


Abstract
Lapatinib, an oral breast cancer drug, has recently been reported to be a mechanism-based inactivator of cytochrome P450 (P450) 3A4 and also an idiosyncratic hepatotoxicant. It was suggested that formation of a reactive quinoneimine metabolite was involved in mechanism-based inactivation (MBI) and/or hepatotoxicity. We investigated the mechanism of MBI of P450 3A4 by lapatinib. Liquid chromatography-mass spectrometry analysis of P450 3A4 after incubation with lapatinib did not show any peak corresponding to irreversible modifications. The enzymatic activity inactivated by lapatinib was completely restored by the addition of potassium ferricyanide. These results indicate that the mechanism of MBI by lapatinib is quasi-irreversible and mediated via metabolic intermediate complex (MI complex) formation. This finding was verified by the increase in a signature Soret absorbance at approximately 455 nm. Two amine oxidation products of the metabolism of lapatinib by P450 3A4 were characterized: N-hydroxy lapatinib (M3) and the oxime form of N-dealkylated lapatinib (M2), suggesting that a nitroso or another related intermediate generated from M3 is involved in MI complex formation. In contrast, P450 3A5 was much less susceptible to MBI by lapatinib via MI complex formation than P450 3A4. In addition, P450 3A5 had a significantly lower ability than 3A4 to generate M3, consistent with N-hydroxylation as the initial step in the pathway to MI complex formation. In conclusion, our results demonstrate that the primary mechanism for MBI of P450 3A4 by lapatinib is not irreversible modification by the quinoneimine metabolite, but quasi-irreversible MI complex formation mediated via oxidation of the secondary amine group of lapatinib.
Introduction
Lapatinib is an oral, small-molecule, reversible inhibitor of both epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases (Rusnak et al., 2001; Xia et al., 2002). In 2007, the U.S. Food and Drug Administration approved lapatinib for use in combination with capecitabine for the treatment of advanced breast cancer or metastatic breast cancer in patients whose tumors overexpress human epidermal growth factor receptor-2 and who have received previous treatment that included an anthracycline, a taxane, and trastuzumab (Medina and Goodin, 2008; Ryan et al., 2008).
Lapatinib has been reported to be a mechanism-based inactivator of cytochrome P450 (P450) 3A4, and clinically significant drug-drug interactions (DDIs) may occur if it is coadministered with drugs that are P450 3A4 substrates (Teng et al., 2010). Some drugs and related compounds are known to be oxidized by P450 3A4 and other P450s to metabolites that can lead to their autocatalytic inactivation either via irreversible or quasi-irreversible mechanisms (Lin and Lu, 1998). The mechanism of irreversible inactivation is proposed to be covalent modifications of either the apoprotein or the heme moiety by reactive metabolites. Quasi-irreversible inactivation is caused by formation of a coordinate covalent bond between the heme iron and certain kinds of metabolites, and the coordination complex is referred to as a metabolic intermediate complex (MI complex).
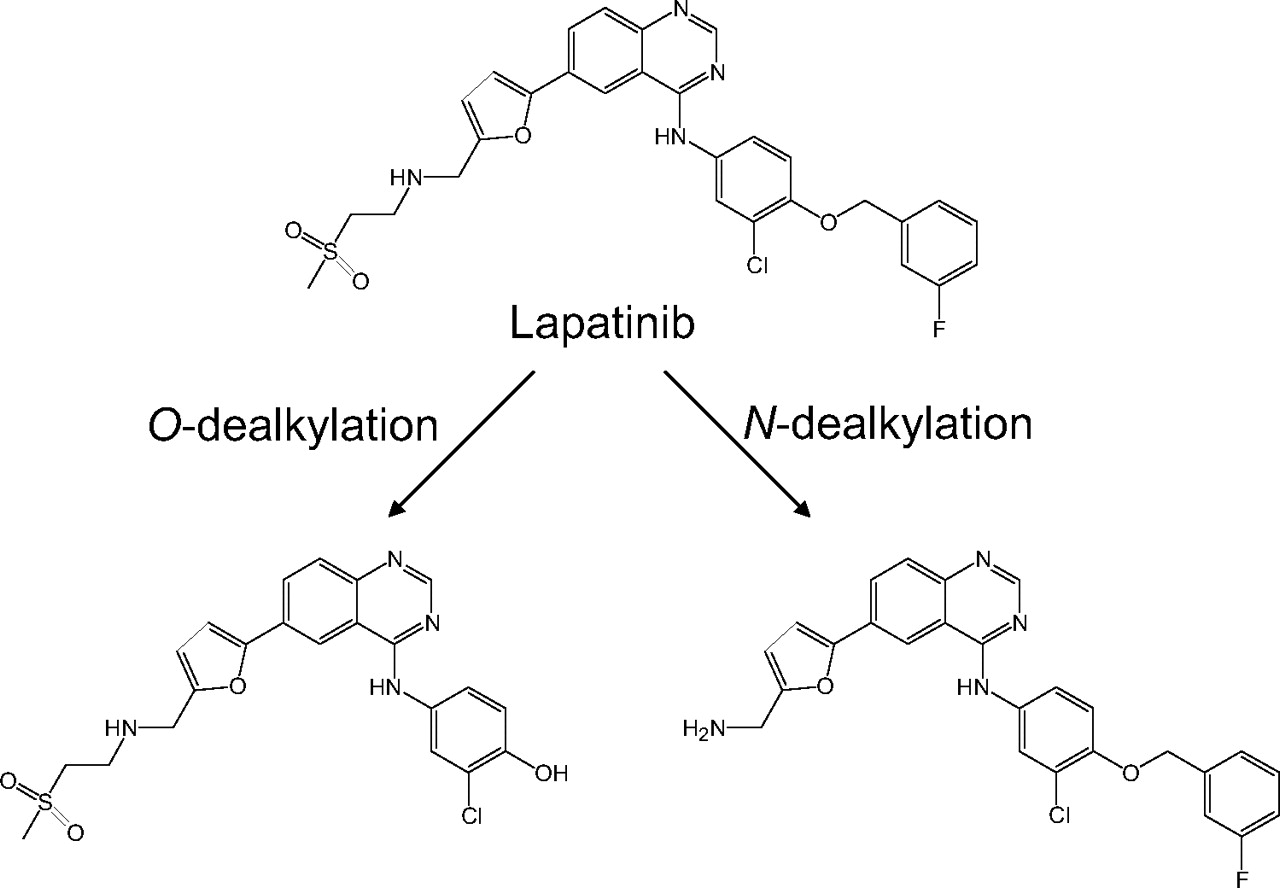
Lapatinib is metabolized mainly by P450 3A4 to form O- and N-dealkylated metabolites (Fig. 1) (Teng et al., 2010). The O-dealkylated metabolite has the structural features of a p-hydroxyanilide, like acetaminophen, that is susceptible to oxidation to reactive quinoneimine species, similar to N-acetyl-p-quinoneimine generated from acetaminophen (Dahlin et al., 1984). In a recently reported study, a glutathione adduct of the reactive quinoneimine derived from O-dealkylated lapatinib was detected and characterized. On the basis of this finding, it was suggested that the MBI of P450 3A4 by lapatinib was due to irreversible covalent modification of its apoprotein and/or heme moiety by a reactive quinoneimine species (Teng et al., 2010).

Chemical structures of lapatinib and its two major metabolites.
