


Abstract
Dexamethasone (DEX) is a potent and widely used anti-inflammatory and immunosuppressant glucocorticoid. It can bind and activate the pregnane X receptor (PXR), which plays a critical role as xenobiotic sensor in mammals to induce the expression of many enzymes, including cytochromes P450 in the CYP3A family. This induction results in its own metabolism. We have used a series of transgenic mouse lines, including a novel, improved humanized PXR line, to compare the induction profile of PXR-regulated drug-metabolizing enzymes after DEX administration, as well as looking at hepatic responses to rifampicin (RIF). The new humanized PXR model has uncovered further intriguing differences between the human and mouse receptors in that RIF only induced Cyp2b10 in the new humanized model. DEX was found to be a much more potent inducer of Cyp3a proteins in wild-type mice than in mice humanized for PXR. To assess whether PXR is involved in the detoxification of DEX in the liver, we analyzed the consequences of high doses of the glucocorticoid on hepatotoxicity on different PXR genetic backgrounds. We also studied these effects in an additional mouse model in which functional mouse Cyp3a genes have been deleted. These strains exhibited different sensitivities to DEX, indicating a protective role of the PXR and CYP3A proteins against the hepatotoxicity of this compound.
Dexamethasone (DEX) is a synthetic glucocorticoid that is used to treat a wide variety of medical conditions. It is a potent anti-inflammatory and immunosuppressant, which is metabolized primarily by the liver and undergoes renal excretion (Minagawa et al., 1986).
DEX induces cytochrome P450 (P450) genes, such as Cyp3a11 and Cyp2b10 in mice and CYP3A4 in humans (Watkins et al., 1985; Wrighton et al., 1985; Strom et al., 1996; Yanagimoto et al., 1997). The mechanism of this induction is complex and is mediated by a number of nuclear receptors. For example, the glucocorticoid receptor (GR) is essential for the DEX-mediated induction of Cyp2b proteins in mouse liver, but at high doses the induction of Cyp3a enzymes is mediated by other transcription factors (Schuetz et al., 2000). In addition, it has been reported that DEX induction of CYP3A4 in human hepatocytes is mediated by several transcription factors (Pascussi et al., 2001). At low (nanomolar) concentrations, CYP3A4 induction is mediated by a GR-dependent elevation of pregnane X receptor (PXR) expression, whereas at high (supramicromolar) concentrations, DEX directly binds to and activates PXR. In addition, submicromolar concentrations of DEX also enhance the expression of the constitutive androstane receptor (CAR) and the retinoid X receptor-α in human hepatocytes (Pascussi et al., 2000a,b). It has been shown that DEX is a more potent ligand for mouse PXR than the human receptor (Meehan et al., 1988; Moore et al., 2000), and hepatic induction of Cyp3a, but not Cyp2b, expression in mice is PXR-dependent (Schuetz et al., 2000; Scheer et al., 2008).
CYP3A plays a central role in DEX disposition in human liver because it catalyzes the formation of the two major metabolites, 6α- and 6β-hydroxy-dexamethasone (Gentile et al., 1996). Indeed, the relatively simple metabolic profile of DEX compared with many other substrates led to the proposal that DEX would be a useful in vivo probe for CYP3A4 activity (Gentile et al., 1996). Therefore, the ability of DEX to activate PXR in different species would be predicted to be a significant factor in determining both the metabolism and circulating levels of this glucocorticoid.
DEX can influence the toxicity of other compounds by enzyme induction. For example, pretreatment of rats with DEX (5–20 mg/kg, p.o.) 24 h before administration of the antitumor drug Yondelis [ecteinascidin 743 (ET-743); PharmaMar, Madrid, Spain] markedly reduced the biochemical and histopathological manifestations of Yondelis-induced liver changes (Donald et al., 2003). This protective effect was speculated to be a consequence of the decreased hepatic exposure to Yondelis linked to induction of metabolism by P450 enzymes.
On the other hand, pretreatment of mice with high doses of DEX (75 mg/kg i.p. for 4 days) increased acetaminophen-induced hepatotoxicity, and at this dose DEX was also hepatotoxic by itself (Madhu et al., 1992). At the therapeutic doses used in humans, DEX is not regarded as hepatotoxic. However, there are some occasional cases in which DEX administration is associated with severe hepatotoxicity. For example, DEX treatment has been associated with methotrexate hepatotoxicity in children in a manner unrelated to changes in methotrexate serum levels (Wolff et al., 1998).
The in vivo factors that determine the metabolism and toxicity of DEX are complex, and because of species differences in its interaction with nuclear receptors, such as CAR, PXR, and GR, studies in rodents might not accurately reflect the human situation. In this study, we have used mice either deleted or humanized at the Pxr gene locus, as well as mice in which seven functional Cyp3a genes have been deleted, to evaluate the roles of the human receptor and Cyp3a proteins in DEX response. As part of this work, we describe the generation of a new and improved humanized PXR model. The data presented show that mouse and human PXRs exhibit marked differences in their in vivo interactions with DEX, and the impact of these differences on DEX hepatotoxicity was analyzed in this study.
Materials and Methods
Animal Husbandry.
Mice were kept in Tecniplast (Buguggiate, Italy) Sealsafe microisolator cages. Food and water were available ad libitum. Light cycles were on a 13:11-h light/dark cycle with the light phasing starting at 6:00 AM. Temperature and relative humidity were maintained between 21 and 23°C and 45 and 65%.
