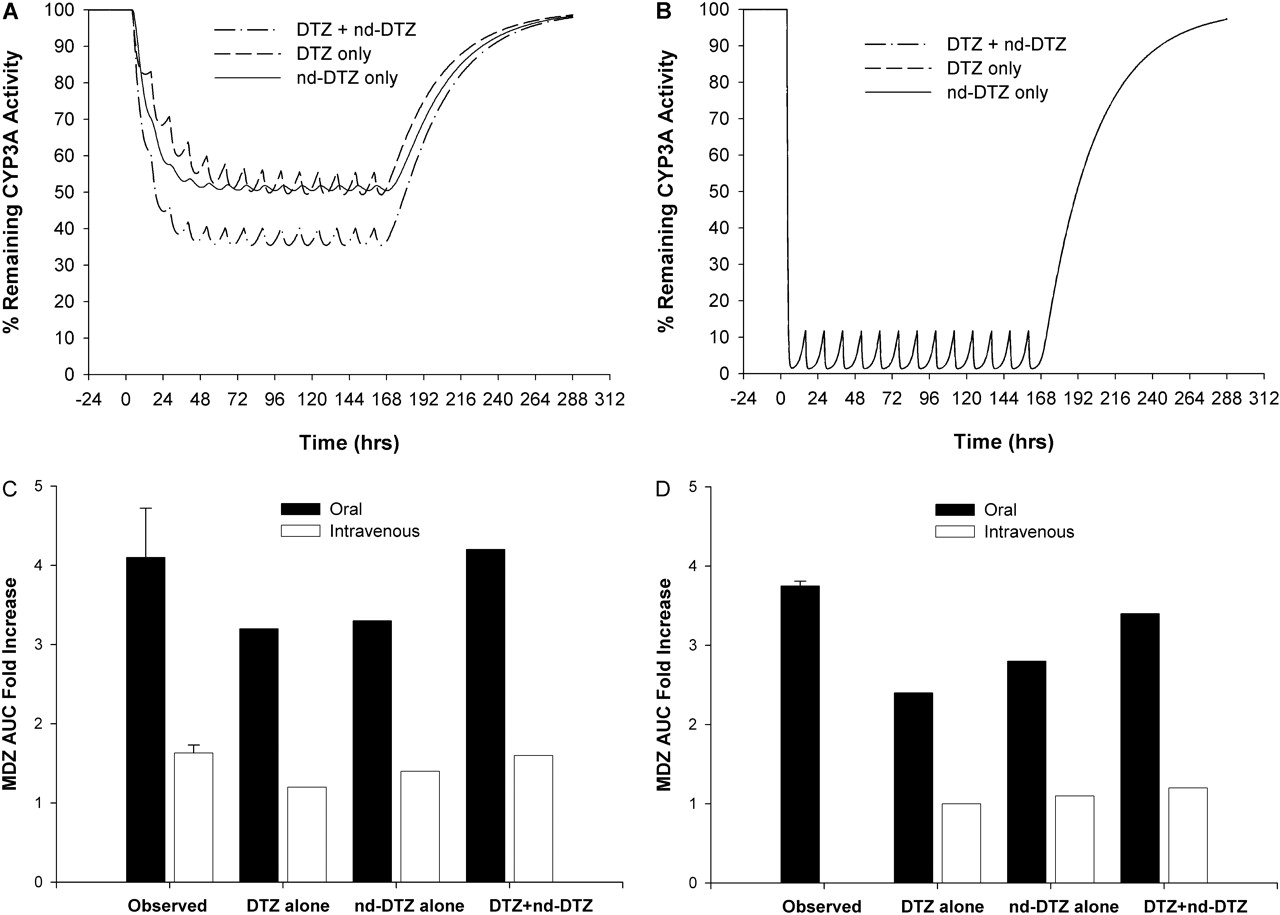
Comparison of the Effects of DTZ and nd-DTZ. The foregoing simulation results are based on the model in which both DTZ and nd-DTZ (only affects liver CYP3A4) contribute to the overall inhibition effect after DTZ administration. The model was also used to simulate the remaining CYP3A4 activity in liver and gut wall and the corresponding MDZ AUC increase, supposing that either DTZ or nd-DTZ acted as the inhibitory species in liver. As shown in Fig. 6A, the model predicted that DTZ or nd-DTZ alone caused approximately 45% maximal inactivation of liver CYP3A4, whereas a maximum of 60% inactivation of liver CYP3A4 was achieved if the effects of DTZ and nd-DTZ were both considered using the additive model. On the other hand, the gut wall CYP3A4 was the same under each situation because nd-DTZ did not reach the gut wall CYP3A4 in our model environment (Fig. 6B).