Formation of reactive metabolites also has toxicological significance, because covalent and noncovalent modifications of biological macromolecules by reactive metabolites are associated with various drug toxicities (Nelson, 1995). Many drugs that form reactive metabolites have been reported to be involved in both MBI and drug-induced toxicities, and, in some cases, the causal mechanisms for MBI and toxicities may share a common metabolic bioactivation pathway (Masubuchi and Horie, 2007). One of the best examples is tienilic acid, a diuretic drug withdrawn from the market because of severe hepatotoxicity (Zimmerman et al., 1984). Tienilic acid undergoes metabolic activation by P450 2C9 to form an oxygenated thiophene metabolite that reacts with P450 2C9, resulting in MBI (Jean et al., 1996). In addition, autoantibodies to P450 2C9 were observed in plasma samples from several patients who experienced liver injury from the drug (Homberg et al., 1984; Beaune et al., 1987). Therefore, mechanistic investigations of MBI from the viewpoint of metabolism are very important not only for DDI but also for drug-induced toxicities.
In the case of lapatinib, serious adverse events, including severe hepatotoxicity and rare cases of liver-related deaths, have been observed in clinical trials and postmarketing surveillance, and a black box warning regarding the risk for hepatotoxicity is now part of the drug labeling (Gomez et al., 2008). Although the mechanism for the hepatotoxicity is unknown, it might be associated with reactive quinoneimine formation as discussed in a recent article (Teng et al., 2010).
P450 3A5 is also known to be involved in the metabolism of lapatinib (European Medicines Agency, Assessment Report for Tyverb, 2008, http://www.emea.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000795/WC500044960.pdf). Whereas P450 3A5 shares 83% amino acid sequence homology with 3A4 and has similar substrate specificity, it is different from P450 3A4 with respect to its susceptibility to inhibition (Aoyama et al., 1989; Thummel and Wilkinson, 1998). P450 3A5 has generally been considered less important than P450 3A4 because of a relatively low level of hepatic and intestinal expression. However, several reports have suggested its importance because the frequency of polymorphic P450 3A5 expression differs among ethnic groups, e.g., P450 3A5 is found at high levels in 10 to 30% of whites and >50% of African Americans (Lamba et al., 2002; Lin et al., 2002; Xie et al., 2004).
In the present study, the mechanism of MBI of P450 3A4 by lapatinib was investigated using several approaches, including a search for P450 3A4 protein adducts by LC-MS, MI complex analysis based on absorption spectra, and structural characterization of metabolites. An investigation of the metabolism of lapatinib by P450 3A5 was performed for comparison.
Materials and Methods
Materials.
Lapatinib (free base) was purchased from LC Laboratories (Woburn, MA). Testosterone, 11α-hydroxyprogesterone, NADPH, potassium ferricyanide, CHAPS, potassium HEPES, glutathione, and diltiazem were purchased from Sigma-Aldrich (St. Louis, MO). Midazolam, 1′-hydroxymidazolam, and 6β-hydroxytestosterone were purchased from Cerilliant Corporation (Round Rock, TX). Salts for potassium phosphate buffer and MgCl2 were purchased from Mallinckrodt Baker, Inc. (Phillipsburg, NJ). D2O was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA). l-α-Dilauroyl-sn-glycero-3-phosphocholine, l-α-dioleoyl-sn-glycero-3-phosphocholine, and l-α-dilauroyl-sn-glycero-3-phosphoserine were purchased from Avanti Polar Lipids (Alabaster, AL). Pooled human liver microsomes (HLMs) and human P450 3A4 and 3A5 Supersomes, coexpressed with cytochrome b5 and cytochrome P450 reductase, were purchased from BD Biosciences (Woburn, MA). Human P450 3A4 was expressed and purified as described previously (Baer et al., 2007), except that Escherichia coli C41(DE3) cells were used instead of E. coli DH5αF'IQ cells. The pCW 3A4-His6 expression vector and C41(DE3) cells were kindly provided by Dr. William Atkins (University of Washington, Seattle, Washington) and Dr. Rheem Totah (University of Washington, Seattle, Washington), respectively. Rat P450 reductase was expressed and purified as described previously (Shen et al., 1989), except that E. coli BL21(DE3) cells from Invitrogen (Carlsbad, CA) were used instead of E. coli C-1A cells. The expression vectors encoding rat P450 reductase and BL21(DE3) were kindly provided by Dr. Allan Rettie (University of Washington, Seattle, Washington). Human cytochrome b5 was purchased from Invitrogen. All other chemicals and reagents were of analytical grade and were available from commercial sources.
Search for Lapatinib Adduction to P450 3A4.
P450 3A4 (500 pmol) was combined with P450 reductase (1.0 nmol), cytochrome b5 (500 pmol), 0.1 mg/ml CHAPS, 20 μg/ml liposomes [l-α-dilauroyl-sn-glycero-3-phosphocholine, l-α-dioleoyl-sn-glycero-3-phosphocholine, and l-α-dilauroyl-sn-glycero-3-phosphoserine (1:1:1, v/v/v)], 3 mM reduced glutathione, 50 mM potassium HEPES (pH = 7.4), 30 mM MgCl2, and 100 μM lapatinib in a total volume of 1.0 ml. The final organic solvent concentrations were 0.9% (v/v) acetonitrile and 0.1% (v/v) dimethyl sulfoxide. The reconstituted mixture was incubated for 3 min at 37°C before the addition of 10 μl of a solution of NADPH in H2O (final concentration, 1 mM) or H2O as a control. After a 30-min incubation at 37°C, the reaction mixture was cooled on ice, followed by centrifugal filtration (2000g, 25 min, 4°C) using Amicon Ultra-4 centrifugal filter units (Millipore Corporation, Billerica, MA). The concentrated samples were analyzed by using a Synapt HDMS system (Waters, Manchester, UK) equipped with a high-performance liquid chromatography system consisting of two Shimadzu LC-10AD pumps with a gradient controller and a Shimadzu SIL-10ADvp autoinjector (Shimadzu Scientific Instruments Inc., Columbia, MD). Analyte separation was achieved using a POROS R1/10 column (100 × 2.1 mm; Applied Biosystems, Foster, CA) at a flow rate of 0.5 ml/min. Solvents A and B were nanopure H2O with 0.1% (v/v) trifluoroacetic acid and LC-MS-grade acetonitrile with 0.1% (v/v) trifluoroacetic acid, respectively. The gradient program was as follows: isocratic at 20% B (0–3 min), linear gradient from 20 to 35% B (3–4 min), linear gradient from 35 to 60% B (4–16.5 min), and isocratic at 60% B (16.5–20 min). The data were acquired in the full-scan mode in a range of m/z 200 to 2000. The MS conditions were as follows: capillary voltage, 3.5 kV; cone voltage, 30 V; source temperature, 120°C; desolvation temperature, 350°C; ionization mode, ESI in the positive ion mode; and analyzer, V mode. The MS spectral data were analyzed and deconvoluted by using MassLynx version 4.1 (Waters).
Reversibility of MBI.
