


Abstract
Metabolism-dependent inhibition (MDI) of cytochrome P450 is usually assessed in vitro by examining whether the inhibitory potency of a drug candidate increases after a 30-min incubation with human liver microsomes (HLMs). To augment the IC50 shift, many researchers incorporate a dilution step whereby the samples, after being preincubated for 30 min with a high concentration of HLMs (with and without NADPH), are diluted before measuring P450 activity. In the present study, we show that the greater IC50 shift associated with the dilution method is a consequence of data processing. With the dilution method, IC50 values for direct-acting inhibitors vary with the dilution factor unless they are based on the final (postdilution) inhibitor concentration, whereas the IC50 values for MDIs vary with the dilution factor unless they are based on the initial (predilution) concentration. When the latter data are processed on the final inhibitor concentration, as is commonly done, the IC50 values for MDI (shifted IC50 values) decrease by the magnitude of the dilution factor. The lower shifted IC50 values are a consequence of data processing, not enhanced P450 inactivation. In fact, for many MDIs, increasing the concentration of HLMs actually leads to considerably less P450 inactivation because of inhibitor depletion and/or binding of the inhibitor to microsomes. A true increase in P450 inactivation and IC50 shift can be achieved by assessing MDI by a nondilution method and by decreasing the concentration of HLMs. These results have consequences for the conduct of MDI studies and the development of cut-off criteria.
Introduction
Inhibition of cytochrome P450 (P450) enzymes is a well recognized cause of drug-drug interactions. This occurs by two general mechanisms: direct inhibition and metabolism-dependent inhibition (MDI). Both can cause clinically significant cytochrome P450 inhibition. For this reason, the U.S. Food and Drug Administration (FDA) and the European Medicines Agency both require an in vitro assessment of the ability of drug candidates to cause direct inhibition and MDI of the seven major drug-metabolizing P450 enzymes in human liver microsomes (HLMs) (FDA draft Guidance for Industry, 2006, http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm080499.htm; Huang et al., 2008; European Medicines Agency Guideline on the Investigation of Drug Interactions, 2010, http://www.ema.europa.eu/ema/pages/includes/document/open_document.jsp?webContentId=WC500090112).
A recent PhRMA publication on cytochrome P450 inhibition focuses on the conduct of in vitro experiments to identify drug candidates that cause MDI (Grimm et al., 2009). The PhRMA consensus paper recommends that MDI be assessed in vitro by examining whether a 30-min incubation of the drug candidate with HLMs in the presence of NADPH increases its inhibitory potency relative to a 30-min incubation in the absence of NADPH. According to the consensus paper, most pharmaceutical researchers use the fold difference between these two values (i.e., the magnitude of the IC50 shift) to decide whether a drug candidate should be evaluated in vivo for its ability to cause clinically significant MDI of cytochrome P450. Surprisingly, industry cut-off values for what constitutes evidence for MDI based on the magnitude of the IC50 shift range from 1.2 to 10 (Grimm et al., 2009). Furthermore, the consensus paper reported that approximately half of the pharmaceutical researchers surveyed incorporate a dilution step in the design of their experiments to evaluate MDI, such that, after being incubated for 30 min with a relatively high concentration of HLMs, the samples are diluted (e.g., 10-fold) before measuring P450 enzyme activity.
The studies described here were prompted by the observation that the magnitude of the IC50 shifts reported with the dilution method were much greater than the magnitude of the IC50 shift we determined with the nondilution method. For example, using a nondilution method (with HLMs at 0.1 mg/ml), we determined that furafylline, an irreversible MDI of CYP1A2, causes a 20-fold shift in IC50 value (Yerino et al., 2007), whereas a 400-fold shift was reported for the dilution method (with an initial concentration of HLMs at 2.0 mg/ml followed by a 10-fold dilution to 0.2 mg/ml) (Perloff et al., 2009). This phenomenon was not unique to furafylline; all MDIs are reported to have lower shifted IC50 values when evaluated by the dilution method compared with the nondilution method, which implies that all MDIs cause more enzyme inactivation when they are incubated with relatively high concentrations of HLMs.
The possibility that all MDIs cause more cytochrome P450 inactivation when the concentration of HLMs is increased is not only counterintuitive but is inconsistent with the results of our studies on the effects of protein concentration on P450 inactivation by MDIs such as 8-methoxypsoralen and furafylline (Yerino et al., 2007; Ogilvie et al., 2008). Accordingly, we sought another explanation for the observation that MDIs have lower shifted IC50 values when assessed by the dilution method. The hypothesis we developed is that the lower shifted IC50 values associated with the dilution method are a consequence of how the data are processed. The basis for this hypothesis is illustrated in Fig. 1, which shows the design of a typical dilution assay in which the initial concentration of HLMs is 1.0 mg/ml and the final concentration, after a 10-fold dilution, is 0.1 mg/ml. Figure 1 shows the conditions for examining direct inhibition either by performing a zero time preincubation (IC50 curve A) or by incubating the inhibitor with HLMs for 30 min in the absence of NADPH (IC50 curve B) and the conditions for examining MDI, which involves incubating the inhibitor with HLMs for 30 min in the presence of NADPH (IC50 curve C). In this scheme, we posit that the IC50 value for direct inhibition (IC50 curves A and B) is governed by the final (postdilution) concentration of inhibitor because direct inhibition can only occur in the presence of a substrate, which is added after the dilution step. In contrast, we posit that the IC50 value for MDI (IC50 curve C) is governed by the initial (predilution) concentration of inhibitor because MDI occurs during the 30-min preincubation of the inhibitor with HLMs, which occurs before the dilution step. If the shifted IC50 values are based on the final concentration, as they commonly are, they will be artificially lowered by the dilution factor, and, correspondingly, the magnitude of the IC50 shift will be artificially increased by a factor of 10.

