


Abstract
The epidermal growth factor receptor tyrosine kinase inhibitor erlotinib (ERL) is approved for treatment of non–small-cell lung cancer. Numerous reports of ERL-associated toxicities are consistent with immune-mediated toxicity, including drug-induced hepatitis, interstitial lung disease, Stevens-Johnson syndrome, and toxic epidermal necrolysis. Although the mechanism of toxicity has not been established, we present evidence that reactive intermediates are formed during the metabolism of ERL, which can covalently conjugate to the cysteine group of the peptide-mimetic GSH. Seven ERL-GSH conjugates were identified in incubations with hepatic microsomes. Cytochrome P450 (P450)-dependent adducts are proposed to be formed via reactive epoxide and electrophilic quinone-imine intermediates. In incubations of human liver microsomes, intestinal microsomes, pulmonary microsomes, and recombinant P450s, CYP3A4 was the primary enzyme responsible for the bioactivation of ERL; however, CYP1A1, CYP1A2, CYP3A5, and CYP2D6 were capable of catalyzing the bioactivation as well. During the metabolism of ERL, CYP3A4 and CYP3A5 are irreversibly inactivated by ERL in a time- and concentration-dependent manner. Inactivation was not dependent on oxidation of the ERL alkyne group to form a reactive oxirene or ketene, as shown by synthesizing analogs where the alkyne was replaced with a cyano group. CYP1A1, CYP1A2, and CYP2D6 were not inactivated despite catalyzing the formation of ERL-GSH adducts.
Erlotinib (ERL) is a reversible inhibitor of the epidermal growth factor receptor tyrosine kinase (HER1/EGFR) and was approved for the second- and third-line treatment of non–small-cell lung cancer in 2005 (Siegel-Lakhai et al., 2005). Clinical trials indicate that ERL provides a survival benefit after failure of first- or second-line chemotherapy as a single agent and in the treatment of advanced pancreatic adenocarcinomas together with chemotherapy (Tang et al., 2006; Moore et al., 2007). Although having therapeutic benefit, treatment with ERL has been associated with life-threatening adverse effects, including drug-induced hepatitis (Liu et al., 2007b; Ramanarayanan and Scarpace, 2007; Saif, 2008; Pellegrinotti et al., 2009), interstitial lung disease (Liu et al., 2007a; Makris et al., 2007), and the severe skin disorders Stevens-Johnson syndrome and toxic epidermal necrolysis (Chou et al., 2006; Lübbe et al., 2008; Bovenschen and Alkemade, 2009). In September 2008, OSI Pharmaceuticals and Genentech (www.fda.gov/downloads/safety/medwatch/safetyinformation/safetyalertsforhumanmedicalproducts/ucm135238) reported a pharmacokinetic study of 15 patients with advanced solid tumors and moderate liver impairment. During the study, one patient died from hepatorenal syndrome and another died because of progressive liver failure, and both deaths were attributed to ERL.
In humans, ERL is extensively metabolized, predominantly by CYP3A4 and to a lesser extent by CYP1A2 and the inducible isoform CYP1A1 (Ling et al., 2006; Li et al., 2007), with metabolites excreted by the biliary system (∼75%). There are three primary routes of ERL metabolism: O-demethylation of the side chain with or without further oxidation to the carboxylic acid, oxidation of the acetylene moiety followed by further hydrolysis to the aryl carboxylic acid, and 4-hydroxylation of the phenyl-acetylene moiety (Ling et al., 2006). 4-Hydroxylation forms a para-hydroxylated metabolite on the phenyl ring, resulting in the formation of a para-hydroxyaniline (Li et al., 2007), which has the potential to undergo cytochrome P450 (P450)-mediated two-electron oxidation to form a reactive quinine-imine.
In this study, we have monitored quinone-imine formation using GSH trapping. Compounds capable of reacting with GSH would be expected to be able to react with other cellular sulfhydryls such as in cysteine residues of protein. To date, ERL has not been reported to be metabolized to form reactive intermediates capable of reacting with biomolecules. We have shown the formation of GSH adducts during the oxidative metabolism of ERL, determined the site of bioactivation, and identified the individual human P450 enzymes involved. ERL was also found to inactivate CYP3A4 and CYP3A5 in a time- and concentration-dependent manner, whereas CYP1A1 and CYP2D6 were not inactivated despite these enzymes forming GSH-reactive metabolites. Finally, we showed that adduct formation and enzyme inactivation were not dependent on oxidation of the acetylene by using synthesized ERL analogs where ethyl and cyano groups replaced the acetylene.
Materials and Methods
Materials.
Pooled human liver microsomes (HLMs), human intestinal microsomes, human pulmonary microsomes (smoker and nonsmoker), and recombinant P450 + reductase Bactosomes were purchased from XenoTech, LLC (Lenexa, KS). Recombinant human microsomal epoxide hydrolase was purchased from BD Gentest (Woburn, MA). Formic acid, dimethyl sulfoxide (DMSO), and acetonitrile were purchased from Thermo Fisher Scientific (Waltham, MA). ERL was purchased from LC Laboratories (Woburn, MA). Midazolam, carbamazepine, α-naphthoflavone, phenacetin, diclofenac, (S)-mephenytoin, and ketoconazole were from Sigma-Aldrich (St. Louis, MO).
Analytical Conditions.
Liquid chromatography/tandem mass spectrometry analyses of GSH adducts were performed on an API 4000 Q-Trap mass spectrometer equipped with a TurboIonSpray source (Applied Biosystems, Foster City, CA) using a negative precursor ion scan of m/z 272 (Dieckhaus et al., 2005) and conditions described previously (Li et al., 2009). Chromatographic separation was achieved by using an Agilent Technologies (Santa Clara, CA) Eclipse XDB C18 column (3.5 μm, 3.0 × 150 mm). High-performance liquid chromatography (HPLC) conditions used a flow rate of 0.4 ml/min with mobile phase A, water with 0.1% formic acid, and mobile phase B, acetonitrile with 0.1% formic acid. A gradient elution was used starting with 5% solvent B for 3 min; then solvent B was rapidly ramped to 10% in 0.5 min, followed by 10 to 50% B in 19.5 min, and 50 to 80% B in 5 min. At 28 min, the column was flushed with 80% B for 2 min and re-equilibrated to initial conditions. Structural information was generated from collision-induced dissociation spectra. Metabolites and GSH adducts were verified by comparing incubated samples with control samples without NADPH, trapping agent, or substrate.
To improve detection sensitivity and specificity, metabolites and GSH adducts were also characterized using multiple reaction monitoring (MRM) triggered enhanced product ion scans (MRM–information-dependent acquisition–enhanced product ion) following preset MRM transitions. The MRM transitions were set to the most intense ion pairs for each adduct, m/z 701.3→428.2, 715.3→442.2, and 717.3→444.2, with the following source settings: declustering potential, 70 V; collision energy, 40 eV; and collision energy spread, ±20 eV. The hydroxylaniline metabolite of ERL was followed using m/z 410.2→294.1, and carbamazepine (m/z 237.3→194.2) was used as an internal standard. NMR analysis was recorded on a BRUKER AXS, Inc. (Madison, WI) AV-400 NMR in deuterated DMSO, and high-resolution mass spectrometry was performed on an Orbitrap mass spectrometer (Thermo Fisher Scientific).