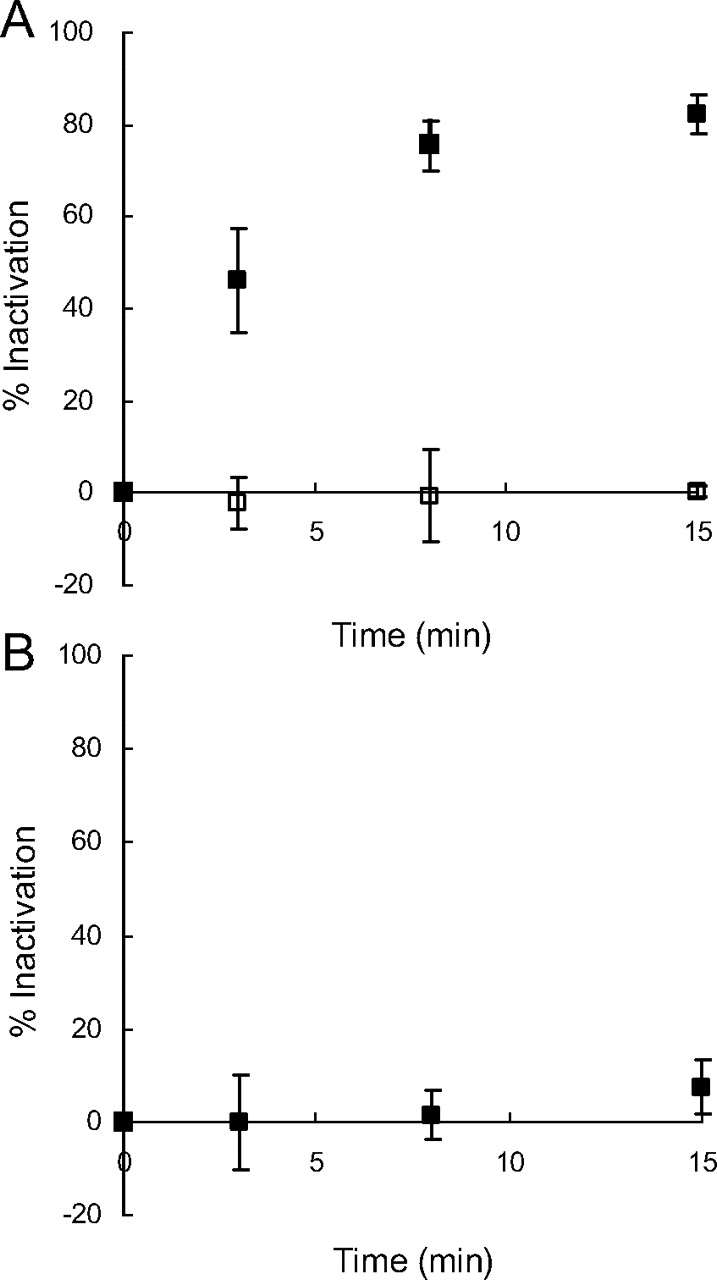
The reversibility of MBI was investigated by oxidation with potassium ferricyanide according to a method reported previously (Watanabe et al., 2007), consisting of three sequential incubations: primary 0- or 30-min incubations with or without lapatinib, secondary 10-min incubations of the primary incubation mixtures with or without potassium ferricyanide, and tertiary 10-min incubations of the secondary incubation mixtures with testosterone. The primary incubation solutions, containing 1.0 mg/ml HLMs in 0.1 M potassium phosphate buffer (pH = 7.4) with or without 50 μM lapatinib, were prepared and kept at 37°C for 3 min. The final organic solvent concentration in the primary incubation solutions was 1% (v/v) acetonitrile. The primary incubation reactions were initiated by the addition of 2.5 μl of a 100 mM solution of NADPH in H2O (final concentration, 1.0 mM). After a 0- or 30-min primary incubation at 37°C, 50 μl of each primary incubation mixture was added to 50 μl of the secondary incubation solutions containing 0.1 M potassium phosphate buffer (pH = 7.4) with or without 2 mM potassium ferricyanide and incubated for 10 min. After a 10-min secondary incubation at 37°C, each secondary reaction mixture was diluted 5-fold with the tertiary incubation solutions, which contained 0.1 M potassium phosphate (pH = 7.4) buffer, 200 μM testosterone, 1% (v/v) acetonitrile, and 1.0 mM NADPH and then were incubated for 10 min. At the end of the tertiary incubation reactions, each tertiary reaction mixture was diluted 2-fold with acetonitrile containing 20 μM 11α-hydroxyprogesterone as an internal standard. Samples were cooled and centrifuged at 9000g for 3 min. The supernatants were transferred to other tubes and kept at −80°C until LC-MS analysis. The samples were analyzed using a Micromass Quattro Micro mass spectrometer (Waters) equipped with a high-performance liquid chromatography system consisting of two Shimadzu LC-10AD pumps with a gradient controller and a Shimadzu SIL-10ADvp autoinjector. Analyte separation was achieved using a Zorbax SB-C18 column (150 × 2.1 mm, 5 μm particle size; Agilent Technologies, Santa Clara, CA) at a flow rate of 0.3 ml/min. Solvents A and B were nanopure H2O with 0.1% (v/v) trifluoroacetic acid and LC-MS-grade acetonitrile with 0.1% (v/v) trifluoroacetic acid, respectively. The gradient program was as follows: isocratic at 20% B (0–2 min), linear gradient from 20 to 90% B (2–5 min), and isocratic at 90% B (5–7 min). The data were acquired in single-ion monitoring mode, and the m/z values of 6β-hydroxytestosterone and 11α-hydroxyprogesterone were 305.2 and 331.2, respectively. The MS conditions were as follows: capillary voltage, 3.5 kV; cone voltage, 25 V; source temperature, 120°C; desolvation temperature, 300°C; and ionization, ESI in the positive ion mode. The chromatographic data were analyzed by using MassLynx version 4.1. The percentage of metabolic activity [percentage control(0 min) and percentage control(30 min)] was calculated for each sample after a 0- or 30-min preincubation with lapatinib and compared with each control sample without lapatinib as follows:

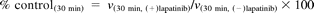
Here, v is the metabolic activity and was calculated from the chromatographic peak area ratios of 6β-hydroxytestosterone/11α-hydroxyprogesterone. The inhibitory effect of potassium ferricyanide on testosterone-6β-hydroxylation by HLMs in the absence of lapatinib was examined by comparing the metabolic activity v of the samples with and without potassium ferricyanide as follows:

Absorption Analysis for MI Complex Formation.
MI complex formation was characterized in P450 3A4 and 3A5 Supersomes by difference spectroscopy using an Olis-modernized Aminco DW-2 spectrophotometer (Olis, Bogart, GA). Sample and reference cuvettes contained 0.11 μM P450 enzyme, 0.1 M KPi buffer (pH = 7.4), and 50 μM lapatinib (or 10 μM diltiazem) in a total volume of 495 μl. The final organic solvent concentration was 1.0% (v/v) acetonitrile. After 3 min of preincubation at 37°C, NADPH (100 mM in H2O, 5.0 μl) and H2O (5.0 μl) were added to the sample and reference cuvettes, respectively. The spectrophotometer was set to scan repetitively from 495 to 430 nm (5-nm intervals) until MI complex formation reached completion. The concentrations of MI complexes were calculated from Absmax and the extinction coefficient for the absorbance difference between 490 and 455 nm, which is 65 cm−1 mM−1 (Franklin, 1974; Liu and Franklin, 1985). The initial rates of MI complex formation (k) and maximal concentration of MI complex (MICmax) were calculated by fitting the data to the standard monoexponential function MICt = MICmax (1 − e−kt), where MICt is the concentration of MI complex at each time point.
Activity Assay for MBI Using Supersomes.