Illustration of a typical IC50 shift experiment incorporating a dilution step. The flow diagram represents the typical sequence of incubations when performing a dilution step as part of an IC50 shift experiment to identify metabolism-dependent inhibitors of cytochrome P450. Initial preincubations are performed with a relatively high concentration of human liver microsomes (typically 1 mg/ml) with 0- and 30-min preincubations (±NADPH). Samples are then diluted (typically 10-fold) to a lower concentration of HLMs (0.1 mg/ml) for incubation with an appropriate P450 marker substrate. When determining IC50 shifts by this method, the metabolism-dependent inhibition component (the 30-min plus NADPH samples) occurs before the dilution step (during the initial preincubation period); accordingly, IC50 curve C should theoretically be based on the initial (predilution) concentration of inhibitor. Conversely, the direct inhibition component [the 0-min (A) and 30-min minus NADPH (B) samples] occurs after the dilution step (during the incubation with the P450 marker substrate); accordingly, IC50 curves A and B should theoretically be based on the final (postdilution) concentration of inhibitor.
In this study, we present experimental evidence to support the hypothesis that the apparent greater sensitivity of the dilution method over the nondilution method is a consequence of data processing. We also show that the dilution method does not result in more MDI of cytochrome P450 but, in many cases, actually results in considerably less MDI. In fact, a true increase in cytochrome P450 inactivation and an increase in the IC50 shift can be achieved by decreasing the concentration of HLMs. These results have important consequences for the conduct of MDI studies and the development of cut-off criteria for MDI.
Materials and Methods
Chemicals and Reagents.
Diltiazem, erythromycin, S-fluoxetine, furafylline, methimazole, mibefradil, mifepristone, paroxetine, ticlopidine, troleandomycin, and verapamil were purchased from Sigma-Aldrich (St. Louis, MO); fluconazole and gemfibrozil glucuronide were purchased from Toronto Research Chemicals Inc. (North York, ON, Canada); tienilic acid was purchased from Cypex (Dundee, UK); azamulin was purchased from BD Biosciences (San Jose, CA). The sources of the other reagents used in this study have been described elsewhere (Robertson et al., 2000; Ogilvie et al., 2006; Paris et al., 2009).
Test System.
Pooled human liver microsomes (n = 16, mixed gender) were prepared from nontransplantable livers and characterized at XenoTech, LLC (Lenexa, KS) as described previously (Pearce et al., 1996; Parkinson et al., 2004).
In Vitro P450 Inhibition.
The effects of known P450 inhibitors were evaluated in IC50 shift experiments with and without a preincubation step (in the presence and absence of NADPH) as described previously (Ogilvie et al., 2008; Paris et al., 2009), either with no dilution or with a 10- to 40-fold dilution step. Conditions for the nondilution method are summarized in Table 1. The inhibitors were incubated at 37°C in 200-μl incubation mixtures containing pooled HLMs (≤0.1 mg/ml), potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM, pH 7.4), an NADPH-generating system (consisting of 1 mM NADP, 5 mM glucose 6-phosphate, and 1 unit/ml glucose-6-phosphate dehydrogenase), and a P450 marker substrate at a concentration approximately equal to its Km, at the final concentrations indicated in Table 1. Reactions were initiated by the addition of an NADPH-generating system and terminated after 5 min by the addition of 200 μl of acetonitrile containing the appropriate internal standard (deuterium-labeled metabolite). Precipitated protein was removed by centrifugation (920g for 10 min at 10°C). Calibration and quality control metabolite standards were prepared in zero time incubations. The analytical procedures are summarized in Table 1. Three IC50 curves were generated: A, a zero time preincubation with an inhibitor to measure direct inhibition; B, a 30-min incubation of the inhibitor with HLMs in the absence of NADPH (a second measure of direct inhibition); and C, a 30-min incubation of the inhibitor with HLMs in the presence of NADPH to measure MDI. Selected inhibitors were also assessed with nondilution IC50 shift experiments conducted at one-tenth the standard concentration of HLMs listed in Table 1.
Experimental conditions for measuring microsomal cytochrome P450 activity for enzyme inhibition studies
Ionization mode indicates the type of ionization (ESI) and the polarity (+ or −).
When the dilution method was used, the initial concentration of HLMs was increased 10-fold above the final concentration of HLMs listed in Table 1. The inhibitors were preincubated at 37 ± 1°C for zero minutes (A), for 30 min in the absence of NADPH (B), for 30 min in the presence of NADPH, or C, after which the samples were diluted 10-fold, mixed with P450 marker substrate (at the final concentrations listed in Table 1), and incubated for 5 min. Reactions were terminated by the addition of an equal volume of acetonitrile containing the appropriate internal standard and analyzed by LC/MS/MS (Table 1). In selected cases, the initial concentration of HLMs was increased 20-, 30-, or 40-fold, and, after the 30-min preincubation, the samples were diluted 20-, 30-, and 40-fold, respectively.