Construction of the Targeting Vector Used for the Generation of an Improved PXR-Humanized Mouse Line.
Introns 6 and 7 of the human PXR gene were amplified by polymerase chain reaction (PCR) from a bacterial artificial chromosome carrying human PXR and subcloned into the targeting vector used for the generation of the PXR-humanized (huPXR) mice described previously (Scheer et al., 2008), to give rise to the targeting vector depicted in Fig. 1.

Strategy used to generate huPXR mice. A human PXR minigene, containing a fusion of exons 2 through 4, genomic sequences between exons 4 and 8, and a fusion of exons 8 and 9, was knocked in onto the translational start ATG of the mouse Pxr WT gene to generate PXR-targeted mice. Mouse exons are indicated in black vertical lines and with small type. Human exons are white boxes with capitals, and the targeting arms are shown as dark and light gray lines. Targeted mice were crossed to a mouse strain expressing the FLPe recombinase to delete the hygromycin selection cassette by FLP-mediated recombination at the FRT sites (white triangles) and to generate huPXR mice. For the sake of clarity, sequences of the targeting vector are not drawn to scale.
Generation and Molecular Characterization of Improved PXR-Humanized Mouse Line Targeted Embryonic Stem Cells.
Culture and targeted mutagenesis of embryonic stem (ES) cells were carried out as described previously (Hogan et al., 1994). The targeting vector was linearized with NotI and electroporated into a C57BL/6 mouse ES cell line. Of 132 hygromycin-resistant and fluorescence-negative ES cell colonies screened by standard Southern blot analyses, 12 correctly targeted clones were identified. Six of these were expanded, and further analysis by Southern blot analyses with different suitable restriction enzymes and 5′ and 3′ external probes and an internal hygromycin probe confirmed that these clones were correctly targeted at both homology arms and did not carry additional random integrations (data not shown).
Generation and Molecular Characterization of Improved PXR-Humanized Mice.
One of the improved PXR-targeted ES cell clones described previously was expanded, injected into BALB/c blastocysts, and transferred into foster mothers as described previously (Hogan et al., 1994). Litters from these fosters were visually inspected, and chimerism was determined by hair color. Highly chimeric animals were used for further breeding in a C57BL/6 genetic background. The hygromycin selection cassette was removed in vivo by crossing to an efficient FLP deleter (FLPe deleter) strain carrying a transgene that expresses FLPe in the germline (Fig. 1). The FLPe deleter has been generated in-house on a C57BL/6 genetic background. The improved PXR-humanized allele was detected by PCR with the primers 5′-GGACTTGCCCATCGAGGAC-3′ and 5′-ACAGGATGGAGGGGCAGC-3′, giving rise to a 332-base pair PCR fragment. PXR knockout (PXR KO) and huPXR mice as described previously (Scheer et al., 2008) were used in addition to the improved PXR-humanized mouse line (huPXRi) mice described here. Homozygously humanized and knockout mice for PXR were used for all the studies, and wild-type (WT) C57BL/6 animals for control experiments were purchased from Harlan UK Limited (Bicester, Oxon, UK).
Animal and Treatments.
All the animal procedures were carried out under a United Kingdom Home Office license, and all the animal studies were approved by the Ethical Review Committee, University of Dundee. Male, sexually mature huPXR, huPXRi, PXR KO, Cyp3a(−/−)/Cyp3a13(+/+), and WT (C57BL/6J) mice were dosed daily by intraperitoneal injection with the vehicle, rifampicin (RIF; 3, 10, 20, 40, or 60 mg/kg/day), or DEX (1, 3, 10, 30, or 60 mg/kg/day) for 4 days and sacrificed 24 h after the last dose.
Quantitative Reverse Transcriptase-PCR.
Human PXR RNA was analyzed by quantitative reverse transcriptase (qRT)-PCR (TaqMan). Total RNA was prepared from the liver using the QIAGEN (Hilden, Germany) RNA mini-kit. cDNA was synthesized from 1 μg of total RNA using the Quantiscript Reverse Transcriptase Kit (QIAGEN). Primers for human PXR were from the Assay-On-Demand Kit Hs00243666_ml (Applied Biosystems, Foster City, CA). qRT-PCR reactions were performed using TaqMan Universal PCR Mastermix in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems). Data were analyzed using comparative cycling time methodology, in which fluorescent output, measured as CT, was directly proportional to input cDNA concentrations. Input cDNA concentrations were normalized to murine β-actin. Calculations were performed by a comparative method (2−ΔΔCT).
Sequencing Analysis.
RT-PCR was performed using RNA isolated from a huPXRi mouse using oligos to amplify the potential full-length transcript. The oligos used were P6-F, 5′-GGA ATT CAA CAT GGA GGT GAG ACC CAA AG-3′; and human PXR (hPXR)-REV_3, 5′-TCA GCT ACC TGT GAT GCC GAA CAA C-3′. One-step RT-PCR reaction was conducted using Superscript RT-PCR Kit (Invitrogen, Carlsbad, CA). Sequence analysis was performed by Lark Technologies Ltd. (Takeley, UK). Alignments were performed using T-COFFEE (http://tcoffee.vital-it.ch/cgi-bin/Tcoffee/tcoffee_cgi/index.cgi) and Contig Express and Align-X programs of Vector NTI 8 software (Invitrogen).
Hepatic Microsomal Preparation.