For the P450 3A4 activity assay, the preincubation solutions containing 0.11 μM P450 3A4, 0.1 M KPi buffer (pH = 7.4), and 50 μM lapatinib were prepared and kept at 37°C for 3 min. The preincubation reactions were initiated by the addition of 2.5 μl of a solution of NADPH in H2O (final concentration, 1.0 mM) or 2.5 μl of H2O as a control. The total volume of each preincubation solution was 250 μl, and the final organic solvent concentration was 1.0% (v/v) acetonitrile. At 0, 3, 8, and 15 min after the initiation, 20 μl of each preincubation solution was transferred to 180 μl of the incubation solution, which contained 0.1 M potassium phosphate (pH = 7.4) buffer, 200 μM testosterone, 1% (v/v) acetonitrile, and 1.0 mM NADPH and then incubated for 10 min. At the end of the incubation reactions, 200 μl of acetonitrile containing 11α-hydroxyprogesterone (20 μM) was added to each reaction mixture. Samples were cooled and centrifuged at 9000g for 3 min, and the supernatants were transferred to other tubes and kept at −80°C until LC-MS analysis. The methods for LC-MS analysis were the same as those described under Reversibility of MBI. Then, metabolic activity at each preincubation time point was calculated from the chromatographic peak area ratio of 6β-hydroxytestosterone/11α-hydroxyprogesterone. The percentage of inactivation was calculated by normalization of the metabolic activity to 0-min preincubation. The initial rate and maximal percentage of inactivation were calculated by fitting the data to the standard monoexponential function. P450 3A5 activity assays were performed using midazolam (25 μM) and alprazolam (1.0 μg/ml) as the substrate and internal standard, respectively.
Structural Elucidation of Lapatinib Metabolites.
The incubation solutions containing 0.2 μM P450 enzyme (Supersomes), 0.1 M KPi buffer (pH = 7.4), and 50 μM lapatinib were prepared and kept at 37°C for 3 min. The incubation reactions were initiated by the addition of 10 μl of a 10 mM solution of NADPH in H2O (final 1.0 mM) or H2O as a control. The total volume of each preincubation solution was 100 μl and the final organic solvent concentration was 1.0% (v/v) acetonitrile. After 30-min incubations at 37°C, the reaction was terminated by the addition of 100 μl of acetonitrile. Samples were cooled and centrifuged at 9000g for 3 min, and the supernatants were transferred to other tubes and kept at −80°C until LC-MS analysis. The samples were analyzed by the same LC-MS system and solvents as those described under Search for Lapatinib Adduction to P450 3A4. In the case of the hydrogen-deuterium exchange experiments, D2O with 0.1% (v/v) trifluoroacetic acid was used for solvent A. Analyte separation was achieved using a Zorbax SB-C18 column (150 × 2.1 mm, 5-μm particle size) at a flow rate of 0.3 ml/min. The gradient program was as follows: isocratic at 25% B (0–3 min), linear gradient from 25 to 65% B (3–7 min), linear gradient from 65 to 95% B (7–10 min), and isocratic at 95% B (10–11 min). The data were acquired in the full-scan and MS/MS modes. The MS conditions were as follows: capillary voltage, 3.5 kV; cone voltage, 35 V; source temperature, 120°C; desolvation temperature, 350°C; ionization, ESI in the positive ion mode; analyzer, W mode; and collision energy for MS/MS, 15, 30 and 45 eV. Data were acquired using an independent reference spray via the LockSpray interface to ensure high mass accuracy; the [M + H]+ ion of leucine enkephalin was used as the reference lock mass (m/z 556.2771). The data were analyzed using MassLynx version 4.1.
Comparison of Metabolite Formation between P450 3A4 and 3A5.
The incubation solutions containing 0.2 μM P450 enzyme (Supersomes), 0.1 M KPi buffer (pH = 7.4), and 50 μM lapatinib were prepared and kept at 37°C for 3 min. The incubation reactions were initiated by the addition of 10 μl of a 10 mM solution of NADPH in H2O (final concentration, 1.0 mM) or H2O as a control. The total volume of each preincubation solution was 100 μl, and the final organic solvent concentration was 1.0% (v/v) acetonitrile. After a 30-min incubation at 37°C, the reaction was terminated by the addition of acetonitrile containing 20 μM 11α-hydroxyprogesterone as an internal standard. Samples were cooled and centrifuged at 9000g for 3 min, and the supernatants were transferred to other tubes and kept at −80°C until LC-MS analysis. The samples were analyzed by the same LC-MS system, solvents, and MS conditions as those used for the reversibility assay. The gradient program was the same as that described under Structural Elucidation of Lapatinib Metabolites. The data were acquired in single-ion monitoring mode and the m/z values for M1, M2, M3, M4, and the internal standard were 475.1, 489.1, 597.1, 597.1, and 331.2, respectively. The peak area of each metabolite was normalized to that of the internal standard. Then, the ratio of metabolite formation (3A5/3A4) was calculated from the normalized peak area of each metabolite.
Results
Search for Lapatinib Adducts to P450 3A4.
A P450 3A4 reconstitution system with P450 reductase and cytochrome b5 was used to detect P450 3A4 protein adducted with reactive metabolites from lapatinib. Reduced glutathione (3 mM) was added to the reconstitution system for maximal catalytic activities of P450 3A4 (Philips and Shephard, 2005). It was previously confirmed that coincubation with reduced glutathione did not affect the rate of P450 3A4 inactivation by lapatinib (Teng et al., 2010). The LC-MS analysis of the sample from the −NADPH control incubation yielded a deconvoluted envelope with a mass number of 57,270, which corresponds to P450 3A4 apoprotein (Fig. 2A). The deconvolution analysis revealed that the +NADPH complete system gave the same spectrum as the control, showing no peaks that could correspond to P450 3A4 apoprotein adducted with reactive metabolites from lapatinib (Fig. 2B). The unmodified heme peak (m/z 616) from P450 3A4 was detected, but there were no peaks corresponding to a heme modified by lapatinib (data not shown). We also analyzed the data to look for an adduct to either P450 reductase or cytochrome b5. The mass spectra after deconvolution exhibited peaks of intact P450 reductase and cytochrome b5, but no mass shifted peak that can be considered as an adduct to them (data not shown). To confirm that P450 3A4 was actually inactivated by lapatinib in this reconstitution system, an activity assay was performed using testosterone as a substrate. Based on this assay, 31% of P450 3A4 enzymatic activity was inactivated after a 30-min incubation with lapatinib in the presence of NADPH.

LC-MS spectra and deconvoluted spectra for P450 3A4 apoprotein after 30-min incubation with lapatinib in the absence (A) and presence (B) of NADPH. The incubation was performed in the reconstitution system including purified P450 3A4 (500 pmol), P450 reductase (1.0 nmol), and cytochrome b5 (500 pmol).
Reversibility of MBI.
To investigate whether MBI by lapatinib is irreversible or quasi-irreversible, human liver microsomal incubations with or without potassium ferricyanide were performed according to a method reported previously (Watanabe et al., 2007). Quasi-irreversible inactivators are known to form MI complexes with P450 enzymes that can be dissociated by oxidation with potassium ferricyanide. The percentages of the metabolic activity [percentage control(0 min) and percentage control(30 min)] with or without treatment with potassium ferricyanide are shown in Table 1. Testosterone-6β-hydroxylation activity was inactivated by a 30-min preincubation with lapatinib in the absence of potassium ferricyanide [percentage control(30 min) = 43.2 ± 4.5% (mean ± S.D.)]. This inactivation was completely restored by treatment with potassium ferricyanide [percentage control(30 min) = 98.8 ± 4.0%], suggesting that MBI by lapatinib is quasi-irreversible and caused by MI complex formation. To investigate the inhibitory effect of the use of ferricyanide on P450 reactions, the percentage of inhibition of testosterone-6β-hydroxylation in the absence of lapatinib by potassium ferricyanide [percentage inhibition(ferricyanide)] was calculated from the metabolic activity of the samples with and without potassium ferricyanide as described under Materials and Methods. As a result, percentage inhibition(ferricyanide) was determined to be 27.4 ± 11.9% (mean ± S.D., n = 3).