Additional P450 inhibition experiments were conducted with selected inhibitors to determine IC50 values for direct inhibition (IC50 curve B) under standard conditions (detailed in Table 1) and at one-fourth the HLMs concentration (e.g., 0.1 mg/ml → 0.025 mg/ml) but at 4 times the substrate incubation time (20 min) to keep the overall extent of substrate metabolism the same.
KI and kinact Determination.
To determine KI and kinact for the inactivation of CYP2C19, various concentrations of S-fluoxetine (3, 10, 30, 60, and 100 μM) were incubated at 37°C for 3, 6, 9, 15, or 30 min with two concentrations of NADPH-fortified pooled HLMs (1.0 and 4.0 mg/ml). After the preincubation step, duplicate samples were diluted 10- or 40-fold (to give a final concentration of 0.1 mg protein/ml) into incubation medium containing 400 μM S-mephenytoin (∼10 × Km) and an NADPH-generating system. The diluted samples were incubated for 5 min and processed to measure residual CYP2C19 activity as summarized in Table 1.
Metabolic Stability of Selected Inhibitors.
The metabolic stability of selected P450 inhibitors, namely ticlopidine (0.2 μM), tienilic acid (0.5 μM), S-fluoxetine (10 μM), paroxetine (0.2 μM), azamulin (0.1 μM), and mibefradil (0.2 μM), was determined at two concentrations of microsomal protein (typically 0.1 or 1.0 mg/ml, unless otherwise noted) under conditions described above for P450 inhibition experiments. To determine inhibitor loss over time, aliquots (100 μl) from a single-vessel incubation were transferred at 1-min intervals for 15 min to an equal volume of acetonitrile containing an internal standard. The samples were analyzed by LC/MS/MS, as summarized in Table 2.
Analytical conditions for measuring inhibitor depletion by LC/MS/MS
Ionization mode indicates the type of ionization (i.e., ESI) and the polarity (+ or −).
Microsomal Binding of Selected Inhibitors.
Selected P450 inhibitors, namely ticlopidine (0.2 μM), tienilic acid (0.5 μM), S-fluoxetine (10 μM), paroxetine (0.2 μM), azamulin (0.1 μM), and mibefradil (0.2 μM), were incubated (500 μl) with pooled HLMs (typically 0.1 or 1.0 mg/ml) in the presence or absence of an NADPH-generating system at 37°C for 30 min. After the 30-min incubation, microsomes were isolated in 20 min by centrifugation at ∼21,000g relative centrifugal force in a 5417R Eppendorf microcentrifuge at room temperature. Identical incubations without HLMs served as a control for nonspecific binding of the inhibitors to the incubation vessel. After centrifugation, the samples were processed three ways. First, to determine the contribution of both nonspecific binding and metabolism to inhibitor depletion, an aliquot (200 μl) of those samples that were incubated with HLMs in the presence of NADPH was transferred to an equal volume of acetonitrile and analyzed by LC/MS/MS as summarized in Table 2. Second, to determine the contribution of microsomal binding alone (fumic) to inhibitor depletion, an aliquot (200 μl) of those samples that were incubated with HLMs in the absence of NADPH (to prevent metabolic loss) was transferred to an equal volume of acetonitrile and analyzed by LC/MS/MS (Table 2). Third, to determine the contribution of metabolism alone to inhibitor depletion, an equal volume of acetonitrile was added directly to postcentrifugation samples that had been incubated with HLMs in the presence of NADPH. This solubilized any inhibitor bound nonspecifically to microsomes or the vessel. The amount of inhibitor remaining was determined by LC/MS/MS (Table 2).
In the case of fluoxetine, the unbound fraction at various concentrations of HLMs (fumic) was estimated both experimentally (as described above) and theoretically as described by Hallifax and Houston (2006) and Austin et al. (2002) (eqs. 1 and 2, respectively).

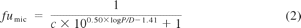
Assessment of MDI Reversibility.
Selected P450 inhibitors, namely ticlopidine (0.2 μM), tienilic acid (0.5 μM), S-fluoxetine (10 μM), paroxetine (0.2 μM), azamulin (0.1 μM), and mibefradil (0.2 μM), were incubated with various concentrations of NADPH-fortified pooled HLMs (0.1, 0.25, 0.5, 1, 2, and 3 mg/ml) at 37°C for 30 min under conditions described above. Incubations with solvent alone (acetonitrile; 0.5%, v/v) were conducted at each concentration of HLMs and served as negative controls. After the 30- min incubation, microsomal protein was reisolated by ultracentrifugation (100,000g for 60 min at 4°C). The supernatant fraction was discarded, and the resultant microsomal pellets were rinsed twice with wash buffer (150 mM potassium chloride and 10 mM EDTA, pH 7.4) to remove residual inhibitor and/or any reversible inhibitory metabolites. The microsomal pellets were resuspended in 250 mM sucrose, and microsomal protein concentration was determined by the Pierce BCA Protein Assay (Pierce Chemical, Rockford, IL). Residual P450 activity was assessed at a final concentration of 0.05 or 0.1 mg protein/ml as described in Table 1.