Microsomal Incubations.
Pooled HLMs and recombinant P450 were thawed on ice. ERL (40 μM from a DMSO stock) was mixed with HLM or recombinant enzyme (2 mg/ml protein for microsomes or 100 pmol/ml for recombinant P450) in 100 mM potassium phosphate buffer, pH 7.4, fortified with 5 mM GSH. The final concentration of organic solvent in the incubations was 0.2% (v/v). Incubations were performed at 37°C in a shaking incubator. After a 4-min preincubation at 37°C, reactions were initiated by the addition of 1 mM NADPH. Reactions were stopped by the addition of an equal volume of acetonitrile (with or without internal standard added, depending on analysis purpose) after 60 min. Control samples containing no NADPH or substrate, or control samples with heat-denatured HLM or blank phosphate buffer were included. Where indicated, ketoconazole (selective CYP3A4/5 inhibitor) at a final concentration of 1 μM, α-naphthoflavone (CYP1A1/2 inhibitor) at 20 μM, or microsomal epoxide hydrolase at 1 mg/ml was added to the incubations. Samples were centrifuged at 10,000g for 10 min at 4°C to pellet proteins, and supernatants were dried down by SpeedVac (Thermo Fisher Scientific) and reconstituted in 100 μl of 30% acetonitrile.
Time- and Concentration-Dependent Inactivation of P450s.
Time- and concentration-dependent loss of CYP3A4 activity in the presence of ERL was determined by midazolam 1′-hydroxylase activity. Primary incubations included ERL (0, 5, 10, 20, and 40 μM), 1 mM NADPH, 0.5 mg/ml HLM, 3 mM MgCl2, and 0.1 M potassium phosphate buffer, pH 7.4. The mixture was incubated in a 37°C shaking incubator for various time points (0, 4, 8, 15, 22, and 30 min). At each preincubation time point, aliquots (10 μl) of the primary incubation mixtures were transferred to a secondary incubation with a final volume of 200 μl. Secondary incubations had a final concentration of 20 μM midazolam, 1 mM NADPH, 3 mM MgCl2, and 0.1 M potassium phosphate, pH 7.4, and were incubated at 37°C for 5 min and stopped by the addition of acetonitrile (1:1, v/v). All the samples were analyzed as described previously (Li et al., 2009). Other major P450s—CYP1A1, CYP1A2, CYP2D6, CYP2C9, and CYP2C19—were similarly evaluated according to validated assays (Perloff et al., 2009) with slight modification.
Nonspecific binding was evaluated by determining the percentage of unbound ERL under incubation conditions. Protein concentrations were chosen to reflect those for the primary incubations in the time-dependent inactivation studies. ERL concentrations were similar to the experimentally determined Ki (HLM, 20 μM; recombinant CYP3A4, 10 μM; and recombinant CYP3A5, 40 μM). ERL and enzyme were allowed to incubate 37°C for 20 min. Triplicate samples were centrifuged at 100,000 rpm (436,000g) in a Beckman Coulter (Fullerton, CA) TLA-120.2 rotor for 240 min at 4°C. Unbound ERL was determined by sampling 1 to 2 mm below the supernatant surface and comparing ERL levels with uncentrifuged samples.
Purification of para-Hydroxyerlotinib.
para-Hydroxyerlotinib was purified from 1-ml incubations of HLMs (1 mg/ml) containing 40 μM ERL and 1 mM NADPH in a final volume of 1 ml. The reaction was stopped after 1 h by addition of 150 μl of 10% trichloroacetic acid. Metabolites were isolated by solid-phase extraction (Strata C18-E, 300 mg/3 ml cartridges; Phenomenex, Torrance, CA) and further purified by manual collection of HPLC peaks.
Plasma and Tissue Concentration after Oral Dosing of ERL.
Tissue distribution of ERL was evaluated in C57BL6 mice (n = 3) dosed orally with ERL, 10 mg/kg. After 2 h, blood, liver, lung, and brain were collected. Tissues were not perfused to reduce the risk that ERL would be eluted from the tissue during the perfusion process. Plasma was generated using standard centrifugation techniques, and the plasma and tissues were frozen at −80°C. Plasma and tissues were mixed with acetonitrile (1:5 v/v or 1:5 w/v, respectively), sonicated with a probe tip sonicator, and analyzed for drug levels by liquid chromatography/tandem mass spectrometry. All the procedures were conducted in the Scripps vivarium, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and were approved by the Scripps Florida Institutional Animal Care and Use Committee.
Synthesis of ERL Analogs.
ERL analogs were prepared using the following general protocol. A mixture of aniline (3-ethyl aniline or 3-cyanoaniline) and commercially available 4-chloro-6,7-dimethoxyquinazoline (American Custom Chemicals Corporation, San Diego, CA) was heated in isopropanol at 90°C overnight. After cooling to 25°C, the precipitates were filtered, washed with isopropanol and ether, and dried in vacuo to give the products as near colorless solids, >95% pure as judged by analytical HPLC and liquid chromatography/mass spectrometry analysis. Products were confirmed by 1H NMR analysis.
Results
ERL Tissue Distribution.
The major site of ERL metabolism is the liver. When dosed in mice, ERL concentrations were 3-fold higher in the liver compared with the plasma. Lung concentrations did not show compound accumulation, and brain levels are significantly below the level in plasma (Table 1). With standard therapy, daily 150-mg oral dose, the clinical ERL Cmax is 6 to 8 μM (Rudin et al., 2008). If the liver/plasma ratio measured in mice is representative of humans, concentrations of 15 to 25 μM would be expected in the liver, and higher levels may be observed in select individuals, including those with hepatic impairment or in patients taking medication that inhibits CYP3A. Most of the experiments presented in this study were conducted using 20 or 40 μM ERL.
ERL tissue distribution
Drug levels were determined 2 h after a single 10-mg/kg oral dose in C57BL/6 mice. Tissues were not perfused to ensure ERL was not washed out during perfusion.
Time- and Concentration-Dependent Inactivation of P450 by ERL.
Preliminary experiments were conducted to investigate time-dependent inhibition of individual P450s in HLMs by 80 μM ERL. Assessment of remaining P450 activity after 30 min showed that CYP3A4/5 activity was sharply decreased during the 30-min incubation with ERL, whereas CYP1A2, CYP2D6, CYP2C9, and CYP2C19 activities were essentially unchanged. More extensive experiments were carried out to evaluate the kinetics of CYP3A4/5 inactivation in HLMs by ERL. Midazolam 1′-hydroxylation was used as a measure of CYP3A4/5 activity. As shown in Fig. 1A, preincubation of ERL with HLM in the presence of NADPH decreased CYP3A4/5 activity in a time- and concentration-dependent manner. The observed first-order rate constants (Kobs) of the inactivation reaction were calculated for specific ERL concentrations. The hyperbolic plot of Kobs versus ERL concentrations is shown in Fig. 1B. The Kinact was calculated to be 0.09/min and KI was 22 μM. Parallel incubations were set up where ERL was preincubated with CYP3A4 in the absence of NADPH. No inactivation was observed in the absence of NADPH, and CYP3A4/5 activity was not protected by the presence of 5 mM GSH or catalase/superoxide dismutase (Fig. 1C). The reversibility of time-dependent inhibition was assessed by dialysis. ERL incubations with HLM ± NADPH were sampled at 0 and 20 min, and 150-μl aliquots were dialyzed against 2 liters of phosphate buffer (0.1 M, pH 7.4) for approximately 8 h (fresh buffer at 4 h) at 4°C with constant stirring. Before and after dialysis, CYP3A4/5 activity was determined for all the conditions using midazolam hydroxylation as a marker for CYP3A4/5 activity. CYP3A4/5 activity was not recoverable by dialysis (data not shown).