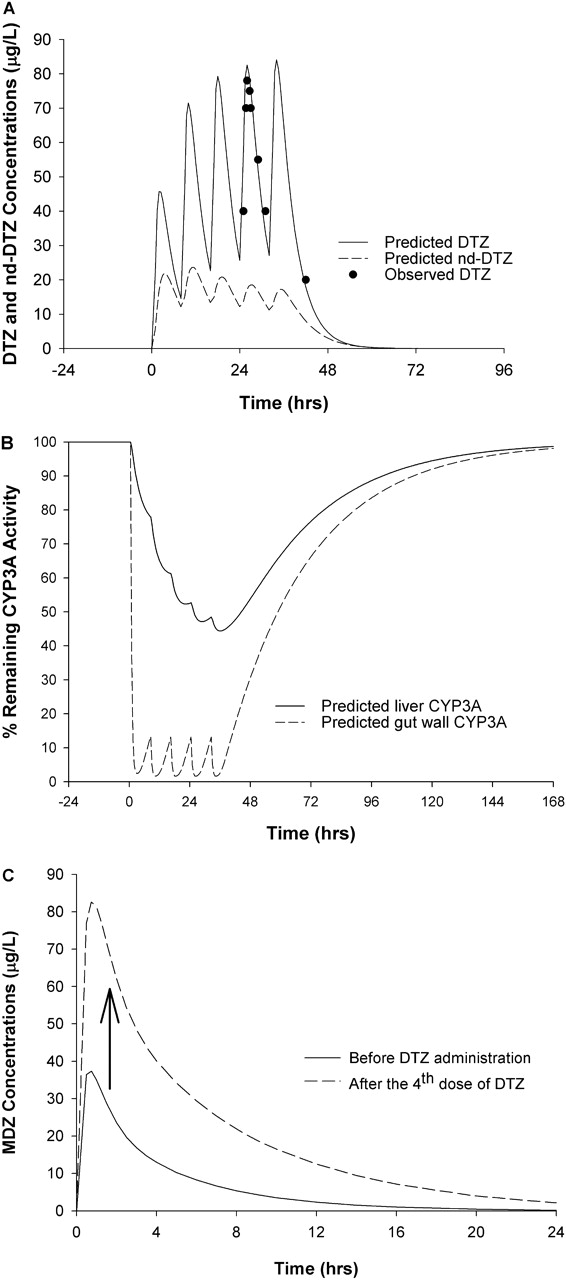
Predicted concentrations of DTZ and nd-DTZ (A), active CYP3A4 content in liver (——) and gut wall (– – –) (B), and MDZ AUC increase (C) after DTZ immediate-release formulation (60 mg three times a day for five doses). •, observed DTZ concentration after the fourth dose of DTZ (Backm994). In C, 10 mg MDZ was given orally before and after the fourth dose of DTZ.