Reversibility of MBI by lapatinib in pooled human liver microsomes
Pooled human liver microsomes (1 mg/ml) were preincubated with lapatinib (50 μM), followed by incubation with [+K3Fe(CN)6] or without [−K3Fe(CN)6] potassium ferricyanide. The percentage of the metabolic activity (percentage control) was calculated from testosterone 6β-hydroxylase activity after incubations with and without lapatinib, as described under Materials and Methods. Data represent the mean ± S.D. (n = 3).
MI Complex Formation.
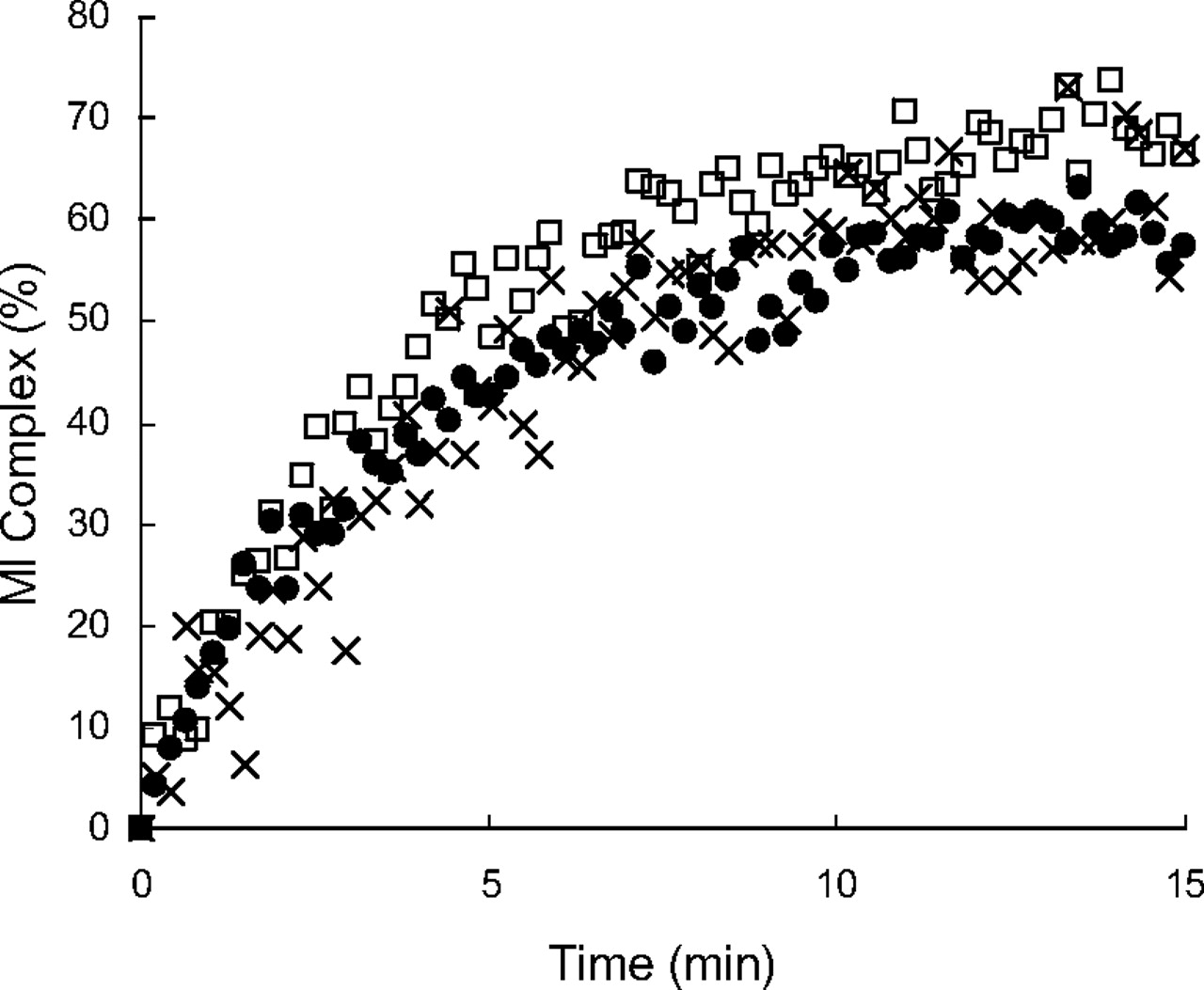
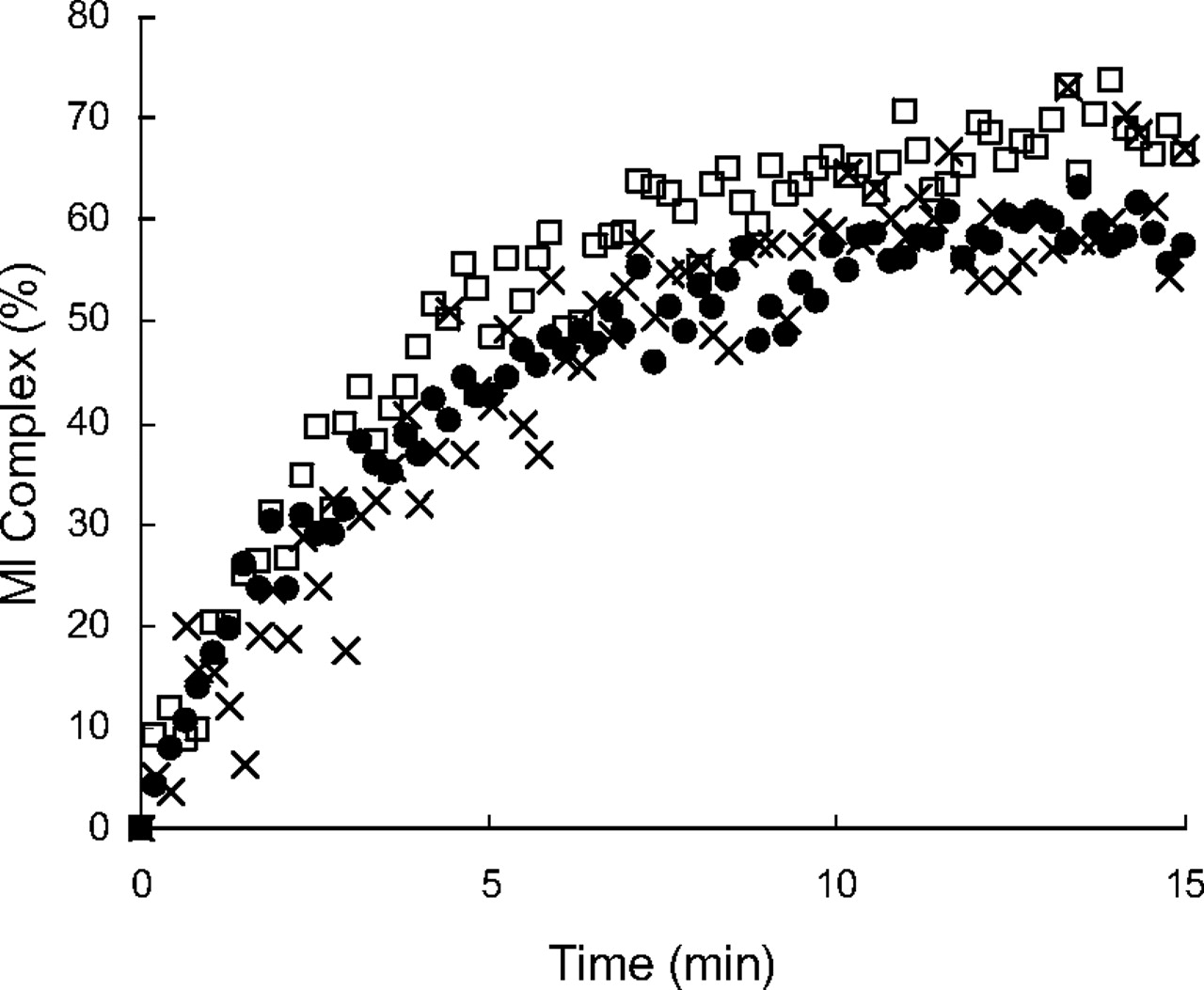
MI complexes are known to exhibit a signature Soret absorbance at approximately 455 nm (Franklin, 1974). To examine MI complex formation, absorption spectra of the incubation mixtures in triplicate of P450 3A4 Supersomes with lapatinib were monitored. The absorbance at approximately 455 nm was enhanced in a time-dependent manner after the addition of NADPH, as shown in representative spectra (Fig. 3A). In this method, diltiazem, a positive control for MI complex formation, also exhibited a similar increase in the absorption spectra (Fig. 3B). For comparison, we also investigated MI complex formation and MBI of P450 3A5 by lapatinib under the same incubation conditions as those for P450 3A4. The absorbance at approximately 455 nm was not enhanced after the addition of NADPH to the incubation mixture of P450 3A5 Supersomes with lapatinib (Fig. 3C). The concentrations of the MI complexes by lapatinib with P450 3A4 calculated from the extinction coefficient of 65 mM−1 cm−1 for the 455 and 490 nm absorbance difference (Liu and Franklin, 1985) were plotted against time after the addition of NADPH (Fig. 4). The time courses of MI complex formation were reproducible, and the concentrations of MI complex reached maximal levels by 15 min after addition of NADPH. Based on these data (n = 3), the initial rate (k) and maximal concentration (MICmax) of MI complex formation between P450 3A4 and lapatinib were calculated to be 0.25 ± 0.04 min−1 (mean ± S.D.) and 65 ± 5% (mean ± S.D.), respectively. In addition, an activity assay for MBI by lapatinib using P450 3A4 Supersomes was performed under the same incubation conditions as those for the absorption analysis for MI complex formation. Time- and NADPH-dependent P450 3A4 inactivation by lapatinib was observed as shown in Fig. 5A. Based on these data from the activity assay (n = 3), the initial rate and maximal percentage inactivation were calculated to be 0.28 ± 0.08 min−1 and 85 ± 2%, respectively. In contrast, the midazolam hydroxylation activity of P450 3A5 was not significantly inactivated by lapatinib (Fig. 5B).

MI complex formation of P450 3A4 by lapatinib at 50 μM (A), P450 3A4 by diltiazem at 10 μM (B), and P450 3A5 by lapatinib at 50 μM (C). P450 Supersomes were used for the incubation. The data represent absorption spectra acquired at 2.5 (□), 5 (▵), 10 (×), and 15 (○) min after the addition of NADPH in P450 Supersomes with lapatinib.
The time course of MI complex formation of P450 3A4 by lapatinib at 50 μM after the addition of NADPH. P450 Supersomes were used for the incubation. Each result in triplicate experiments is represented by a different symbol (□, ●, or ×).
The time-dependent inactivation of P450 3A4 (A) and 3A5 (B) after preincubation with lapatinib at 50 μM. P450 Supersomes were used for the incubation. The presence (■) and absence (□) of NADPH in the preincubation mixtures are shown. The data shown represent the mean ± S.D. from three separate experiments.
Structural Elucidation of Lapatinib Metabolites.
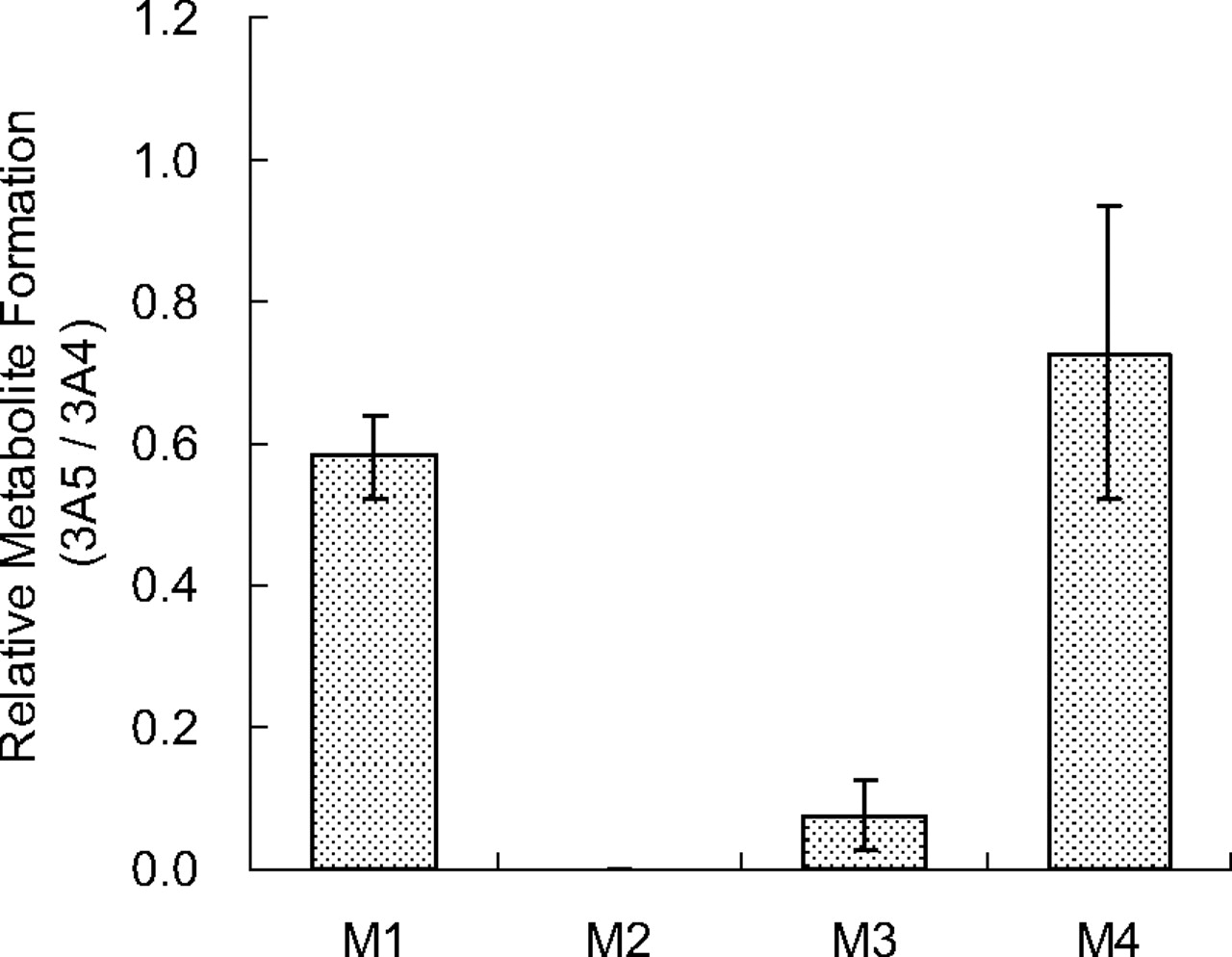