Time- and concentration-dependent inactivation of CYP3A4 by ERL. Incubations containing 0.5 mg/ml HLM and 1 mM NADPH in 100 mM phosphate buffer, pH 7.4, were incubated with the following ERL concentrations: 0, 5, 10, 20, and 40 μM. A, at the indicated time points, the remaining CYP3A4 activity was measured by a midazolam hydroxylation assay. Each point represents the mean of triplicate incubation. The observed inactivation rate constants, Kobs, were calculated from the slopes of the regression lines in A. B, the hyperbolic plot of Kobs versus ERL concentration was used to calculate kinetic constants. Potential preservation of CYP3A4 activity by the addition of 5 mM GSH or 1000 units of superoxide dismutase and catalase to incubations containing 0.5 mg/ml HLM, 1 mM NADPH, and 20 μM ERL was evaluated (C).
Recombinant CYP3A4, CYP3A5, and CYP1A1 were used to further assess ERL-induced P450 inactivation. Recombinant CYP3A4 was inactivated and had similar kinetic constants as observed in HLMs: Kinact = 0.10/min and KI = 9 μM. CYP3A5 inactivation was more complicated and appeared to have two phases. The initial phase corresponded to Kinact = 0.18/min and KI = 40 μM, but at approximately 30% remaining activity, the rate of inactivation was greatly decreased despite more than 70% of the initial ERL concentration remaining. CYP1A1 showed no evidence of inactivation.
Nonspecific binding was determined under conditions similar to the primary time-dependent incubations for HLM and recombinant CYP3A4 and CYP3A5 with ERL concentrations approximately equal to the determined KI, 20, 10, and 40 μM, respectively. The fraction of free ERL was 12% for HLM, 14% for recombinant CYP3A5, and 35% for recombinant CYP3A4.
GSH Adducts of ERL.
GSH was used as a representative cellular nucleophile. Seven GSH adducts were identified in NADPH-supplemented HLM incubations containing ERL and GSH, as shown in Fig. 2A. Four of the adducts were generated by P450 and were NADPH-dependent, whereas three adducts (ERL-G3, ERL-G4, and ERL-G6) were also detected in control incubations and even when ERL and its metabolites were incubated with GSH in buffer (Fig. 2B).

Formation of ERL-GSH adducts. A, ERL-GSH adducts were detected in 1-h incubations containing 2 mg/ml HLM, 1 mM NADPH, 40 μM ERL, and 5 mM GSH in 100 mM phosphate buffer, pH 7.4. Parallel incubations to those in A were prepared without GSH; after 1 h, the solution was directly applied to a solid-phase extraction column. The column was washed with 20 bed volumes of water and eluted with acetonitrile. The acetonitrile was evaporated, and ERL and its metabolites were reconstituted in phosphate buffer with 5 mM GSH and incubated at 37°C for 4 h (B).
Identification of Enzyme-Dependent ERL-GSH Adducts.
Four ERL-GSH adducts—ERL-G1 (m/z 715.3), ERL-G2 (m/z 701.3), ERL-G5 (m/z 715.3), and ERL-G7 (m/z 715.3)—were detected in incubations containing HLM and NADPH but were not identified in control incubations, suggesting these adducts were derived from NADPH-dependent bioactivation of ERL. Three adducts had m/z of 715.3, and all had similar fragmentation patterns. The spectrum for the most abundant adduct, ERL-G5, is shown in Fig. 3. The molecular ion of m/z 715.3 is consistent with attachment of the glutathionyl moiety to the monooxygenated metabolite of ERL (ERL + GSH + O-2H). Characteristic −129 Da fragments, resulting from the neutral loss of pyroglutamate, were observed for all the adducts. The major GSH adduct, ERL-G5, had a molecular ion mass of 715.241, which is consistent with the mass of the proposed metabolite ion, 715.240 (data not shown). Previous in vitro and human clinical studies report a major para-hydroxylated metabolite (Ling et al., 2006), and the three monohydroxylated-GSH adducts are likely caused by displacement of the three aromatic hydrogen.

Tandem mass spectrometry spectrum of the MH+ ion m/z 715.3 of ERL-G5. The origins of the characterized ions are as indicated.
ERL-G2 had a molecular ion mass of 701.3 and is proposed to be a demethylated secondary metabolite related to adducts G1, G5, and G7. Although only one peak was detected, it is likely that there are demethylated secondary metabolites of all the GSH adducts. It is possible that some of the secondary metabolites were below the level of detection or they were not chromatographically resolved.
Identification of ERL-GSH Adducts Independent of Enzyme and NADPH.
Two adducts, ERL-G3 (m/z 717.3) and ERL-G6 (m/z 701.3), were identified in incubated and control incubations without NADPH or when the microsomes were heat-inactivated by boiling for 10 min. The molecular ion of ERL-G6, m/z 701.3, suggested the direct addition of GSH (ERL + GSH), and high-resolution mass spectroscopy found a mass of m/z 701.263, which is consistent with the theoretical m/z 701.262 (data not shown). Sample processing methods had an obvious effect on the formation of enzyme-independent adducts. If samples containing ERL and GSH were directly concentrated using a SpeedVac (Thermo Fisher Scientific), the amount of adduct was significantly higher than in samples where the concentration of GSH was depleted by solid-phase extraction before SpeedVac (data not shown). Likewise, overnight incubation of 4 mg/ml ERL and 200 mM GSH at 37°C in the absence of enzyme resulted in the formation of a GSH-ERL adduct with identical mass and retention time as ERL-G6 in sufficient quantities for an H NMR spectrum (Fig. 4). GSH appeared to be covalently conjugated to the alkyne group forming cis/trans isomers, which led to extensive splitting of the vinyl protons (the NMR signal for the alkyne hydrogen of unconjugated ERL is a singlet). A second GSH-ERL adduct consistent with ERL-G3, m/z 717.3 (ERL + GSH + O), may be the result of oxidation of the sulfur of the attached glutathionyl moiety, but levels of ERL-G3 were too low for NMR analysis.

H NMR spectrum of ERL-G6.
Although ERL-G6 and ERL-G3 could be formed in the absence of enzyme and NADPH, both adducts were formed in greater amounts when enzyme and NADPH were included in the incubation. This was particularly true with recombinant P450s and may be caused by a nonspecific free radical reaction because addition of superoxide dismutase/catalase inhibited the formation of this adduct (see Fig. 5). It is unclear whether the alkyne group of ERL would react with nucleophiles in a biological setting or whether this is an in vitro artifact.