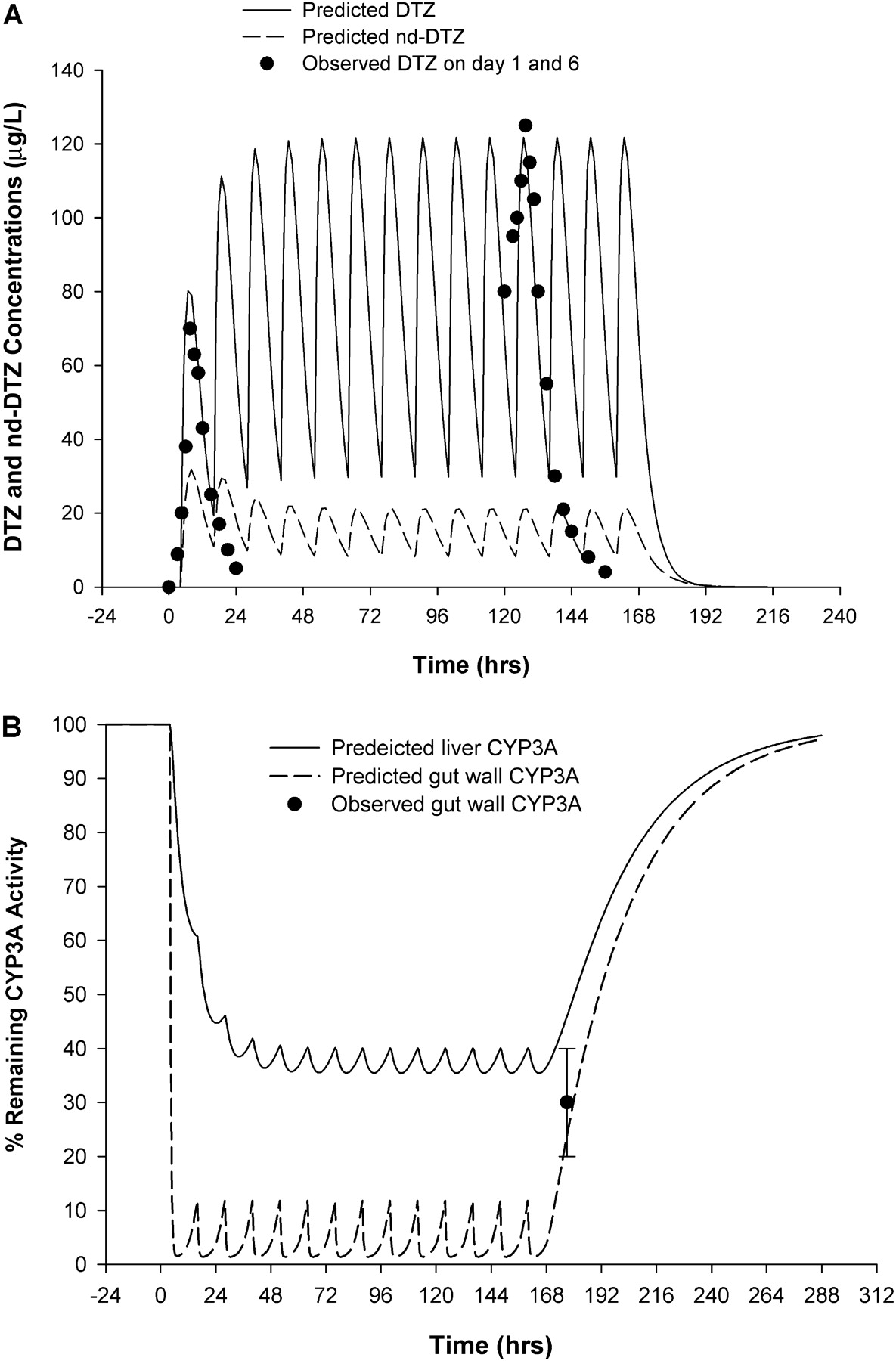
Predicted and observed concentration profiles of DTZ (——) and nd-DTZ (– – –) (A) and active CYP3A4 in liver (——) and gut wall (– – –) (B) after DTZ sustained-release formulation (120 mg twice a day for 7 days). Closed circles in A are observed DTZ concentrations on day 1 and day 6 of DTZ treatment (Lefebvre et al., 1994). Closed circle with error bar in B is observed CYP3A4 activity on the morning of the 8th day (Pinto et al., 2005).