Structural analysis by LC-MS of metabolites after incubation of lapatinib with P450 3A4 Supersomes was performed to investigate the mechanism of MI complex formation. With the use of full-scan conditions, four metabolites related to oxidations of the secondary amine moiety of lapatinib, M1 [retention time (Rt) = 7.15 min, m/z 475], M2 (Rt = 8.34 min, m/z 489), M3 (Rt = 8.12 min, m/z 597), and M4 (Rt = 6.93 min, m/z 597), were detected as shown in mass chromatograms (Fig. 6 A). These peaks were not detected in the control sample without NADPH (data not shown). The molecular compositions were estimated to be C26H20 ClFN4O2 for M1, C26H18ClFN4O3 for M2, and C29H26ClFN4O5S for M3 and M4 by accurate mass measurements. Hydrogen-deuterium exchange measurements using D2O as an eluent displayed the molecular ions [M + D]+ at m/z 479 (M1), 492 (M2), 600 (M3), and 601 (M4), indicating that the numbers of exchangeable protons of the metabolites (M1–4) are 3, 2, 2, and 3, respectively. The MS/MS spectra and proposed structures based on the fragmentation patterns are shown in Fig. 6B. The molecular composition and mass fragmentation of M1 are consistent with the structure of N-dealkylated lapatinib. M2 is proposed to be the oxime form of N-dealkylated lapatinib based on its molecular composition, fragmentation pattern, and the number of exchangeable protons (2) described above. Both M3 and M4 were shown to be monooxygenated metabolites of the secondary amine side chain of lapatinib. On the basis of these findings along with the numbers of exchangeable protons (2), M3 is proposed to be a hydroxylamine of lapatinib. The formation of these metabolites by P450 3A5 was also examined and compared with that of with 3A4. Metabolite formation in a 30-min incubation with P450 3A5 relative to that observed in incubations with P450 3A4 is shown in Fig. 7. Significant differences were found for the formation of M2 and M3 between P450s 3A4 and 3A5. M2 was not detected in the incubation samples with P450 3A5, and the peak area of M3 for P450 3A5 was less than one-tenth of that for P450 3A4.

LC-MS chromatograms (A) and MS/MS spectra and proposed structures (B) of the metabolites in the sample after a 30-min incubation of lapatinib at 50 μM with P450 3A4 Supersomes in the presence of NADPH.

Ratios of metabolite formation catalyzed by P450 3A5 versus P450 3A4 after a 30-min incubation with lapatinib at 50 μM. P450 Supersomes were used for the incubation. The data shown represent the mean ± S.D. from three separate experiments. M2 was not detected in the P450 3A5 incubation sample.
Discussion
On the basis of recent findings (Teng et al., 2010), we anticipated that reactive metabolites of lapatinib would covalently bind to P450 3A4. However, no adducts of lapatinib to this enzyme were detected by LC-MS analysis (Fig. 2). In several studies, adducts of reactive metabolites of MBIs to some P450s have been detected (Blobaum et al., 2002; Baer et al., 2007; Yukinaga et al., 2007). In these cases, the mass spectra after deconvolution exhibited peaks of modified P450 apoprotein with a mass shift due to adduction, in addition to the intact P450 apoprotein peak. In the case of P450 3A4 incubated with lapatinib in the reconstitution system, the mass spectra did not exhibit any mass shifted peak that could be considered as P450 3A4 apoprotein or heme modified by lapatinib metabolites. Some minor peaks detected at approximately 57,400 Da in both the complete and control spectra are thought to be P450 3A4 apoprotein adducted with Na or K. From the lack of detection of an adduct to P450 reductase or cytochrome b5, the possibility that the adduction to these proteins contributes to the loss of P450 3A4 activity could be ruled out. Because 31% of the enzymatic activity of P450 3A4 in the reconstitution system was actually inactivated by lapatinib under the same conditions, a similar fraction of P450 3A4 would be expected to be modified in some way. Therefore, we speculated that the modification to P450 3A4 might not be irreversible but either was unstable or quasi-irreversible.