Data Processing and Statistical Analysis.
All IC50 values were determined by nonlinear regression with XLfit3 (version 3.0.5; ID Business Solutions Ltd., Guildford, Surrey, UK) or by GraFit (version 4.0.21; Erithricus Software Ltd., Horley, Surrey, UK), as detailed previously (Paris et al., 2009). Data from the KI and kinact determinations were processed with a validated Laboratory Information Management System (Galileo version 3.3; Thermo Fisher Scientific, Waltham, MA). To determine the rate of enzyme inactivation at each inhibitor concentration tested, the data were analyzed by a two-step method incorporating nonlinear regression. The first step calculated the apparent slope of enzyme inactivation (kobs) for each inhibitor concentration based on the following formula (eq. 3):
 For this equation, based in part on a method described by Kitz and Wilson (1962), the natural log of the ratio of the residual activity (Et) to the control activity (E0) (where the residual activity is the rate after a defined preincubation period with the test article) is plotted against preincubation time for each concentration of inhibitor.
For this equation, based in part on a method described by Kitz and Wilson (1962), the natural log of the ratio of the residual activity (Et) to the control activity (E0) (where the residual activity is the rate after a defined preincubation period with the test article) is plotted against preincubation time for each concentration of inhibitor.
In the second step, KI and kinact were calculated by solving the nonlinear equation (eq. 4) described by Jones et al. (1999):
 This equation is analogous to the Michaelis-Menten equation, where kobs represents the rate of enzyme inactivation at each inhibitor concentration, [I] is the initial (predilution) inhibitor concentration, KI is the inhibitor concentration that produces half the maximum rate of enzyme inactivation (analogous to Km), and kinact represents the maximum rate of enzyme inactivation (analogous to Vmax). This equation assumes there is negligible change in inhibitor concentration during the incubation period and that the loss of enzyme activity is due solely to enzyme inactivation.
This equation is analogous to the Michaelis-Menten equation, where kobs represents the rate of enzyme inactivation at each inhibitor concentration, [I] is the initial (predilution) inhibitor concentration, KI is the inhibitor concentration that produces half the maximum rate of enzyme inactivation (analogous to Km), and kinact represents the maximum rate of enzyme inactivation (analogous to Vmax). This equation assumes there is negligible change in inhibitor concentration during the incubation period and that the loss of enzyme activity is due solely to enzyme inactivation.
Results
Most of the P450 inhibition experiments described below involved measuring three IC50 curves (Fig. 1, designated A–C). Regardless of whether the assessment of P450 inhibition was based on the nondilution or dilution method, IC50 curve A (no preincubation) and IC50 curve B (a 30-min preincubation in the absence of NADPH) represent measures of direct inhibition, whereas IC50 curve C (a 30-min preincubation in the presence of NADPH) represents a measure of MDI. For all the inhibitors used in this study, we observed only negligible differences between IC50 curves A and B. For simplicity, we present below only the results for IC50 curve B (as the measure of direct inhibition) and IC50 curve C (as the measure of MDI). For simplicity, we also describe the concentration of HLMs in the standard nondilution method as 0.1 mg/ml and the concentration of HLMs in the preincubation phase of the dilution method as 1.0 mg/ml even though, as shown in Table 1, the concentrations of HLMs for assays with midazolam (CYP3A4) and amodiaquine (CYP2C8) were one-half and one-eighth the so-called standard concentration, respectively.
Data Processing for Direct-Acting Inhibitors.
We hypothesized that, when P450 inhibition is assessed by the dilution method, the IC50 value for direct inhibition should be based on the final (postdilution) concentration of inhibitor because direct inhibition can occur only in the presence of substrate, which is added after the dilution step. This hypothesis was tested with fluconazole, a direct-acting inhibitor of CYP3A4 (Tran et al., 2002), and the results are shown in Table 3 and Fig. 2 (left). Figure 2 (top left) shows the results of a nondilution experiment, in which fluconazole was preincubated with HLMs (0.05 mg/ml) and NADPH for 0 or 30 min before the addition of midazolam to measure CYP3A4 activity. Having confirmed that fluconazole is a direct-acting acting inhibitor of CYP3A4 with an IC50 value of ∼6 μM (with no shift in IC50), we evaluated the inhibition of CYP3A4 by fluconazole with the dilution method, which incorporated a 10-, 20-, 30-, or 40-fold dilution step. The IC50 data were processed based on both the initial and final concentrations of fluconazole. As predicted, when the data were processed based on the final concentration, fluconazole inhibited CYP3A4 with an IC50 value of 5 to 8 μM, which matched that determined by the nondilution method (Table 3; Fig. 2, bottom left). In contrast, when the data were processed based on the initial concentration of fluconazole, the IC50 values increased as the fold dilution increased (Table 3; Fig. 2, middle left).