Correspondingly, the oral MDZ AUC was predicted to increase by 3.2-, 3.3-, and 4.2-fold; and the intravenous MDZ AUC was predicted to increase by 1.2-, 1.4-, and 1.6-fold when DTZ alone, nd-DTZ alone, or both were considered as inhibitors, respectively, for DTZ SR (Fig. 6C). Compared with the observed 4.1-fold increase of MDZ AUC as observed in the three patients, the model with both DTZ and nd-DTZ as inhibitors best predicted the DTZ-MDZ interaction. The same holds true for DTZ IR administration as shown in Fig. 6D.
Sensitivity Analysis. The sensitivity of the model prediction to the key model parameters was evaluated by varying individual parameters 5-fold from the value used for the prediction (an overall variation of 10-fold) using the model with DTZ as the inactivator as an example. For liver CYP3A4, a greater kinact or a smaller KI led to greater extent of CYP3A4 inactivation, whereas the greater the kdeg, the less the extent of CYP3A4 inactivation and the faster the CYP3A4 was restored to its initial level (Fig. 7A, C, and E). Regarding CYP3A4 in the gut wall, the overall pattern was similar to that of inactivation of liver CYP3A4; however, the effect of a 10-fold variation in kinact, KI, or kdeg on the CYP3A4 level in the gut wall was obscured because of extensive inactivation at this site (Fig. 7, B, D, and F).
Discussion
The current study illustrated the development and validation of a semi-PBPK model for the prediction of the interaction between the mechanism-based inhibitor, DTZ, its major metabolite, nd-DTZ, and the prototypical CYP3A4 substrate, MDZ. The interaction model, takes into consideration the temporal change in inhibitor concentrations, gut wall interaction, and contribution from nd-DTZ to successfully predict the nonlinear disposition of DTZ and the interaction between DTZ and MDZ. The simulation results suggested that both DTZ and nd-DTZ contributed to the overall inhibitory effect observed after the administration of DTZ. The sensitivity analysis suggested that the in vitro-estimated enzyme inhibition parameters (kinact, KI, and Ki) and the CYP3A4 degradation rate constant, kdeg, are critical for the model prediction (Fig. 7).
Efforts have been made to predict in vivo DDIs involving mechanism-based inhibition with varying degrees of success and failure (Kanamitsu et al., 2000; Mayhew et al., 2000; Takanaga et al., 2000; Ito et al., 2003; Wang et al., 2004; Venkatakrishnan and Obach, 2005). The core interaction model used in these approaches considered the changes in the amount of active enzyme in the presence of a mechanism-based inhibitor, which, in turn, determined the nonlinear elimination of inhibitors and the corresponding DDIs (Jones and Hall, 2002). Successful predictions have been reported for several mechanism-based inhibitors of CYP3A4. In the example of the MDZ/verapamil interaction, the intrinsic clearance of gut wall CYP3A4 was applied in an attempt to account for the gut wall metabolism, but only a single inhibitor concentration (the unbound average plasma concentration of the inhibitor at the steady state) was used in this study (Wang et al., 2004). The MDZ/macrolides, triazolam/erythromycin, and 5-fluorouracil/sorivudine interactions were successfully predicted using a semi-PBPK model, in which the temporal change of inhibitors was adequately addressed (Kanamitsu et al., 2000; Ito et al., 2003). However, the gut wall metabolism and interaction were accounted for in a relatively simplistic method by incorporating an Fg term (fraction of the dose that is metabolized in gut wall) with a fixed value. The results of the current study indicate the importance of incorporating the temporal disposition of the inhibitor and DDIs at the gut wall into the model development.