One of the methods to distinguish between irreversible and quasi-irreversible inactivation modes is a comparative activity assay of incubation samples with or without an oxidizing agent, potassium ferricyanide. Quasi-irreversible MI complexes can be dissociated by oxidation with potassium ferricyanide, and, thus, the enzymatic activity of P450 can be restored. In contrast, the enzymatic activity inactivated by irreversible binding should not be restored (Lin and Lu, 1998). One study (Watanabe et al., 2007) has provided an assay method using HLMs that distinguishes between irreversible and quasi-irreversible inactivation of P450 3A4 using 17 typical mechanism-based inactivators. Seven of them were well known quasi-irreversible inactivators: diltiazem, verapamil, nicardipine, amlodipine, erythromycin, clarithromycin, and troleandomycin. The testosterone-6β-hydroxylation activity inactivated after a 30-min incubation with the quasi-irreversible inactivators was restored by >20% with the addition of potassium ferricyanide. In contrast, the activity inhibited by the irreversible inactivators was not restored by potassium ferricyanide treatment (Watanabe et al., 2007). We used this method for evaluating the reversibility of lapatinib inhibition of P450 3A4. According to the published criterion for quasi-irreversible binding of >20% restoration as a percentage of control(30 min) with the addition of potassium ferricyanide (Watanabe et al., 2007), the major mechanism for MBI by lapatinib is quasi-irreversible and mediated by MI complex formation with P450 3A4. As shown in Table 1, a 32% loss of enzymatic activity by lapatinib was observed in the 0-min preincubation system without potassium ferricyanide treatment [i.e., percentage control(0 min) = 68.3 ± 3.0%]. The incubation mixtures for calculation of percentage control(0 min) were not subjected to primary incubations (i.e., 0-min primary incubation) but were subjected to 10-min secondary and 10-min tertiary incubations as described under Materials and Methods. These secondary and tertiary incubation mixtures contained the carryover of lapatinib from the primary mixtures. The maximal concentrations of lapatinib in the secondary and tertiary incubation mixtures are calculated to be 25 and 5 μM, respectively. It is probable that MI complex formation occurs during the 10-min secondary incubations in the presence of 25 μM lapatinib. Therefore, we presume that the 32% loss of enzymatic activity by lapatinib in the absence of potassium ferricyanide was mainly due to MI complex formation during the secondary incubation. This consideration is consistent with no significant loss of enzymatic activity (i.e., percentage control(0 min) = 93.1 ± 5.8%) by lapatinib in the presence of potassium ferricyanide because the MI complex formed during secondary incubations can be dissociated by potassium ferricyanide. According to a previous study (Watanabe et al., 2007) that established this reversibility assay, all seven well known quasi-irreversible inactivators mentioned above showed a similar tendency in percentage control(0 min) values. One concern about the use of ferricyanide is that it may affect P450 reactions because it is an oxidizing agent and can accept electrons from P450 reductase. It was found in our study that potassium ferricyanide did decrease the rates of testosterone-6β-hydroxylation in HLMs (by ∼27%) by comparing the metabolic activity in the absence of lapatinib between the samples with and without potassium ferricyanide. Therefore, it should be noted that ferricyanide has an inhibitory effect on P450 reactions. Although this assay may not be sufficient for a rigorous kinetic analysis, its utility in distinguishing between irreversible and quasi-irreversible binding was demonstrated in a previous study (Watanabe et al., 2007) as mentioned above. In addition, only the reversibility assay in the present study used HLMs. Because HLMs include the contributions of other P450 enzymes and the content of P450 reductase and cytochrome b5 in HLMs is not identical to that in Supersomes, values of inactivation in HLMs cannot be directly compared with those in Supersomes.