Experimentally determined IC50 values based on the initial or final inhibitor concentration and the ratio of IC50 values determined with dilution (10–40-fold) versus no dilution for fluconazole, a direct-acting inhibitor of CYP3A4, and methimazole and diltiazem, metabolism-dependent inhibitors of CYP3A4 based on midazolam 1′-hydroxylation (4 μM)
All values were rounded to two significant figures. [HLM] indicates microsomal protein concentration before and after dilution. [Initial] refers to initial or predilution concentrations of inhibitor; [Final] refers to final or postdilution concentrations.

The effect of data processing on IC50 values determined by the nondilution and dilution methods for fluconazole, a direct-acting inhibitor of CYP3A4, and methimazole and diltiazem, metabolism-dependent inhibitors of CYP3A4. Fluconazole (left), methimazole (middle), and diltiazem (right) were evaluated as CYP3A4 inhibitors based on a nondilution method (top graphs) and a dilution method that incorporated a 10-, 20-, 30-, or 40-fold dilution, as described under Materials and Methods. When the dilution method was used, the IC50 values were calculated based either on the initial (predilution) concentration of inhibitor (middle graphs) or the final (postdilution) concentration of inhibitor (bottom graphs). The IC50 values are listed in Table 3.
Data Processing for Metabolism-Dependent Inhibition.
The experiment conducted with the direct-acting inhibitor fluconazole was repeated with two MDIs of CYP3A4, namely methimazole and diltiazem. Figure 2 shows the results obtained with the nondilution method for methimazole (top middle) and diltiazem (top right). As expected, preincubating methimazole and diltiazem with HLMs (0.05 mg/ml) for 30 min in the presence of NADPH increased their inhibitory potency toward CYP3A4 as evidenced by the leftward shift in IC50 values (570 → 80 μM for methimazole; 89 → 18 μM for diltiazem).
To test the hypothesis that, when cytochrome P450 inhibition is assessed by the dilution method, the IC50 value for MDI (IC50 curve C) should be based on the initial (predilution) concentration of inhibitor (Fig. 1), we evaluated the MDI of CYP3A4 by methimazole and diltiazem with the dilution method, which incorporated a 10-, 20-, 30-, or 40-fold dilution. As predicted, when the data for MDI (IC50 curve C) were processed based on the initial inhibitor concentration, methimazole and diltiazem inhibited CYP3A4 with an IC50 value that closely matched (generally within a factor of 2) the IC50 value determined by the nondilution method (Table 3; Fig. 2). In contrast, when the data were processed based on the final inhibitor concentration (as is commonly done), the IC50 values decreased as the fold dilution increased. Obach et al. (2007) and Perloff et al. (2009) previously evaluated diltiazem as an MDI of CYP3A4 with an experimental design that incorporated a 10-fold dilution step and in which CYP3A4 activity was measured with both midazolam and testosterone. We performed the same 10-fold dilution experiment with both substrates and processed the data for MDI (IC50 curve C) from all three research groups based on the final concentration of diltiazem [like Obach et al. (2007) and Perloff et al. (2009)] and the initial concentration of diltiazem (which was shown in Table 3 and Fig. 2 to be the appropriate method for MDI). We also performed the MDI experiment by the nondilution method. The results are shown in Fig. 3.

Interlaboratory comparison of IC50 values determined by the dilution and nondilution methods for diltiazem, a metabolism-dependent inhibitor of CYP3A4. Diltiazem was evaluated as a metabolism-dependent inhibitor of CYP3A4 (measured with midazolam and testosterone) by a nondilution method (XenoTech: No dilution) and by a dilution method that incorporated a 10-fold dilution step (XenoTech: 10-fold dilution), as described under Materials and Methods. When the dilution method was used, the IC50 values were calculated based either on the final (postdilution) concentration of inhibitor (B) or the initial (predilution) concentration of inhibitor (C). The magnitude of the IC50 shift was based on the ratio of the IC50 values for direct inhibition (A) and the IC50 value for MDI based on the final inhibitor concentration (A/B) or initial inhibitor concentration (A/C). The corresponding data from Perloff et al. (2009) and Obach et al. (2007), both of whom used a 10-fold dilution method, are shown for comparative purposes. The IC50 values for all three research groups are shown in Table 4.
Figure 3A shows that the IC50 values for direct inhibition (IC50 curve B) were similar between the nondilution and dilution methods and were similar among all three groups, all of which appropriately based the values for IC50 curve B on the final concentration of diltiazem. The values for IC50 curve B reported by Obach et al. (2007) were slightly lower than those determined by Perloff et al. (2009) and us, which will be discussed later. Figure 3B shows that the IC50 values for MDI determined by the nondilution method were approximately 10 times greater than those determined by the dilution method when the data were processed based on the final concentration of diltiazem. This 10-fold difference is also apparent from the data reported by Obach et al. (2007) and Perloff et al. (2009). Figure 3C shows that this 10-fold difference between the nondilution and dilution methods essentially disappeared when the data for MDI determined by the dilution method were processed based on the initial concentration of diltiazem. When the magnitude of the IC50 shift was calculated (i.e., the ratio of the IC50 value for direct inhibition and the IC50 value for MDI), the same trend was observed, as shown in Fig. 3A/B and A/C. The magnitude of the IC50 shift determined by the dilution method (by all three groups) was approximately 10 times greater than that for the nondilution method when the data for MDI were processed based on the final concentration of diltiazem (Fig. 3A/B), but this difference largely disappeared when the data for MDI were processed based on the initial concentration of diltiazem (Fig. 3A/C).