Mouse livers used were freshly harvested, blotted, and weighed. The liver was scissor-minced in ice-cold KCl (1.15%; w/v) and then homogenized in ice-cold sucrose/EDTA/Tris-HCl (SET) buffer (0.25 M sucrose, 5 mM EDTA, and 20 mM Tris-HCl, pH 7.4) to make a 10% (w/v) homogenate solution (9 ml of SET buffer/1 g of liver). Microsomes were prepared by centrifugation first at 6441g [Heraeus 6445 rotor/Sorvall (Newton, CT) legend RT centrifuge] for 10 min at 4°C, and then the supernatant was spun at 13,864g (Sorvall F28/13 rotor) for 15 min at 4°C. The resulting supernatant was spun at 82,187g (Sorvall F28/13 rotor) for 90 min at 4°C, and the microsomal pellets were resuspended in ice-cold SET buffer and stored at −70°C.
Immunoblot Analysis.
For Western blot analysis, microsomal protein (5 μg) from pooled mouse samples (n = 3) was separated by SDS-polyacrylamide gel electrophoresis (PAGE), electrophoretically transferred to nitrocellulose membranes, and probed using an anti-rat CYP3A1 polyclonal antibody and anti-rat CYP2B1/2 polyclonal antibody (Biomedical Research Institute, University of Dundee) (Forrester et al., 1992) used at 1:2000 dilutions. The detection of mouse Cyp3a11 or Cyp2b10 by the corresponding rat antibodies was determined by Western blots using recombinant Cyp3a11 and Cyp2b10 proteins. The secondary antibody was anti-rabbit horseradish peroxidase conjugate, used at a dilution of 1:2000 (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Detection of immunoreactive proteins was performed by an enhanced chemiluminescence blot detection system (GE Healthcare). Chemiluminescence was captured by Gel Doc XR (Bio-Rad Laboratories, Hercules, CA). Protein bands were automatically detected, and their intensity was quantified by Quantity One software (version 4.60; Bio-Rad Laboratories).
P450 Enzyme Activity Assays.
Activity of Cyp2b10 in hepatic microsomes was monitored using the preferred Cyp2b10 substrate pentoxyresorufin. Pentoxyresorufin dealkylation (PROD) was monitored in a 1-cm3 cuvette containing 66 mM Tris-HCl buffer, pH 7.4, microsomes (20–50 μg), and 1 mM pentoxyresorufin. The reactions were initiated by the addition of 50 μM NADPH, and the rate of fluorescence was measured. As an internal control, each microsomal mixture was spiked with 10 μM resorufin.
Dealkylation of 7-benzyloxyquinoline (BQ; 20 mM), a Cyp3a11 preferred substrate, was measured in liver microsomes (20–50 μg) suspended in 66 mM Tris-HCl buffer, pH 7.4. Assays were initiated by the addition of 50 μM NADPH to the microsomal reaction and followed using a Hitachi (Tokyo, Japan) F-4500 fluorescence spectrophotometer with FL Solution 2.0 software. 7-Hydroxyquinoline (1 mM) was used as the internal standard.
Protein concentrations were measured using a modified version of the method of Lowry et al. (1951) with bovine serum albumin standards. Values were calculated and expressed as nanomoles per minute per milligram of protein or picomoles per minute per milligram of protein for the BQ and PROD assays, respectively.
Plasma Clinical Chemistry.
Blood was collected by cardiac puncture into lithium/heparin-coated tubes for plasma. After removal into suitable tubes for plasma preparation, venous samples were mixed on a roller for 10 min and cooled on ice. Red blood cells were removed by centrifugation (2000–3000 rpm for 10 min at 8–10°C). The supernatant (plasma) was stored at −70°C before analysis. Plasma samples were analyzed for alanine aminotransferase (ALT) using the COBAS Integra 400+ (Roche Diagnostics, Basel, Switzerland).
Liver Histopathology.
Two samples of liver, one from the left lobe and one from the median lobe, from different mice were taken and preserved in 4% neutral buffered formaldehyde. The tissue samples were subsequently embedded in paraffin wax and sectioned at a nominal thickness of approximately 5 μm. All the sections were stained with hematoxylin and eosin. The microscopic analysis and interpretation were carried out by an independent veterinary pathologist.
DEX Pharmacokinetics.
Concentrations of DEX in whole blood were measured by liquid chromatography/tandem mass spectrometry. Calibration standards were prepared in whole blood/water (1:1, v/v; 95 μl) by adding an appropriate amount of DEX in methanol/H2O (1:1, v/v; 5 μl). Then 10 mM ammonium acetate in methanol (80 μl) was added to 20 μl of the test samples and the calibration standards. The mixture was vortexed for 30 s and centrifuged at 13,000g for 6 min. The supernatant was transferred to a 96-well plate, and the concentrations of DEX were measured by liquid chromatography/tandem mass spectrometry from the calibration curve.
Statistics.
Statistical significance was assessed to determine differences between mouse groups using a two-tailed, paired, Student's t test. The criterion for statistical significance was p < 0.05.
Results
Generation of the New huPXR Mouse Model.