nd-DTZ has been shown to be more potent than its parent drug, as both a reversible and irreversible inhibitor of CYP3A4 in vitro (Ki is 11-fold lower and kinact/KI is 4-fold higher than those for DTZ) (Sutton et al., 1997; Mayhew et al., 2000). Zhao et al. (2007) studied CYP3A4 inactivation by DTZ in human hepatocytes and suggested that the loss of CYP3A4 activity incubated with DTZ may be largely attributed to nd-DTZ. However, the role of nd-DTZ in vivo remains unclear because of several factors. First, it is unlikely that nd-DTZ will be extensively excreted from the systemic circulation into the intestinal lumen and exert inhibitory effect on CYP3A4 in the enterocytes. Second, the level of nd-DTZ in the systemic circulation is only approximately one-third of that of DTZ (Hermann and Morselli, 1985). In the current study, the model predicted that DTZ or nd-DTZ had similar effects on liver CYP3A4 activity (Fig. 6A), but neither DTZ nor nd-DTZ alone can fully account for the observed increase in MDZ AUC. Therefore, simulation with the drug and metabolite PBPK model provides a valuable tool for studying in vivo DDIs involving metabolites that would otherwise be difficult to identify. With the additive model incorporated into the semi-PBPK model for the simultaneous inactivation by DTZ and nd-DTZ, the -fold increase of MDZ AUC after oral and intravenous administration was accurately predicted. A detailed evaluation of the additive model for the extent of inhibition in the presence of multiple inhibitors was presented in a previous study by our group (Zhang et al., 2009). One point needs to be clarified: DTZ must be first metabolized to nd-DTZ to inactivate the enzyme. The inactivation parameters derived for DTZ will include the possibility that some nd-DTZ is formed but does not leave the active site and leads to the terminal inactivating species. Some nd-DTZ leaves the active site and enters the systemic circulation such that equilibrium is reached between hepatic and systemic unbound concentrations. This metabolite will then reenter the enzyme active site, after competing with DTZ and inactivate with the intrinsic potency of the metabolite per se.

Simulated percentage of the remaining CYP3A4 activity in liver (A) and gut wall (B) with DTZ alone (– – –), nd-DTZ alone (——), and DTZ + nd-DTZ (— · —) as inhibitors and comparison between observed MDZ AUC -fold increases and predicted MDZ AUC -fold increases with DTZ alone, nd-DTZ alone, and DTZ + nd-DTZ as inhibitors after DTZ sustained-release formulation (C) and DTZ immediate-release formulation (D) treatments with oral and intravenous MDZ administration.
The expression of cytochromes P450 in enterocytes results in significant presystemic intestinal metabolism of drugs and possible gut wall DDIs after oral administration (Schwenk, 1988; Kaminsky and Fasco, 1991; Paine and Oberlies, 2007). The results of the current clinical study showed that DTZ treatment led to a much higher AUC increase for oral MDZ (4.1-fold) than for intravenous MDZ (1.6-fold), suggesting that the interaction between DTZ and MDZ occurred mainly during the first pass for this low-extraction-ratio drug. This conclusion was also supported by the observation that the terminal half-life of MDZ was not affected by DTZ treatment. The prediction of DDIs at the level of the gut wall remains challenging because of the added uncertainty in the effective inhibitor concentration at this site. In the current model, the drug concentration in the gut wall is the amount of drug presented in the gut wall compartment in a volume of 250 ml, which is the volume of water a patient would take with the drug. A relatively high DTZ concentration was predicted in the gut wall by this approach. When the inhibitor concentration (It) is much higher than KI, It in eq. 13 is insignificant and inactivation occurs at its maximal rate kinact. Although the gut wall concentration may not be identical to this value, it is undoubtedly high enough to result in the kinact condition. Thus, the duration of exposure rather than the precise gut wall concentration determines the extent and duration of the gut wall inhibition. This translated into 10 to 20% CY3A4 activity remaining in the gut wall at steady state after DTZ treatment for 6 days and effectively predicted the saturation in gut wall metabolism during first-pass elimination and the corresponding increase in MDZ AUC after oral administration. The enzyme parameters (kinact, KI, Ki, Km, and Vmax) for gut wall CYP3A4 were assumed to be equivalent to those for liver CYP3A4 in the current study. This assumption is supported by the evidence that although a difference in Vmax for liver and gut wall CYP3A4 has been reported when it was studied in vitro, once these data are normalized in terms of P450 expression level and method of isolation, they are in fact very close (Galetin and Houston, 2006). However, further studies on a possible discrepancy in the intrinsic clearance and inhibition parameters of drugs for liver and gut wall enzymes are needed.