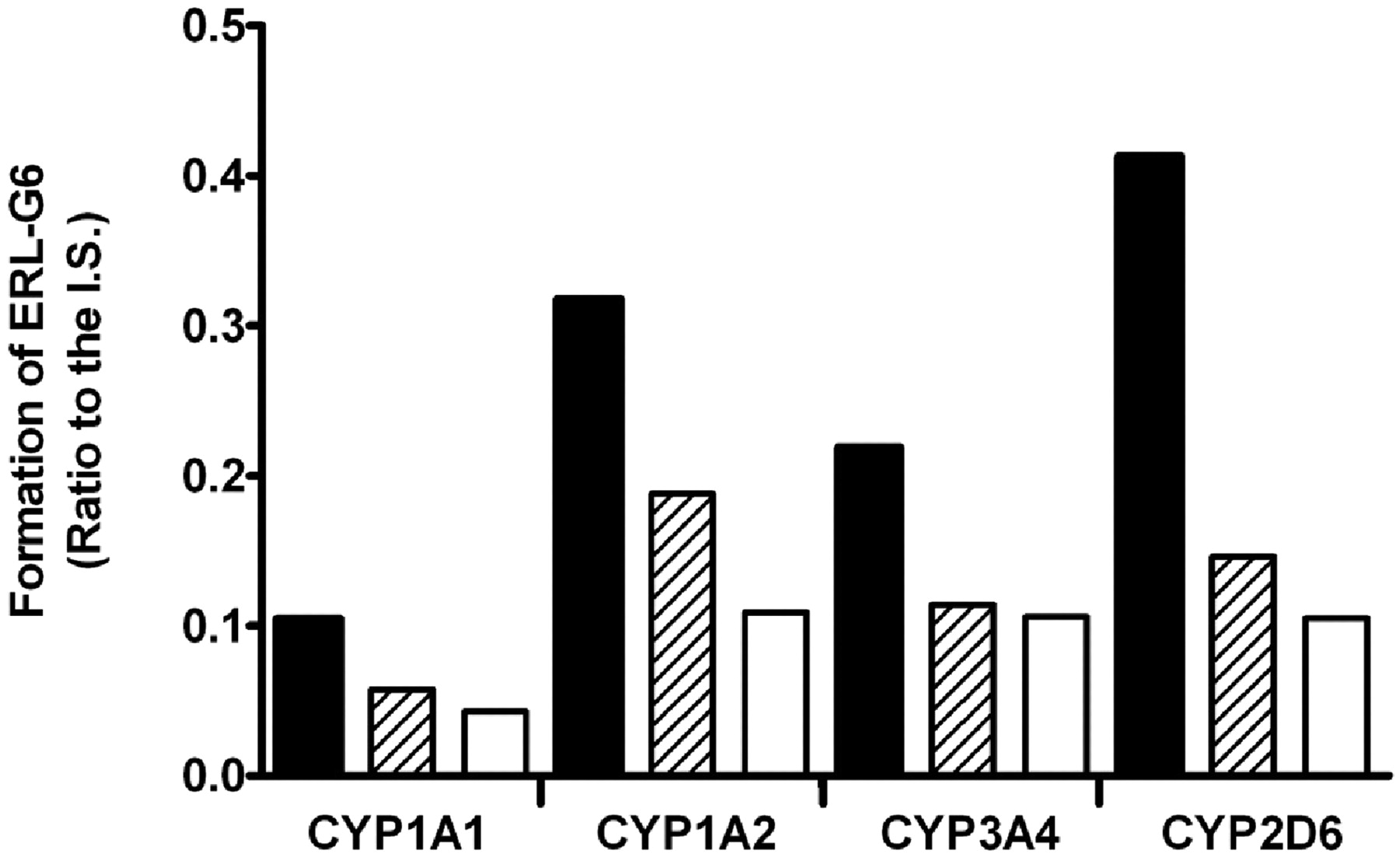
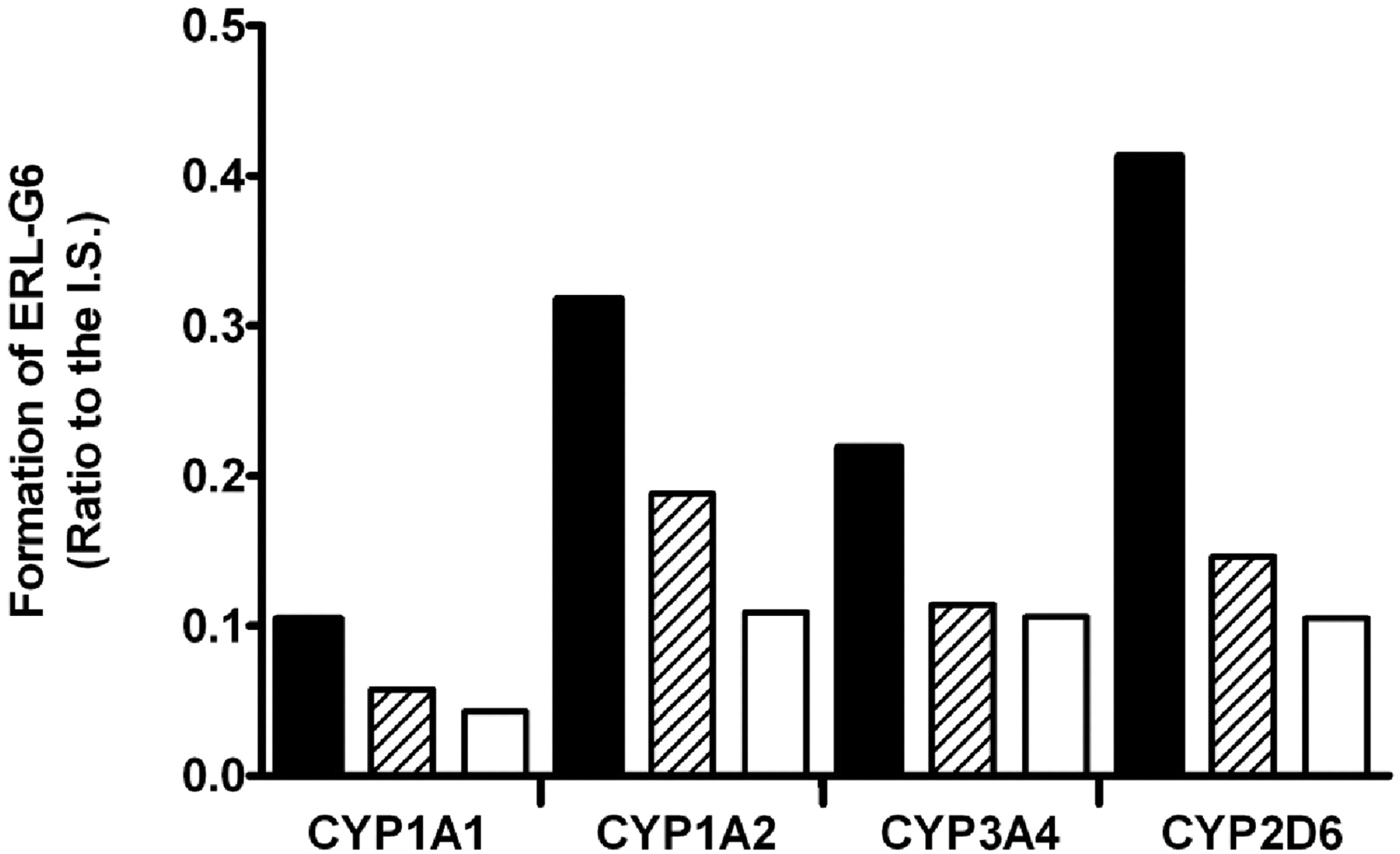
Effect of NADPH and superoxide dismutase plus catalase on the formation rate of ERL-G6 in recombinant P450 incubations. All the incubations had 100 pmol/ml P450, 5 mM GSH, and 20 μM ERL in 100 mM phosphate buffer, pH 7.4. Filled bars contained 1 mM NADPH; open bars did not have NADPH; and hashed bars had 1 mM NADPH plus 1000 units of superoxide dismutase and 1000 units of catalase.