We recently described the generation of a huPXR mouse expressing the human receptor under control of the mouse Pxr promoter (Scheer et al., 2008). The DNA cassette used contained a fusion of exons 2 through 4, intron 4, exon 5, intron 5, and a fusion of exons 6 through 9 of the human PXR gene. This huPXR mouse expressed a functional human PXR protein and exhibited the predicted species differences in its interaction with drugs such as RIF and DEX. Furthermore, we showed that both the full-length and the most abundant splice variants identified in humans, hPXR SV2 (or hPXR.2), were also expressed (Dotzlaw et al., 1999; Hustert et al., 2001; Fukuen et al., 2002). However, in this model we found that an as-yet unidentified splice variant was detected. Further investigation showed that a splice acceptor site was created by the fusion of exons 7 and 8, which led to a variant where exon 5 was spliced out of frame to exon 8, resulting in a premature stop codon in exon 8. Because most of the ligand-binding domain is altered, these transcripts either code for a nonfunctional protein or, although unlikely, maybe even a dominant-negative protein interfering with the normally spliced PXR receptor. They also might be degraded by nonsense-mediated decay (see Schell et al., 2002 for review). Because it is difficult to measure how much of the truncated transcript is degraded, we cannot estimate the total amount of transcript wasted by aberrant splicing. Although it is not possible to quantify this effect, a fraction of the human PXR transcript will not code for a functional protein, potentially reducing the sensitivity of the model to enzyme inducers.
To avoid this possible problem, we have generated a new huPXR mouse line by altering the original targeting vector to include the two additional human introns 6 and 7. The new huPXR mouse line was generated by a knock-in strategy as shown in Fig. 1, such that a chimeric construct containing the cDNA representing exons 2 through 4 of hPXR missing the start CTG, genomic sequences between exons 4 and 8 and again cDNA of exons 8 and 9 followed by an SV40 polyA signal was introduced at the ATG of the mouse Pxr gene. This approach allowed the expression of the human receptor under control of the mouse Pxr promoter, and it deletes the endogenous Pxr gene. This model is referred to as huPXRi.
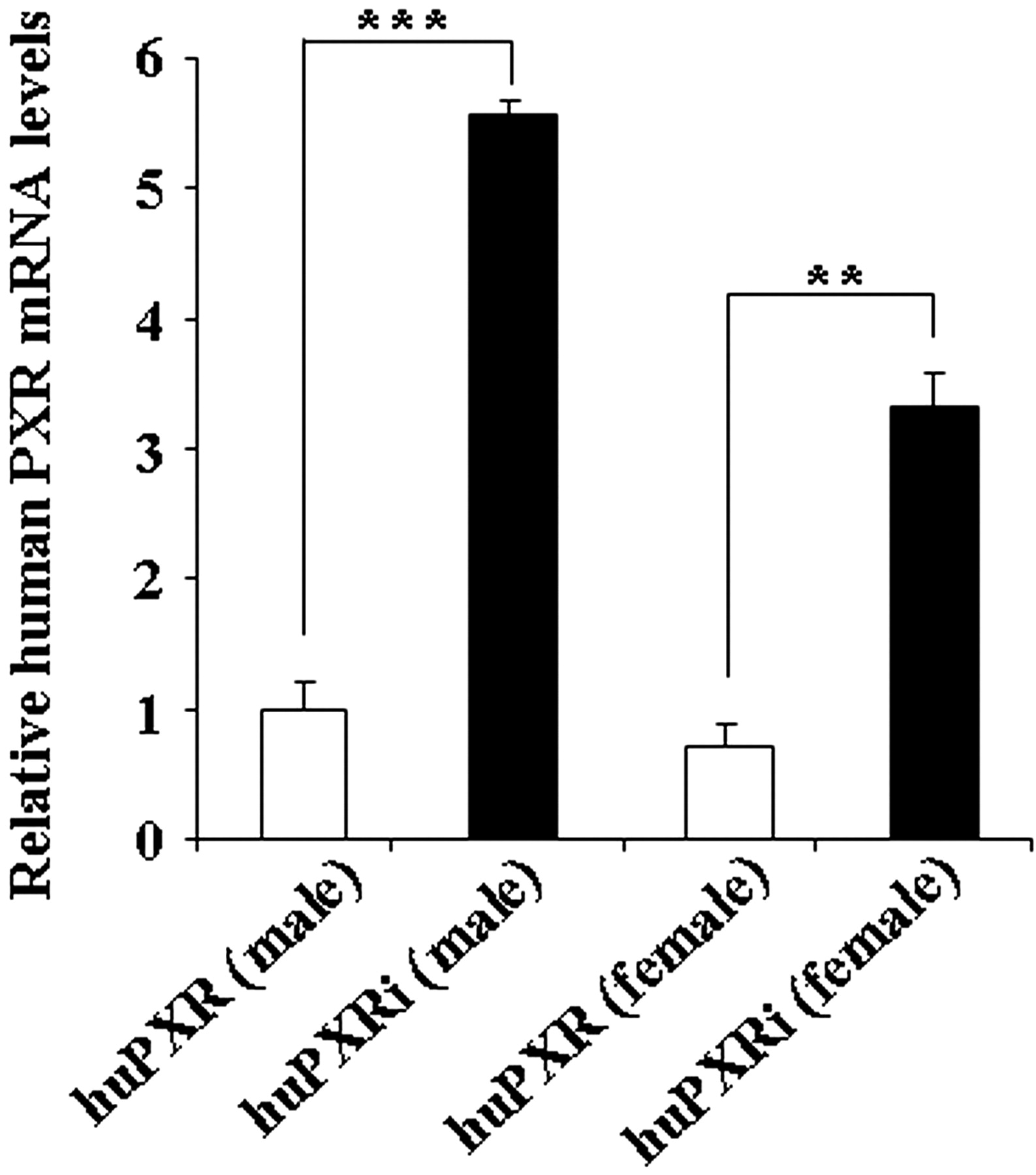
Comparison of the huPXR and huPXRi sequences obtained by RT-PCR from hepatic mRNA showed that both lines expressed the full-length and most abundant human splice isoform hPXR SV2 in the liver. However, the previously observed variant where splicing had occurred between exon 5 and exon 8 was not detected in the huPXRi line. In addition, relative expression levels of hPXR transcripts in male and female livers were quantified by RT-PCR (Fig. 2). Human PXR mRNA expression levels were increased by 5.6-fold in male huPXRi mice compared with the huPXR males. In females, the expression levels were slightly lower for both genotypes, and in huPXRi females, relative expression of human PXR mRNA was increased by 4.6-fold relative to huPXR mice of the same sex. The lower transcript levels in the huPXR mouse line might reflect the degradation of a fraction of the transcripts in this mouse line by nonsense-mediated decay. It was not possible to quantify the amount of full-length PXR protein because no suitable antibody is available.