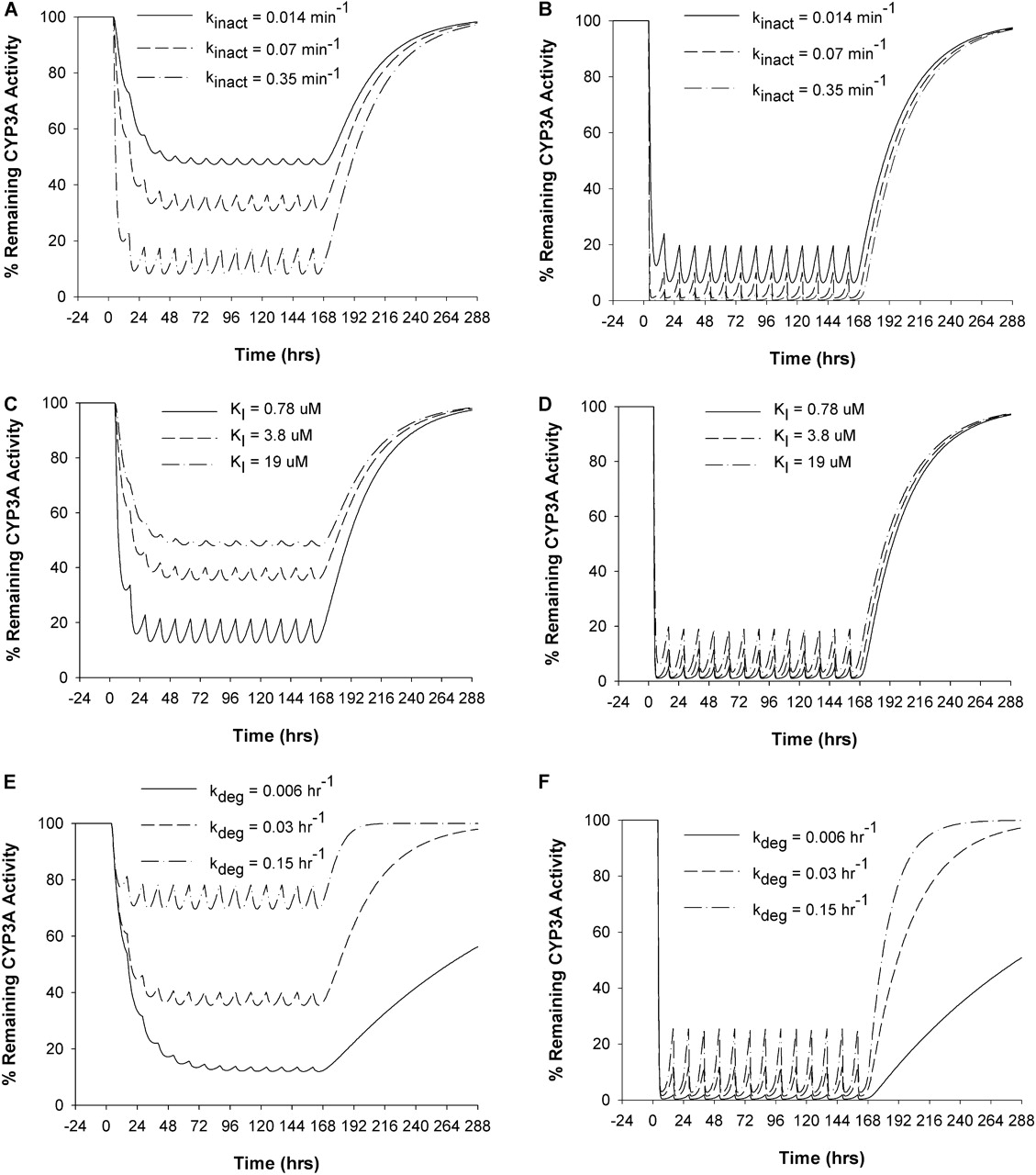
Effects of model parameters, kinact (A and B), KI (C and D), and kdeg (E and F) on the time course of percentage of the remaining enzyme activity in liver (A, C, and E) and gut wall (B, D, and F) during and after DTZ sustained-release formulation administration. The values of each parameter were varied 5-fold from that used in the prediction in this study.