Dilution Versus Nondilution for Multiple MDIs and P450 Enzymes.
Table 4 shows the results of experiments in which 14 known MDIs were evaluated by the nondilution and dilution methods as inhibitors of the seven major P450 enzymes (namely CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A4). When the dilution method was used (with a 10-fold dilution in all cases), the data for direct inhibition (IC50 curve B) were processed based only on the final inhibitor concentration. However, the data for MDI (IC50 curve C) were processed based on both the final inhibitor concentration (as is commonly done) and the initial concentration. Table 4 also contains corresponding data from comprehensive and well documented studies by two other research groups, namely those by Obach et al. (2007) and Perloff et al. (2009). These two groups evaluated the same cytochrome P450 enzymes with many of the same MDIs and many of the same P450 substrates as were used in the current study. Both groups used the dilution method, which in most, but not all, cases involved a 10-fold dilution step. Both groups reported IC50 values for direct and MDI based on the final concentration of inhibitor. In Table 4, we added an additional column to the data from Obach et al. (2007) and Perloff et al. (2009) to show the IC50 values for MDI based on the initial concentration of inhibitor.
Experimentally determined IC50 values for various metabolism-dependent inhibitors of human liver microsomal P450 enzymes with and without a 10-fold dilution based on the initial and final concentrations of inhibitor
[Initial] refers to processing the IC50 values based on an initial or predilution concentration of inhibitor, whereas [Final] refers to processing the IC50 values based on a final or postdilution concentration.
Table 4 shows that the IC50 values for direct inhibition (IC50 curve B) determined by the nondilution method agreed well (generally within a factor of 2) with those determined by the dilution method, and they also agreed well (generally within a factor of 2) with the IC50 values for direct inhibition reported by Perloff et al. (2009). Without exception, the IC50 values for direct inhibition reported by Obach et al. (2007) were slightly lower than those determined in the current study and those reported by Perloff et al. (2009), and an explanation for this small but systematic difference is provided later. Table 4 also shows that, when the dilution method is used, the IC50 values for MDI (IC50 curve C) determined in the current study agreed reasonably well (generally within a factor of 2) with those determined by Obach et al. (2007) and Perloff et al. (2009), although the latter values were once again uniformly lower (with one exception).
Shifted IC50 Values: Dilution Versus Nondilution.
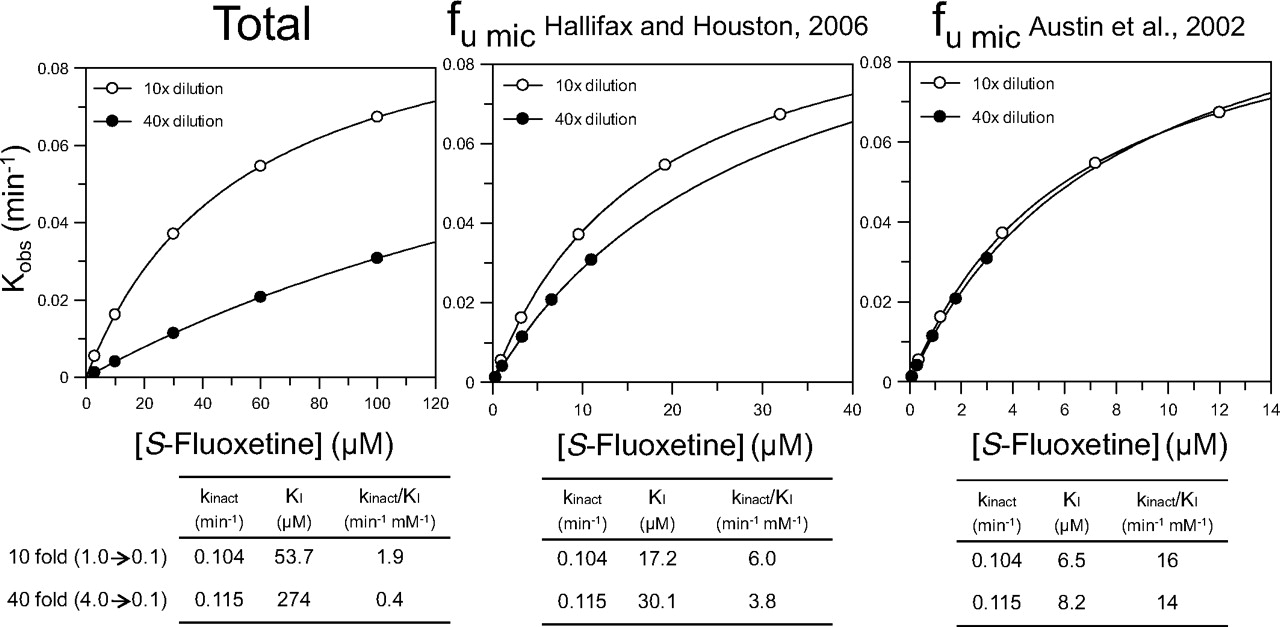
Figure 4 compares the shifted IC50 curves (IC50 curve C) determined by the nondilution method with those determined by the dilution method for 11 MDIs. In the latter case, the data were processed based on the initial inhibitor concentration (which is the appropriate concentration for MDI). The MDIs in Fig. 4 are divided into two groups. Those shown on the left include furafylline (CYP1A2), gemfibrozil glucuronide (CYP2C8), and three MDIs of CYP3A4, namely diltiazem, troleandomycin, and verapamil. In all of these cases, the two shifted IC50 curves (IC50 curve C from the dilution assay and IC50 curve C from the nondilution assay) were essentially superimposable, suggesting that the degree of P450 inactivation that occurred in the dilution and nondilution assay was similar. The MDIs shown in Fig. 4 (right) include ticlopidine (CYP2B6), tienilic acid (CYP2C9), S-fluoxetine (CYP2C19), paroxetine (CYP2D6), azamulin (CYP3A4), and mibefradil (CYP3A4). In these cases, the two shifted IC50 curves were not superimposable, and in all cases, IC50 curve C determined by the dilution method was shifted to the right of that determined by the nondilution method, suggesting that less P450 inactivation occurred with the dilution method.