Relative mRNA expression of hepatic human PXR in huPXR and huPXRi mice. RNA was isolated from liver samples of male and female huPXR and huPXRi mice (n = 3 per genotype and sex), and relative human PXR mRNA levels were assessed as described under Materials and Methods. Values represent the mean expression level ± S.E.M. relative to huPXR males. The human PXR mRNA expression levels were normalized to murine β-actin. A Student's t test (two-sided) was performed on the results, with *, **, and *** statistically different from control at p < 0.05, 0.01, and 0.001, respectively.
Induction Profiles of P450 Cyp3a and Cyp2b Proteins in RIF-Treated WT, huPXR, and huPXRi Mice.
To test whether the new PXR model had an altered sensitivity to PXR ligands, we treated WT, huPXR, and huPXRi mice with RIF, a compound showing higher affinity for the human versus the mouse receptor (Xie et al., 2000a). In agreement with our previous findings (Scheer et al., 2008), a dose of 10 mg/kg b.wt. resulted in a consistent Cyp3a11 induction in the huPXR mice but only a marginal induction in the WT mice (Fig. 3A). In the huPXRi mice, however, the induction at this dose was more pronounced than in both WT and huPXR mice. To quantify these differences, we measured the band intensities of Cyp3a11 in animals treated with 10 mg/kg RIF relative to the untreated control of the corresponding genotype. The increase in band intensity was 1.8-fold in the WT, 4.5-fold in the huPXR mouse line, and 7.7-fold in huPXRi animals. Furthermore, at a maximal dose of 60 mg/kg, the Cyp3a11 band intensity in the huPXRi mouse line was 1.9-fold higher compared with huPXR mice and 2.7-fold higher compared with the WT. Therefore, the huPXRi mouse line has an increased sensitivity to RIF-mediated Cyp3a11 induction compared with the huPXR mice both in the dose response and the magnitude of induction. The dose dependence exemplifying the difference in sensitivity between WT and huPXRi mice is shown in Fig. 3B. The differences observed by Western blotting were also reflected in the metabolism of the Cyp3a11 substrate BQ; a significant increase in metabolism was observed at a 10-mg/kg dose of RIF in huPXRi compared with WT mice (Fig. 3C). Namely, the increase of BQ metabolism at this dose was 1.5-fold in the WT, 1.9-fold in the huPXR mice, and 2.9-fold in huPXRi animals compared with the untreated controls of the corresponding genotype. As reported previously, a slight increase in the basal level of Cyp3a11 expression in both huPXR and huPXRi mice was observed (Scheer et al., 2008).

Effect of RIF on hepatic Cyp3a11 and Cyp2b10 expression in WT, huPXR, and huPXRi mice. Pooled liver microsomes (n = 3) from WT, huPXR, huPXRi (A) and WT and huPXRi mice (B), treated with either RIF or the vehicle, were analyzed for both Cyp3a11 and Cyp2b10 expression by immunoblotting. Microsomal protein (5 μg) was separated on 7.5% SDS-PAGE gel and subsequently immunoblotted with polyclonal Cyp3a11 and Cyp2b10 antibodies as described under Materials and Methods. C, BQ and PROD assays in pooled WT, huPXR, or huPXRi mouse liver microsomes (n = 3 mice). Values are expressed as mean ± S.D.; n = 3 mice for all the experiments. A Student's t test (two-sided) was performed on the results, with*, **, and *** statistically different from control at p < 0.05, 0.01, and 0.001, respectively.
A marked induction of Cyp2b10 expression was observed in huPXRi mice at a dose of 10 mg/kg RIF and a much higher induction at 60 mg/kg (Fig. 3A). The induction of this protein was substantiated by the marked increase in the Cyp2b10-specific PROD in the huPXRi mouse line (Fig. 3C). Consistent with the expression observed in Cyp2b10 protein, no induction of PROD activity was measured in WT and huPXR mice at 10 mg/kg RIF, and only a marginal increase is observed at 60 mg/kg. These data show the higher sensitivity of the new PXR model to induction, and consistent with the literature, RIF has been reported to be an inducer of CYP2B6 expression in human hepatocytes (Goodwin et al., 2001; Pichard-Garcia et al., 2002).
Comparison of P450 Induction by DEX in WT and Humanized Mice.