The CYP3A4 degradation rate constant, kdeg, is of great importance for the prediction as shown by the sensitivity analysis, in which kdeg was shown to influence every stage of the CYP3A4 inactivation recovery process (time to reach maximal inactivation, the extent of maximal inactivation, and time for recovery) (Fig. 7). However, kdeg is characterized by considerable uncertainty because of difficulties in estimating the value in vivo. A variety of approaches, including the CYP3A turnover rate in CYP3A4-expressing Caco-2 cells (Malhotra et al., 2001), primary human hepatocytes (Pichard et al., 1992), liver slices (Renwick et al., 2000), or rats (Correia, 1991), the time course of recovery of CYP3A4 activity after inactivation by grapefruit juice in vivo (Greenblatt et al., 2003), the time course of de-induction of rifampin and carbamazepine in vivo (Lai et al., 1978; Fromm et al., 1996), and autoinduction after ritonavir in vivo (Hsu et al., 1997), have been applied in an attempt to obtain an accurate estimate of this parameter. The values for kdeg estimated with these approaches vary from 0.005 to 0.07 h–1. A value of 0.03 h–1 was used for kdeg in both liver and gut wall in the current study based on a clinical study by our group in which MDZ/clarithromycin data were fitted into the inactivation model (S. K. Quinney, X. Zhang, A. Lucksiri, J. C. Gorski, L. Li, and S. D. Hall, submitted for publication). This value is also consistent with the previous estimate for the intestinal CYP3A4 (Greenblatt et al., 2003).
Studies have suggested that the uptake and efflux transporters in liver or gut wall might play important roles in the disposition of many compounds, but the effect of transporters was not incorporated in the current model development (Faber et al., 2003). MDZ is not transported at the gut wall or in the liver (Franke et al., 2008). DTZ has been identified as a P-glycoprotein substrate (Katoh et al., 2006), but because it is highly water-soluble (solubility in water is 56.6 g/100 ml) and highly lipophilic (logP = 2.3), transport is not expected to be an important modulator of intracellular concentration. Therefore, metabolism, not liver or gut uptake, is likely to be the rate-limiting step of the overall disposition of DTZ. Nevertheless, drug transporter effects should be considered in future modeling and simulation studies, especially for the many class 3 drugs (high solubility and low permeability with elimination primarily as unchanged drug in humans) for which disposition could be limited by gut or hepatic uptake.
In summary, the clinical study indicated that DTZ administration significantly elevated MDZ exposure after oral but not intravenous administration, suggesting that DDIs in gut wall play an important role in the interaction between DTZ and MDZ. The semi-PBPK model incorporating DDIs at the gut wall and the effect of nd-DTZ successfully predicted nonlinear disposition of DTZ and its interaction with MDZ. Furthermore, model simulation suggested that both DTZ and nd-DTZ contributed to the overall inhibitory effect after DTZ administration and that the values of the in vitro estimated inhibition parameters and CYP3A4 turnover rate are critical for the prediction.
Acknowledgments
We acknowledge Dr. Aroonrut Lucksiri for contributing to this manuscript.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.109.026658.
-
ABBREVIATIONS: DTZ, diltiazem; nd-DTZ, N-desmethyldiltiazem; AUC, area under the plasma concentration-time curve; AUC0–∞, AUC from 0 to infinity; MDZ, midazolam; DDI, drug-drug interaction; PBPK, physiologically based pharmacokinetic; IR, immediate-release; SR, slow-release; PK, pharmacokinetic; HLM, human liver microsome.
-
↵1 Current affiliation: Department of Drug Disposition, Eli Lilly and Company, Indianapolis, Indiana.
-
↵2 Current affiliation: Division of Biostatistics, Indiana University School of Medicine, Indianapolis, Indiana.
-
↵3 Current affiliation: Mylan Pharmaceuticals, Inc., Morgantown, West Virginia.
- Accepted May 1, 2009.
- Received January 10, 2009.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}