The final detected adduct, ERL-G4, was formed through the nonenzymatic conjugation of GSH to hydroxylated ERL. This was verified by incubating ERL with HLM and NADPH and isolating the metabolites by solid-phase extraction. Addition of GSH to the metabolites resulted in an adduct with the same mass and elution time as ERL-G4 (Fig. 2B).
Identification of the P450 Enzymes Responsible for ERL Bioactivation in Human Microsomes.
ERL-G5 was the predominant enzyme-catalyzed GSH adduct. For simplicity, only ERL-G5 is shown in the phenotyping studies, but similar trends were seen with all three of the primary adducts. Bactosomes prepared from Escherichia coli membranes coexpressing recombinant human P450 and P450 reductase were used. As shown in Fig. 6A, CYP3A4 had the highest level of ERL-G5 formation in incubations containing 100 pmol/ml recombinant P450 and 40 μM ERL. Adduct formation by CYP1A1 was approximately 36% of CYP3A4. CYP2D6, CYP3A5, and CYP1A2 also catalyzed ERL-G5 formation, but the levels of adduct formation range from 9 to 16% of that formed by CYP3A4.
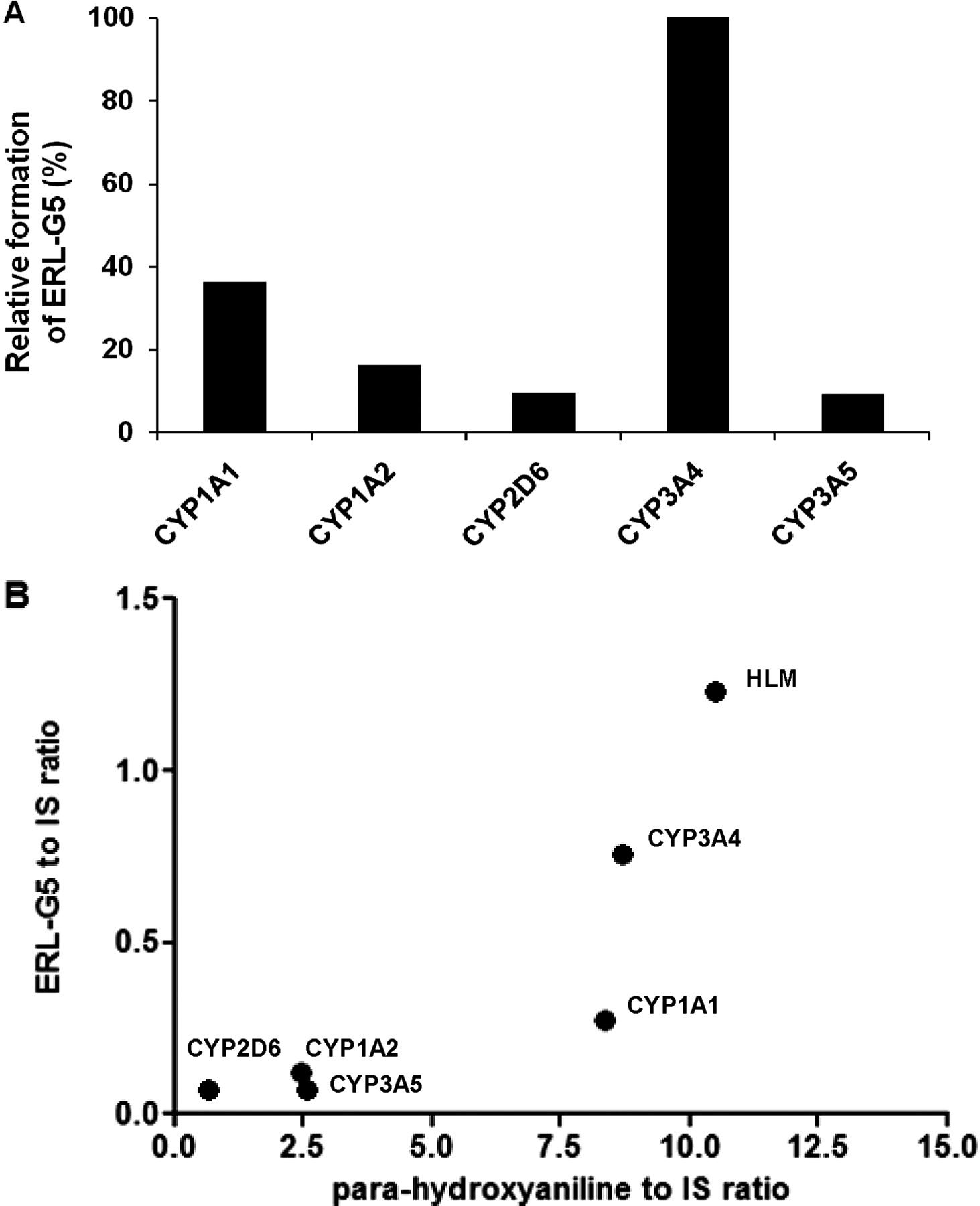
ERL-G5 formation by recombinant P450 enzymes. ERL-G5 generation was compared in incubations containing 100 pmol/ml P450, 1 mM NADPH, 5 mM GSH, and 40 μM ERL (A). Relative ERL-G5 concentration was determined by comparison of the peak area of ERL-G5 to an internal standard. The enzyme activities were expressed as the percentage of CYP3A4 activity and are an average of two measurements. Correlation analysis of ERL-G5 formation to formation of the para-hydroxyaniline metabolite in HLM and recombinant P450 is shown in plot B.