Characterization of the interaction of DEX with PXR is shown in Fig. 4. Cyp3a11 induction in WT mice was observed at much lower doses than in huPXRi mice (Fig. 4A). For example, at a dose of 3 mg/kg, a significant induction of Cyp3a11 was observed in WT animals but not in huPXRi mice. However, a similar level of induction was found at a 10-fold higher dose of 30 mg/kg. This similarity in the level of induction between the WT and huPXRi mice was also reflected in the rate of metabolism of the Cyp3a11 substrate BQ at the 30-mg/kg dose (Fig. 4B). We noted that relative to our original huPXR mice, treatment with DEX resulted in a much more marked consistent induction of Cyp3a11 in the new model. At a dose of 10 mg/kg, Cyp3a11 induction was only observed in huPXRi but not in huPXR mice, and at higher doses, such as 30 mg/kg, induction in huPXRi mice was more profound (see also Scheer et al., 2008). Therefore, in addition to an increased sensitivity and different pattern of induction in response to RIF, huPXRi compared with huPXR mice were also more sensitive to the more potent mouse inducer, DEX. The above effects were shown to be mediated by PXR because no induction of Cyp3a11 was seen in the PXR-null animals (Fig. 4A). In agreement with previous findings, Cyp2b10 induction by DEX is comparable between WT, huPXRi, and PXR KO mice and therefore is independent of PXR (Fig. 4A) (Schuetz et al., 2000; Scheer et al., 2008).

Effect of dexamethasone on hepatic Cyp3a11 expression in WT, huPXRi, and PXR KO mice. A, pooled liver microsomes (n = 3) from WT, huPXRi, and PXR KO mice, treated with either DEX or the vehicle, were analyzed for Cyp2b10 and Cyp3a11 expression by immunoblotting. Microsomal protein (5 μg) was separated on 7.5% SDS-PAGE gel and subsequently immunoblotted with polyclonal Cyp3a11 and Cyp2b10 antibodies as described under Materials and Methods. B, measurement of Cyp3a-related activity in pooled WT, huPXRi, or PXR KO mouse liver microsomes. Values are expressed as mean ± S.D.; n = 3. A Student's t test (two-sided) was performed, with*, **, and *** values statistically different from control at p < 0.05, 0.01, and 0.001, respectively.
Differential Expression of Markers for Hepatotoxicity in Control and Transgenic Mice.
In preliminary studies, we observed that DEX was more hepatotoxic in huPXR and PXR KO compared with WT mice, which suggests that PXR plays a role in the hepatotoxic effects of DEX (unpublished data). We have further investigated this observation in WT, PXR KO, huPXR, and huPXRi mice. To evaluate the role of the P450s, we also included a new mouse line in which the murine Cyp3a57, Cyp3a16, Cyp3a41, Cyp3a44, Cyp3a11, Cyp3a25, and Cyp3a59 genes have been deleted. These genes are in close proximity to each other on mouse chromosome 5 and distant from Cyp3a13, which is the only remaining functional Cyp3a gene in this mouse line. In agreement with the literature, we have found Cyp3a13 expression in mouse liver to be marginal compared with the most abundant Cyp3a11 protein (Yanagimoto et al., 1997; Anakk et al., 2003). Furthermore, it should be mentioned that a transcription profiling study of Cyp3a(−/−)/Cyp3a13(+/+) versus WT mice did not show any significant compensatory change of Cyp3a13 expression in the Cyp3a(−/−)/Cyp3a13(+/+) mouse line (data not shown). A more detailed analysis of this Cyp3a(−/−)/Cyp3a13(+/+) mouse line will be described elsewhere (manuscript in preparation). DEX administration (30 mg/kg i.p. daily for 4 days) significantly increased ALT levels compared with the corresponding untreated controls in all the mouse lines (Fig. 5A). The sensitivity to DEX-induced hepatotoxicity was in the order WT ≤ huPXRi < huPXR < PXR KO < Cyp3a(−/−)/Cyp3a13(+/+). At this dose, the average ALT levels were increased in the different mouse lines relative to the treated WT mice 1.2-fold (huPXRi), 2.2-fold (huPXR), 4.9-fold (PXR KO), and 8.1-fold [Cyp3a(−/−)/Cyp3a13(+/+)], respectively. However, because of the high variance between individuals from one group treated with 30 mg/kg DEX, statistical significance compared with the treated WT animals was only reached for the huPXR and Cyp3a(−/−)/Cyp3a13(+/+) mice. At a lower (10 mg/kg) dose of DEX, increases in ALT values were also observed with the same trend in sensitivity as described above. The Cyp3a(−/−)/Cyp3a13(+/+) mice were not tested at this dose. The same trend as for the ALT values was also seen for aspartate aminotransferase, although the variance between individuals was even higher, and, as a consequence, statistical significance was lower (data not shown).

Hepatotoxic effects of dexamethasone in WT, humanized, and knockout mice. A, plasma ALT levels in control animals or mice treated with either 10 or 30 mg/kg DEX. Group sizes are as follows: untreated [WT, n = 7; huPXRi, n = 6; huPXR, n = 4; PXR KO, n = 9; Cyp3a(−/−)/Cyp3a13(+/+), n = 4], 10 mg/kg DEX [WT, n = 6; huPXRi, n = 3; huPXR, n = 3; PXR KO, n = 3; Cyp3a(−/−)/Cyp3a13(+/+), not tested], 30 mg/kg DEX [WT, n = 10; huPXRi, n = 10; huPXR, n = 10; PXR KO, n = 12; Cyp3a(−/−)/Cyp3a13(+/+), n = 4]. Values are expressed as mean ± S.D. A Student's t test (two-sided) was performed on the results, with *, **, and *** statistically different from untreated control in each group at p < 0.05, 0.01, and 0.001, respectively. In addition, for equally treated mice, the dotted lines highlight the statistical difference between the corresponding genotypes. B, hematoxylin and eosin staining of representative liver sections (5 μm) taken from WT, huPXRi, huPXR, PXR KO, and Cyp3a(−/−)/Cyp3a13(+/+) mice treated with 30 mg/kg DEX. For all the images, a 20× objective lens was used.