A comparison of the shifted IC50 curves (IC50 curve C) determined by the dilution and nondilution methods for several metabolism-dependent inhibitors of cytochrome P450. Eleven known metabolism-dependent inhibitors were evaluated as MDI of various P450 enzymes by the nondilution method (gray circles) and by a dilution method that incorporated a 10-fold dilution step (black circles), as described under Materials and Methods. The shifted IC50 values (IC50 curve C) for the dilution assay are based on the initial (predilution) concentration of inhibitor. Shifted IC50 values for both the nondilution and dilution assays are shown in Table 4. Cytochrome P450 marker substrates are listed in Table 1.
For all the MDIs shown in Fig. 4 (left), the ratio of the two shifted IC50 curves (i.e., the shifted IC50 value for the nondilution method divided by the shifted IC50 value for the dilution method) was close to unity, as illustrated in Fig. 5. Assuming values that agreed within a factor of 2 can be considered the same, the results in Fig. 5 suggest that furafylline (CYP1A2), gemfibrozil glucuronide (CYP2C8), and several MDIs of CYP3A4, namely verapamil, diltiazem, troleandomycin, methimazole, and erythromycin, caused the same degree of P450 inactivation regardless of whether the dilution or nondilution method was used. In contrast, for all the MDIs shown in Fig. 4 (right), the ratio of the two shifted IC50 curves was less than unity, and the values fell outside of the 2-fold range (Fig. 5). These results suggest that those MDIs shown in Fig. 4 (right; i.e., those MDIs with a ratio of less than 0.5 in Fig. 5) caused less P450 inactivation when assayed by the dilution method, which suggests they caused less P450 inactivation when incubated with a relatively high concentration of HLMs.

The ratio of the shifted IC50 value (IC50 curve C) determined by the nondilution method and the dilution method for multiple metabolism-dependent inhibitors. IC50 ratios were calculated as the shifted IC50 value without a dilution step divided by the shifted IC50 value with a 10-fold dilution step, which was based on the initial (predilution) concentration of inhibitor. The corresponding IC50 curves are shown in Fig. 4, and the individual IC50 values are shown in Table 4.
Effect of Protein Concentration on the Extent of P450 Inactivation.
To investigate why the dilution method can lead to less P450 inactivation, we examined the effects of protein concentration on the extent of P450 inactivation by ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil based on an ultracentrifugation method (Buckley et al., 2008). Ticlopidine (0.2 μM), tienilic acid (0.5 μM), S-fluoxetine (10 μM), paroxetine (0.2 μM), azamulin (0.1 μM), and mibefradil (0.2 μM) were incubated with six concentrations of HLMs (from 0.1 to 3 mg/ml) at concentrations of inhibitor known to cause extensive, irreversible P450 inactivation when incubated with HLMs at 0.1 mg/ml. After a 30-min incubation in the presence of NADPH, the microsomes were isolated by ultracentrifugation, rinsed, resuspended, and assayed for cytochrome P450 activity. The results are shown in Fig. 6. All six MDIs showed the same trend; as the concentration of HLMs increased, the extent of P450 inactivation progressively decreased. Mibefradil (0.2 μM), for example, caused ∼70% inactivation of CYP3A4 when incubated with HLMs at 0.1 mg/ml but caused less than 10% inactivation at 1.0 mg/ml. At 2.0 and 3.0 mg/ml, mibefradil caused no detectable inactivation of CYP3A4. At 3.0 mg/ml, none of the MDIs examined caused more than 15% inactivation of cytochrome P450 at inhibitor concentrations that caused extensive P450 inactivation in HLMs at 0.1 mg/ml.

The effect of microsomal protein concentration on the degree of P450 inactivation by various metabolism-dependent inhibitors. Ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil were incubated (at the concentrations shown) with various concentrations of NADPH-fortified pooled HLMs (0.1, 0.25, 0.5, 1, 2, and 3 mg/ml) at 37°C for 30 min. The extent of irreversible P450 inhibition was assessed after reisolation of the microsomes by ultracentrifugation, as described under Materials and Methods, followed by measurement of residual P450 activity at 0.1 mg/ml HLMs, as described in Table 1.
Decreased P450 Inactivation with the Dilution Method.