Based on the results from recombinant P450 experiments, selective inhibitors of CYP1A1/2 and CYP3A4/5, α-naphthoflavone and ketoconazole, respectively, were evaluated in human hepatic, intestinal, and pulmonary microsome (smoker and nonsmoker). As shown in Fig. 7A, the addition of ketoconazole decreased ERL-G5 formation by 69 and 76% in liver and intestinal microsomes but had no effect in pulmonary microsomes. Although CYP3A4 appears to be the predominant catalyst of ERL bioactivation in the liver and intestine, other enzymes are likely responsible for 20 to 40% of adduct formation. The formation of ERL-G5 in pulmonary microsomes was low, but there was a large difference between smokers and nonsmokers. CYP1A1/2 are likely to be the major enzymes responsible for ERL bioactivation in the lung because the addition of the CYP1A1/2 inhibitor α-naphthoflavone inhibited ERL-G5 formation by 54% (Fig. 7B).
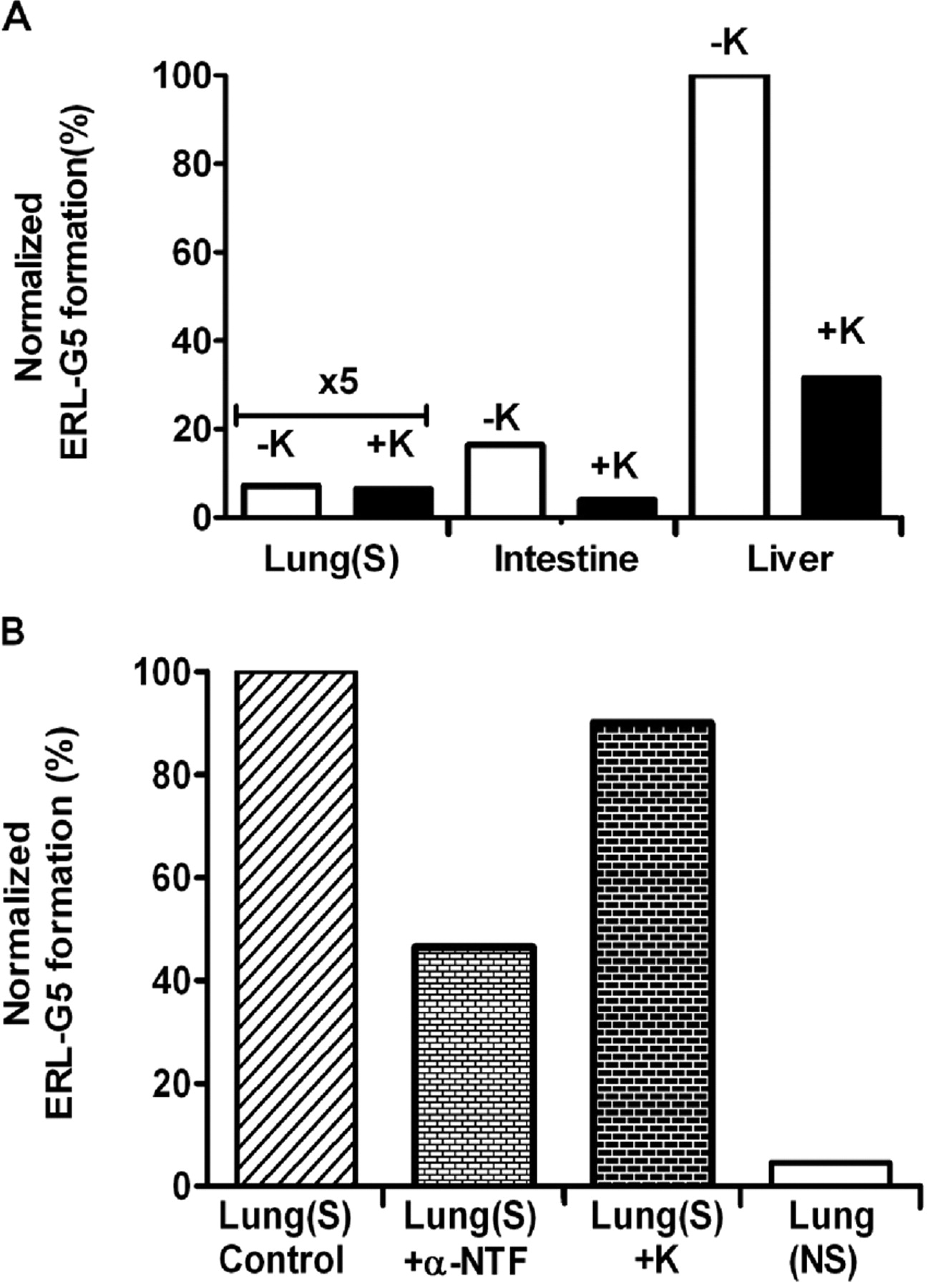
CYP3A and CYP1A inhibition on ERL-G5 formation. A, ERL-G5 production was measured in incubations where the CYP3A inhibitor ketoconazole (1 μM) was added to human pulmonary (smoker), intestinal, and hepatic microsomes. B, inhibition of CYP3A (1 μM ketoconazole) and CYP1A (20 μM α-naphthoflavone) was tested in incubations containing pulmonary microsomes from smokers (S) and nonsmokers (NS). All the incubations contained 2 mg/ml microsomal protein, 40 μM ERL, 5 mM GSH, and 1 mM NADPH in 100 mM phosphate buffer, pH 7.4. The values were an average of two replicates.
Mechanism of P450-Dependent Formation of ERL-GSH Adducts.