The hepatotoxic effect of high doses of DEX (30 mg/kg) was also apparent after histological examination of liver sections in all the strains. Macrovesicular hepatocellular vacuolation that was partly associated with a peripheral dislocation of the usually centrally placed hepatocytic nuclei was observed in most treated mice of the different lines (Fig. 5B), and some individuals displayed necrosis of scattered single cells or subcapsular liver necrosis. Single hepatocyte necrosis in the treated group was not observed in any of the analyzed WT and huPXRi mice but in one huPXR mouse, two PXR KO, and three Cyp3a(−/−)/Cyp3a13(+/+) mice (n = 4 for each group), which strengthens the observation of different susceptibilities to DEX-induced hepatotoxicity as measured by ALT values.
DEX Pharmacokinetics in WT, PXR KO, and Cyp3a(−/−)/Cyp3a13(+/+) Mice.
In the above studies, DEX toxicity was observed after 4 days of administration, suggesting that the increased sensitivity of PXR KO and Cyp3a(−/−)/Cyp3a13(+/+) mice could be explained by an increased metabolism and detoxification in the WT mice as a result of a DEX-mediated induction of Cyp3a proteins in a PXR-dependent fashion. To test this possibility, a pharmacokinetic study was carried out in WT, PXR KO, and Cyp3a(−/−)/Cyp3a13(+/+) mice after 4 days of DEX treatment. In all three mouse strains relatively slow DEX absorption after intraperitoneal administration was observed, probably because of the low water solubility of this compound. Clear differences in DEX pharmacokinetics were observed in the different mouse lines, with an increase in maximal DEX serum concentrations in PXR KO and Cyp3a(−/−)/Cyp3a13(+/+) mice (Fig. 6A). Statistically significant differences in areas under the concentration versus time curve (AUCs) between WT, PXR KO, and Cyp3a(−/−)/Cyp3a13(+/+) mice were also observed (Fig. 6B). It has been shown that after intraperitoneal administration of an isoform-specific P450 substrate, the ratio of the expression level of the corresponding P450 in the liver is inversely proportional to the ratio of the AUCs of the substrate (Lukas et al., 1971; Gibaldi and Perrier, 1982). Thus, the AUC difference suggested that after induction with DEX, enzymes responsible for DEX metabolism were expressed at a lower level in the Cyp3a(−/−)/Cyp3a13(+/+) and PXR KO mice compared with the WT. These results are consistent with the lack of induction of Cyp3a proteins in the DEX-treated PXR KO mice (Fig. 4). These data also show that CYP3A enzymes play an important role in the metabolism of DEX. The changes in pharmacokinetics are also consistent with the hypothesis that the higher susceptibility of the PXR KO and Cyp3a(−/−)/Cyp3a13(+/+) lines to DEX-induced hepatotoxicity is a result of an increased exposure to the parent compound.

DEX pharmacokinetics in WT, PXR KO, and Cyp3a(−/−)/Cyp3a13(+/+) mice. A, plasma concentration curve after four daily treatments with DEX at 30 mg/kg i.p. B, total AUC calculated from the DEX pharmacokinetics. Experimental details are given under Materials and Methods. Data are mean ± S.D. [n = 3 for Cyp3a(−/−)/Cyp3a13(+/+); n = 4 for other strains]. AUCs were compared using an unpaired t test (two-tailed p values), with *, **, and *** statistically different from the WT at p < 0.05, 0.01, and 0.001, respectively.
Discussion
As part of this report we have compared the properties of two mouse lines humanized for the transcription factor PXR that only differ in that in the original model (Scheer et al., 2008) a cryptic splice acceptor site was generated as part of the vector construction. Although the full-length human PXR mRNA was expressed in both models and the expression of hPXR was driven off identical promoter sequences, there were some significant differences between these mouse lines in both the level of PXR expression and its capacity to regulate downstream genes in response to ligands such as RIF and DEX. This result suggests that alternative splicing of PXR can be an important factor in determining the level of functional PXR receptor (Hustert et al., 2001; Fukuen et al., 2002). What was also particularly interesting was that in the new huPXR model RIF was a good inducer of Cyp2b10 and that this protein was not induced in either WT or the huPXR mouse line. This result indicates that the differential expression of PXR will not only affect the amplitude of induction but also which genes are induced. The induction of Cyp2b genes by RIF is consistent with previous studies showing the induction of CYP2B6 in human hepatocytes by this compound (Goodwin et al., 2001; Pichard-Garcia et al., 2002). Although DEX elevates Cyp3a11 expression in a PXR-dependent manner, Cyp2b10 induction by this compound does not require PXR and might at least in part be the consequence of an enhanced expression of CAR and the retinoid X receptor-α in response to DEX (Pascussi et al., 2000a,b). Taken together, the data show that regulation of Cyp2b10 (and possibly CYP2B6 in humans) is complex, and it involves several transcription factors such as CAR (Honkakoski et al., 1998; Wei et al., 2000), GR (Schuetz et al., 2000), and PXR (Xie et al., 2000b; Wagner et al., 2005). Furthermore, depending on the compound, the activation of a given receptor such as PXR can either be required for Cyp2b10 induction (RIF) or nonessential (DEX).