Absorption analysis of the incubation mixtures of P450 3A4 Supersomes with lapatinib was performed to confirm MI complex formation. The observed time- and NADPH-dependent increase of absorbance at approximately 455 nm demonstrated that lapatinib formed an MI complex with P450 3A4. The analyses, run in triplicate, showed good reproducibility, and the increase in absorption at approximately 450 nm by the positive control, diltiazem, was quite similar to that observed in previous studies (Ma et al., 2000; Mayhew et al., 2000). These observations support the validity of our experimental conditions. This result is not consistent with the previous report, in which an increase in absorption by lapatinib was not observed (Teng et al., 2010). One of the differences in procedures between the present and previous studies is the order of the addition of reaction components. In the present study, NADPH in water was added in the final step to incubation mixtures that were already equilibrated with lapatinib. In contrast, lapatinib dissolved in an organic solvent was added in the final step to incubation mixtures that already contained NADPH in the previous study. To investigate the cause of this discrepancy, we performed an additional absorption analysis for MI complex formation using the same instrument (Infinite M200 Tecan microplate reader) and the same conditions as those used in the previous study, except that NADPH in water was added to incubation mixtures already equilibrated with lapatinib. As a result, the absorbance corresponding to MI complex formation was enhanced in a time-dependent manner, which is consistent with the observation in the present study. The addition of organic solvent to aqueous incubation mixtures just before scans were started may have affected the turbidity of the mixtures and the baseline of absorbance in the initial state, which is considered as a possible cause for the lack of MI complex formation in the previous study. The initial rate (k) and maximal concentrations (MICmax) of MI complex formation were comparable to those for MBIs from the activity assays performed under the same conditions. These results indicate that the MBI of P450 3A4 by lapatinib is mostly due to quasi-irreversible MI complex formation, which is consistent with the results of the reversibility assay using potassium ferricyanide. Whereas chemical oxidation can dissociate MI complexes and reactivate P450 enzymes, MI complexes are so stable in vivo that quasi-irreversible inactivators such as lapatinib might cause clinically significant drug-drug interactions (Mayhew et al., 2000). From a toxicological standpoint, on the other hand, the quasi-irreversible MI complex formation is thought to be less significant than formation of reactive quinoneimine metabolites that can modify proteins irreversibly.
From the viewpoint of chemical structures, compounds including methylenedioxyphenyl, alkylamines, and alkylhydrazines are known to undergo metabolic activation by P450 enzymes and form MI complexes (Murray, 1997; Lin and Lu, 1998; Correia and Ortiz de Montellano, 2005). These moieties are metabolized to form carbene-iron complexes, nitroso-iron complexes, and nitrene-iron complexes, respectively. As shown in Fig. 1, lapatinib has a secondary amine between the furan and methanesulfonyl moieties that we hypothesize is sequentially metabolized to a nitroso intermediate that then forms an MI complex with P450 3A4. This hypothesis is supported by the structural analyses of lapatinib metabolites, which provide evidence for formation of the oxime metabolite (M2) by P450 3A4. Alkylnitroso intermediates are generally unstable and tautomerize to more stable oxime forms (Mansuy et al., 1977). In addition, the observation that the oxime metabolite was not generated by P450 3A5 is consistent with the lack of MI complex formation by lapatinib with P450 3A5. With regard to the reaction sequence from secondary amines to nitroso intermediates, two pathways based on initial metabolic reactions have been proposed, namely, oxidation of secondary amines to primary amines (N-dealkylation pathway) or to hydroxylamines (N-hydroxylation pathway) (Lindeke et al., 1979; Cerny and Hanzlik, 2005). Although the N-dealkylation pathway has often been cited as the major pathway, a recent study using desipramine, (S)-fluoxetine, and N-desmethyldiltiazem demonstrated that the major pathway from these secondary amines arises from N-hydroxylation rather than from N-dealkylation (Hanson et al., 2010). In the case of lapatinib, one of the metabolites by P450 3A4 was characterized to be a hydroxylamine metabolite (M3) from its molecular composition, MS/MS fragmentation, and deuterium exchange experiments. Moreover, P450 3A5 was shown to have a significantly lower ability to generate M3 compared with P450 3A4. These data suggest that the generation of M3, namely N-hydroxylation of lapatinib, is the initial step in the pathway to MI complex formation and responsible for the difference between P450 3A4 and 3A5. Several studies have found that typical quasi-irreversible inactivators of P450 3A4, such as erythromycin, diltiazem, nicardipine, and verapamil, exhibit no discernible MI complex formation with P450 3A5 and have weaker inactivation effects on P450 3A5 than on 3A4 (McConn et al., 2004; Wang et al., 2005). Whereas the mechanism responsible for the significant differences of MI complex formation between P450 3A4 and 3A5 is not fully understood, it might be explained in their different abilities to form secondary hydroxylamine metabolites. Although this study focuses on the mechanism of MBI via the N-dealkylation pathway, the O-dealkylation pathway is another major pathway of P450 3A4 metabolism of lapatinib. Because the proportion of N- and O-dealkylation pathways may be diverse in the population because of P450 3A5 polymorphisms, further studies on the contribution of P450 isoforms to each metabolic pathway are important to clarify the clinical relevance of these bioactivation pathways in terms of both DDIs and liver injury.
In conclusion, our results demonstrate that MBI of P450 3A4 by lapatinib is mainly due to quasi-irreversible MI complex formation and not adduction of a reactive metabolite to P450 3A4 apoprotein or heme. MI complex formation is proposed to be mediated via N-hydroxylation of the secondary amine group in lapatinib, followed by formation of a reactive nitroso intermediate. Taken together with recent findings, our results contribute to an understanding of lapatinib metabolism in which there are at least two different bioactivation pathways as shown in Fig. 8. Bioactivation of the secondary amine group is considered to be important in terms of DDIs, whereas bioactivation to form the quinoneimine metabolite may initiate reactions that lead to liver injury.

Proposed metabolic pathway of lapatinib.
Authorship Contributions
Participated in research design: Takakusa, Wahlin, Hanson, and Nelson.
Conducted experiments: Takakusa, New, and Chan.
Contributed new reagents or analytic tools: Zhao.
Performed data analysis: Takakusa.
Wrote or contributed to the writing of the manuscript: Takakusa, Wahlin, Hanson, and Nelson.
Acknowledgments
We acknowledge Chelsea Stewart for help in expression and purification of rat P450 reductase.
Footnotes
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [GM32165]; the Eli Lilly Foundation (to M.D.W.); and Daiichi Sankyo Co., Ltd. (to H.T.).
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.037531.
-
ABBREVIATIONS:
- P450
- cytochrome P450
- DDI
- drug-drug interaction
- MI complex
- metabolic-intermediate complex
- LC
- liquid chromatography
- MBI
- mechanism-based inactivation
- LC
- liquid chromatography
- MS
- mass spectrometry
- ESI
- electrospray ionization
- HLMs
- human liver microsomes
- KPi
- potassium phosphate
- MS/MS
- tandem mass spectrometry.
- Received December 3, 2010.
- Accepted February 28, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}