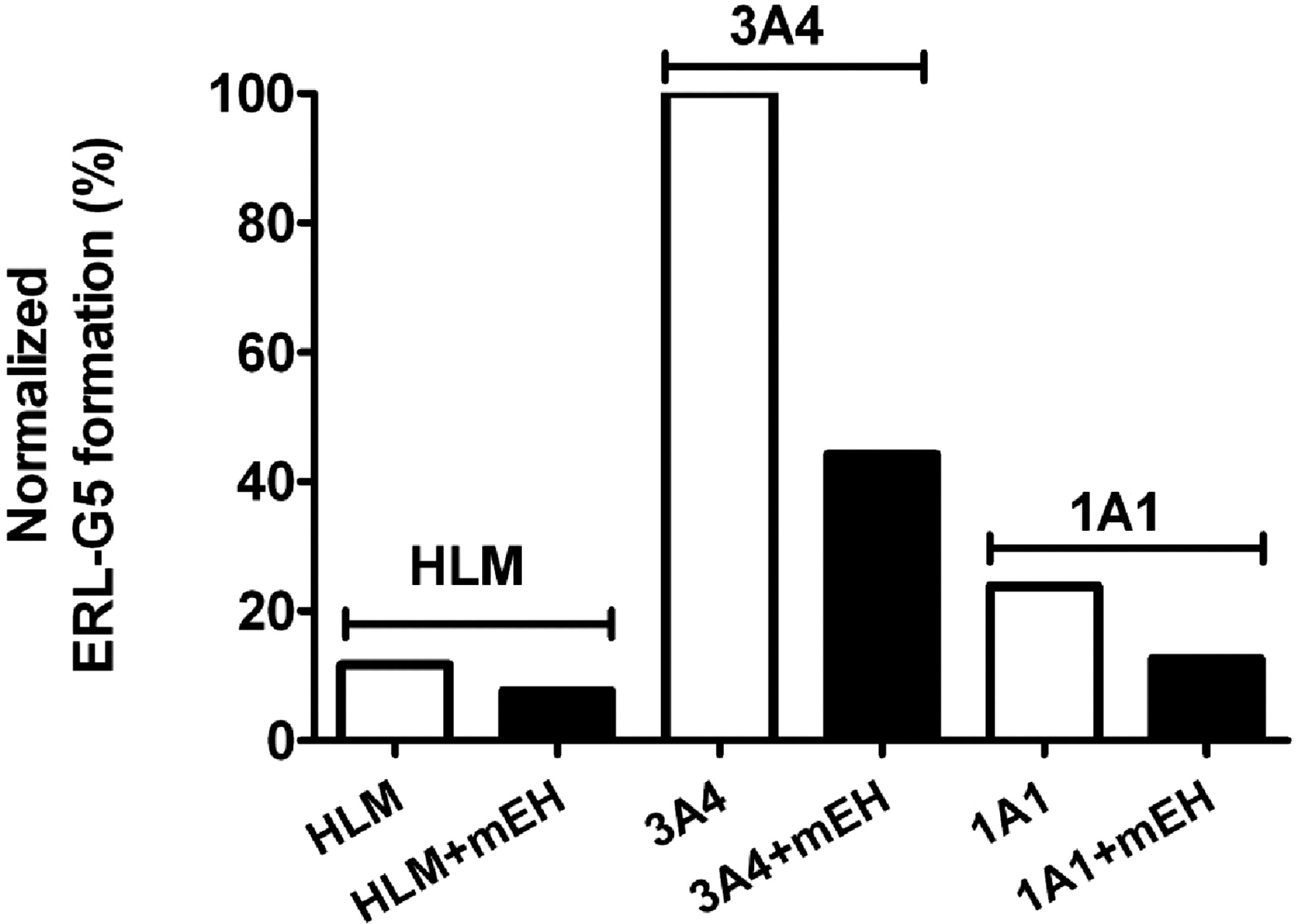
Based on the detected GSH adducts, bioactivation is likely to proceed through the formation of a reactive epoxide and/or through the formation of a quinone-imine. To test this hypothesis, recombinant human microsomal epoxide hydrolase was added to reactions containing HLMs or recombinant P450 with ERL, GSH, and NADPH. The formation of ERL-G5 decreased by 35, 55, and 47% in incubations of HLMs and recombinant CYP3A4 and CYP1A1, respectively (Fig. 8). Similar results were observed for other P450-dependent adducts. The addition of recombinant human microsomal epoxide hydrolase did not decrease the activity of CYP1A1/2 and CYP3A4/5 assayed by phenacetin demethylation and midazolam hydroxylation, respectively (data not shown).
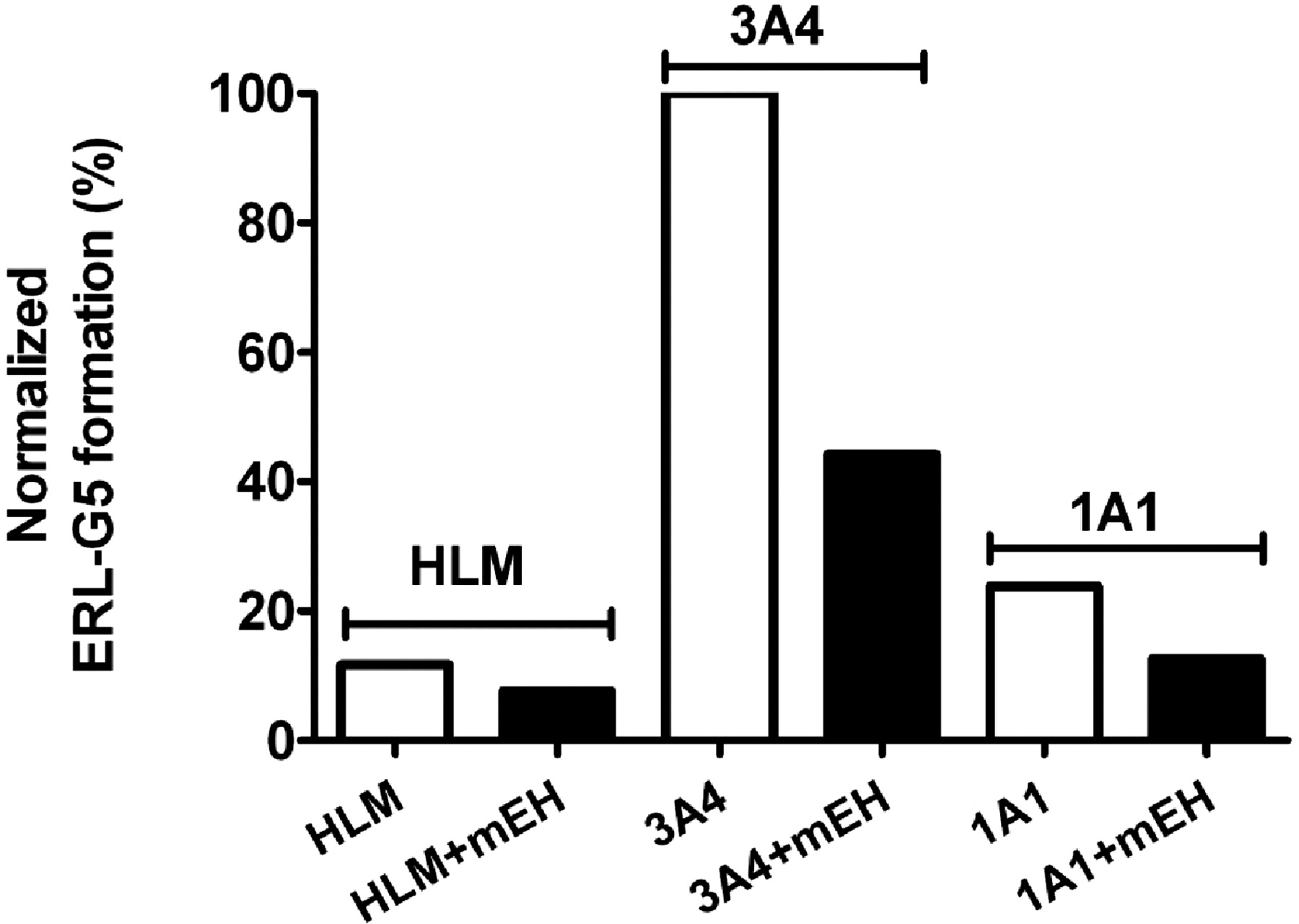
Influence of human microsomal epoxide hydrolase on the formation of ERL-G5. The addition of 1 mg/ml human microsomal epoxide hydrolase was evaluated in incubations of HLM and recombinant CYP3A4 and CYP1A1. Incubations containing 2 mg/ml HLM or 100 pmol/ml recombinant P450, 1 mM NADPH, 5 mM GSH, and 40 μM ERL in 100 mM phosphate buffer, pH 7.4. The values were an average of two replicates.