The presented studies revealed a hepatotoxic effect of high doses of the glucocorticoid DEX in mice. At 30 mg/kg, a statistically significant increase in ALT levels could be observed in WT, huPXR, huPXRi, PXR KO, and Cyp3a(−/−)/Cyp3a13(+/+) mice compared with their corresponding untreated controls. The liver-damaging effect was also confirmed by the induction of macrovesicular hepatocellular vacuolation and different degrees of necrosis in histological examination. This effect is in agreement with the previously observed hepatotoxicity of high doses of DEX in mice (Madhu et al., 1992).
There is currently significant interest in the potential use of humanized and knockout animal models as more reliable predictors of pathways of drug metabolism and chemical safety in humans (Cheung and Gonzalez, 2008; Lin, 2008; Scheer et al., 2008). This interest is based on the increasing evidence that there are species differences in both nuclear receptors that regulate the expression of drug-metabolizing enzymes and the enzymes themselves. There is also evidence that these differences can lead to both increased sensitivity to some molecules or reduced sensitivity to others, such as certain nongenotoxic carcinogens (Zhang et al., 2002; Cheung et al., 2004; Morimura et al., 2006; Gonzalez and Shah, 2008).
One important facet of chemical safety screening is that molecules, as a consequence of their interaction with nuclear receptors such as PXR and CAR, can induce their own metabolism or that of other molecules administered concomitantly. This induction of metabolism can result in reduced pharmacological efficacy or, if the parent molecule is toxic, reduced toxicity or, if the parent molecule is metabolized to a reactive metabolite, increased toxicity. Therefore, species differences in the capacity of compounds to activate rodent or human receptors can result in marked differences in their pharmacological or toxicological effects.
Using a relatively small number of transgenic animals, significant insights into the pathways regulating DEX metabolism and toxicity could be obtained. The use of mice deleted at the Cyp3a gene locus showed the importance of the P450 metabolic pathway in DEX disposition and toxicity. The increased toxicity and reduced metabolic clearance of DEX in Cyp3a(−/−)/Cyp3a13(+/+) mice provide evidence that metabolism of this compound results in detoxification and that Cyp3a enzymes are involved. The fact that similar results regarding toxicity and metabolism were obtained in PXR KO mice also strengthened this evidence and showed that enzyme induction via PXR is important. Therefore, DEX induces its own metabolism, leading to an increased rate of detoxification in mice.
This observation then raised the question whether hPXR interacts in a similar manner to murine PXR? In this regard, DEX was a weaker inducer of Cyp3a protein expression in huPXRi mice than in WT animals. With regard to hepatotoxicity, there was no difference between WT and huPXRi mice at the 30-mg/kg doses of DEX, which led to strongly increased ALT levels in the Cyp3a(−/−)/Cyp3a13(+/+) and PXR KO mice. The reason is probably, at least in part, because induction of Cyp3a proteins at this dose was almost the same in WT and huPXRi mice.
However, the doses of DEX required to induce hepatotoxicity are not representative of the therapeutic doses used in humans. Even in shock therapy, usually only 4 to 8 mg is injected intravenously, and if necessary this is repeated to a total maximal dose of 24 mg. Therefore, the dose of up to 30 mg/kg b.wt. used in this study exceeds the maximal human dose by 2 orders of magnitude. In addition, differences in the hepatic blood flow rates between mice and humans might contribute to potential variations in hepatic sensitivity to this glucocorticoid. Nevertheless, these studies exemplify the fact that species differences in the interaction of compounds with PXR can alter the metabolism and potentially the toxicological profile of drugs and chemicals, which could result in differences in the pathways of metabolism and susceptibility to hepatotoxicity between animals and humans.
Acknowledgments.
We thank Sylvia Krüger, Anja Müller, Oliver Dahlmann (TaconicArtemis, Cologne), and Sarah Waugh (CXR Biosciences, Dundee).
Footnotes
This work was supported by ITI Life Sciences, Scotland.
Conflict of interest: N.S. and A.R. receive income from TaconicArtemis, and J.R., Y.K., and C.R.W. receive income from CXR Biosciences. The authors have declared that no conflict of interest exists.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.109.031872.
-
ABBREVIATIONS:
- DEX
- dexamethasone
- P450
- cytochrome P450
- GR
- glucocorticoid receptor
- PXR
- pregnane X receptor
- CAR
- constitutive androstane receptor
- ET-743
- ecteinascidin 743
- huPXR
- humanized pregnane X receptor mice
- PCR
- polymerase chain reaction
- ES cell
- embryonic stem cell
- FLPe
- efficient FLP
- PXR KO
- pregnane X receptor knockout mice
- huPXRi
- improved humanized pregnane X receptor mice
- WT
- wild-type
- RIF
- rifampicin
- qRT
- quantitative reverse transcriptase
- hPXR
- human pregnane X receptor
- PAGE
- polyacrylamide gel electrophoresis
- PROD
- pentoxyresorufin dealkylation
- BQ
- 7-benzyloxyquinoline
- ALT
- alanine aminotransferase
- AUC
- area under the concentration versus time curve
- SET
- sucrose/EDTA/Tris-HCl.
- Received December 18, 2009.
- Accepted March 30, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}