Formation of para-hydroxyerlotinib (para-hydroxylation on the aromatic aniline ring) was published by Ling et al. (2006) (metabolite M16). This metabolite was determined to be the major metabolite in human plasma and accounted for approximately 10% of the total excreted dose (Ling et al., 2006). The two-electron oxidation of the para-hydroxyaniline moiety would generate a quinone-imine, which is electrophilic and can potentially react with cellular nucleophiles. A positive correlation (Pearson r = 0.85) was found between generation of para-hydroxyerlotinib and ERL-GSH adducts (Fig. 6B).
Hydroxyerlotinib was purified from microsomal incubations and used in reactions with recombinant P450 to test whether CYP3A4 and CYP1A1 are capable of oxidizing the hydroxyerlotinib to form a quinone-imine (as seen during the oxidation of acetaminophen) (Dahlin et al., 1984). Similar monohydroxylated GSH adducts were found with hydroxyerlotinib as with ERL. If the formation of an epoxide was prerequisite for adduct formation, the observed adduct would be expected to incorporate an additional oxygen and be detected as a dihydroxylated adduct.
N-Acetyl-lysine Trapping.
N-Acetyl lysine is better suited to trap hard electrophiles that may be produced through oxidation of the alkyne group (oxirene and ketene intermediates). No N-acetyl-lysine-ERL adducts were detected.
ERL Analogs.
To evaluate the role of the alkyne group in GSH adduct formation and in P450 inactivation, two analogs were synthesized. The alkyne group was replaced by an ethyl and a cyano group. Both analogs formed GSH adducts in a P450- and NADPH-dependent manner. The addition of 1 μM ketoconazole inhibited the formation of GSH adducts by 90 and 97% for the ethyl and cyano analogs, respectively, indicating that CYP3A4/5 were primarily responsible for adduct formation in HLM. The cyano analog was found to inactivate CYP3A4 in a time- and concentration-dependent manner with a KI approximately 4 times higher than ERL (KI, 78.4 μM; Kinact, 0.063/min). CYP3A4 inactivation was not observed for the ethyl analog.
Discussion
CYP3A4, CYP3A5, and to a lesser degree CYP1A1 and CYP1A2 had been previously implicated in the metabolism of ERL (Ling et al., 2006; Li et al., 2007). The current study shows that these same enzymes are capable of the metabolic bioactivation of ERL to form a reactive intermediate, capable of covalently binding to cellular nucleophiles. In addition to potential toxicity associated with covalent adduction of ERL to biomolecules, pharmacokinetic drug-drug interactions may be caused as a result of the inactivation of CYP3A4 and CYP3A5 during the metabolism of ERL.
Smoking has been shown to have a large effect on oral ERL pharmacokinetics. Smokers required 300 mg of ERL to achieve similar Cmax and area under the curve to nonsmokers given a 150-mg dose (Hamilton et al., 2006). Tobacco smoke has been shown to increase the level of CYP1A1 mRNA and protein in human liver and lung (Hukkanen et al., 2002; Thum et al., 2006). The increased expression of CYP1A1 in the lungs of smokers may increase their risk of ERL-associated interstitial lung disease. Although we are not aware of a study examining the rates of interstitial lung disease in smokers versus nonsmokers being treated with ERL, there seems to be a relationship between smoking and interstitial lung disease with the structurally related drug gefitinib (Takano et al., 2004).
The proposed mechanism for the formations of ERL-GSH adducts is depicted in Scheme 1. The oxidation of ERL by P450 appears to form an epoxide that can react with nucleophiles such as the sulfhydryl group of cysteine. An additional route of adduct formation is through the generation of a reactive quinone-imine (reactive intermediate associated with acetaminophen). A quinone-imine can be generated by oxidation of para-hydroxyerlotinib. In this study, we found that CYP1A1 and CYP3A4 could oxidize hydroxyerlotinib to catalyze the formation of ERL-GSH adducts, presumably through forming a quinone-imine; however, other cellular oxidases may also be able to catalyze this reaction. Although reactive epoxides and quinone-imine metabolites cannot diffuse significant distances from the organ where they were formed, para-hydroxyerlotinib is in general circulation and is the major ERL metabolite in human plasma. This may diffuse to other tissues where it is oxidized to generate the reactive quinone-imine. Redox cycling of quinone-imines is implicated in a number of drug-related adverse effects and has been shown to produce oxidative stress (Hinson et al., 2004) and covalently modify cellular proteins (Guengerich and MacDonald, 2007; Tang, 2007). This potentially contributes to the clinically observed toxicities.

Proposed mechanism of ERL-GSH adduct formation.
Little is known about ERL-induced Stevens-Johnson syndrome and toxic epidermal necrolysis, but these syndromes are considered immunogenic, are commonly drug-induced, and drugs commonly associated with initiating these toxicities do not share common targets (Khan et al., 2006). Many compounds that are reported to induce Stevens-Johnson syndrome are associated with metabolic activation to form reactive intermediates, such as carbamazepine, diclofenac, penicillin, modafinil, acetaminophen, ibuprofen, and phenytoin (Neuman and Nicar, 2007; Locharernkul et al., 2008; Levi et al., 2009).
ERL also contains a terminal alkyne group. The oxidation of terminal alkynes by P450 has been shown to form reactive oxirene and ketene intermediates that can react with water or other cellular nucleophiles, and has been proposed to be involved in inactivation of P450 through formation of protein adducts and through oxidation of the heme group of the P450 enzyme (Ortiz de Montellano and Kunze, 1980, 1981; Ortiz de Montellano, 1985). We did not see evidence that reactive oxirene or ketene intermediates were released from the enzyme active site or that they were involved in enzyme inactivation. The alkyne group of ERL was oxidized by P450 to form a carboxylic acid (a known metabolite) (Ling et al., 2006), but several recombinant P450 enzymes were capable of formation of the carboxylic acid metabolite, and this did not correlate with either formation of GSH adducts or enzyme inactivation (data not shown).
We did not observe ERL adducts when a second trapping agent better suited for the expected reactive intermediates generated by alkyne oxidation, N-acetyl lysine, was used. This alone did not sufficiently show that oxidation of the alkyne was not responsible for the observed CYP3A4/5 inactivation. To address this possibility, the alkyne group of ERL was replaced with an ethyl and a cyano group. Both analogs formed GSH adducts when oxidized by CYP3A4. In addition, the cyano analog was found to be a time-dependent inhibitor of CYP3A4, showing that enzyme inactivation was not dependent on the alkyne substituent, but we cannot conclude that the alkyne does not act as a second route for CYP3A4 inactivation.
In summary, the current study identifies ERL as a mechanism-based inactivator of CYP3A4 and CYP3A5. CYP3A4 is the primary hepatic and intestinal enzyme responsible for the catalysis of reactive ERL metabolites, and CYP1A1/2 are primarily responsible for pulmonary adduct formation. ERL bioactivation is proposed to be through the formation of a reactive epoxide and quinone-imine, which may contribute to some of the clinical adverse drug reactions.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.109.030361.
-
ABBREVIATIONS:
- ERL
- erlotinib
- EGFR
- epidermal growth factor receptor
- P450
- cytochrome P450
- HLM
- human liver microsome
- DMSO
- dimethyl sulfoxide
- HPLC
- high-performance liquid chromatography
- MRM
- multiple reaction monitoring.
- Received September 28, 2009.
- Accepted April 9, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}