To explain why certain MDIs (namely ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil) cause less P450 inactivation when assessed by the dilution method, we postulated that incubating these particular MDIs with a relatively high concentration of HLMs is associated with a decrease in the free concentration of inhibitor (due to nonspecific binding of the inhibitor to microsomes) or due to inhibitor depletion (due to extensive metabolism), or a combination of both. To test this hypothesis, we examined the metabolic stability of each inhibitor with a low concentration of HLMs (0.1 mg/ml for CYP2B6, 2C9, 2C19, and 2D6 assays and 0.05 mg/ml for CYP3A4 assays) and at 10 times this concentration (0.5 or 1.0 mg/ml); the former are the incubation conditions for the nondilution assay, and the latter are those for the dilution assay. For this experiment, we selected concentrations of inhibitor known to cause a substantial degree of P450 inactivation at 0.05 or 0.1 mg/ml (they were the same concentrations used in Fig. 6), and we measured the concentration of inhibitor every minute over a 15-min period. The results are shown in Fig. 7. S-Fluoxetine was essentially metabolically stable at both 0.1 and 1.0 mg/ml. In contrast, substantial inhibitor depletion was observed with the five other MDIs, and the extent of inhibitor depletion increased with increasing concentrations of HLMs. Ticlopidine was the least stable. At 1.0 mg/ml, ticlopidine was completely metabolized after 2 min, and complete metabolism was also observed after a 15-min incubation with 0.1 mg/ml.
An evaluation of the metabolic stability of selected metabolism-dependent inhibitors at a low and high concentration of human liver microsomes in the presence of NADPH. The metabolic stability of ticlopidine (A), tienilic acid (B), S-fluoxetine (C), paroxetine (D), azamulin (E), and mibefradil (F) (at the concentrations shown) was evaluated in human liver microsomes at a relatively low protein concentration (0.05 or 0.1 mg/ml; filled squares) and high protein concentration (0.5 or 1.0 mg/ml; open circles), as described under Materials and Methods. Disappearance of each inhibitor was monitored at 1-min intervals for 15 min by LC/MS/MS as summarized in Table 2.
For the study depicted in Fig. 7, metabolic stability was evaluated over a 15-min incubation period. However, with both the nondilution and dilution assays, the usual preincubation time is 30 min. We also evaluated the metabolic stability of ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil after a 30-min incubation with HLMs at 0.1 and 1.0 mg/ml (or 0.05 and 0.5 mg/ml for the CYP3A4 assays), and the results are summarized in Table 5. Once again, S-fluoxetine was the most stable compound; only 21% was consumed after 30 min regardless of the concentration of HLMs, making it the only MDI in this group to meet the FDA's recommendation that less than 30% of the inhibitor should be consumed during the incubation period (FDA draft Guidance for Industry, 2006). After a 30-min incubation with HLMs at 0.5 or 1.0 mg/ml, ticlopidine, tienilic acid, azamulin, and mibefradil were completely metabolized (≥99%), and 88% of paroxetine was metabolized. Less metabolism was observed at 0.05 or 0.1 mg/ml, but even at these relatively low concentrations of HLMs, extensive metabolism (>30%) was observed for ticlopidine, paroxetine, and mibefradil.
Effects of microsomal protein concentration on the unbound (free) concentration of selected metabolism-dependent inhibitors and the extent of inhibitor depletion
All values were rounded to two significant figures.
Table 5 also shows the results of experiments to assess the degree of nonspecific binding of ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil to HLMs at 0.1 and 1.0 mg/ml (or 0.05 and 0.5 mg/ml for the CYP3A4 inhibitors). In all cases, binding to microsomes increased with increasing concentrations of HLMs. Tienilic acid bound to microsomes the least (<15%), followed by azamulin (<25%). Ticlopidine, S-fluoxetine, paroxetine, and mibefradil all bound extensively (78–86%) to HLMs at 0.5 or 1.0 mg/ml; substantial binding (30–64%) was also evident at 0.05 or 0.1 mg/ml.
The final column in Table 5 shows the combined effect of nonspecific binding and metabolism on the percentage loss of inhibitor. When incubated with NADPH-fortified HLMs at 0.5 or 1.0 mg/ml for 30 min, metabolism and nonspecific binding caused dramatic decreases in the free concentration of ticlopidine (100%), tienilic acid (100%), S-fluoxetine (91%), paroxetine (98%), azamulin (100%), and mibefradil (100%). The corresponding values obtained at 0.05 or 0.1 mg/ml were as follows: ticlopidine, ∼100%; tienilic acid, ∼28%; S-fluoxetine, 52%; paroxetine, 74%; azamulin, 11%; mibefradil, 97%.
When binding to microsomes is the chief cause for the decrease in cytochrome P450 inactivation, it should be possible to reconcile the shifted IC50 values obtained by the dilution and nondilution methods by basing the IC50 values on fumic, the free concentration of inhibitor. In contrast, when inhibitor depletion is the chief cause for the decrease in cytochrome P450 inactivation, it should be possible to obtain lower shifted IC50 values (and hence an increase in the magnitude of the IC50 shift) by decreasing the concentration of HLMs. As shown below, both these predictions were borne out experimentally.
Correcting Shifted IC50 Values for Microsomal Binding.
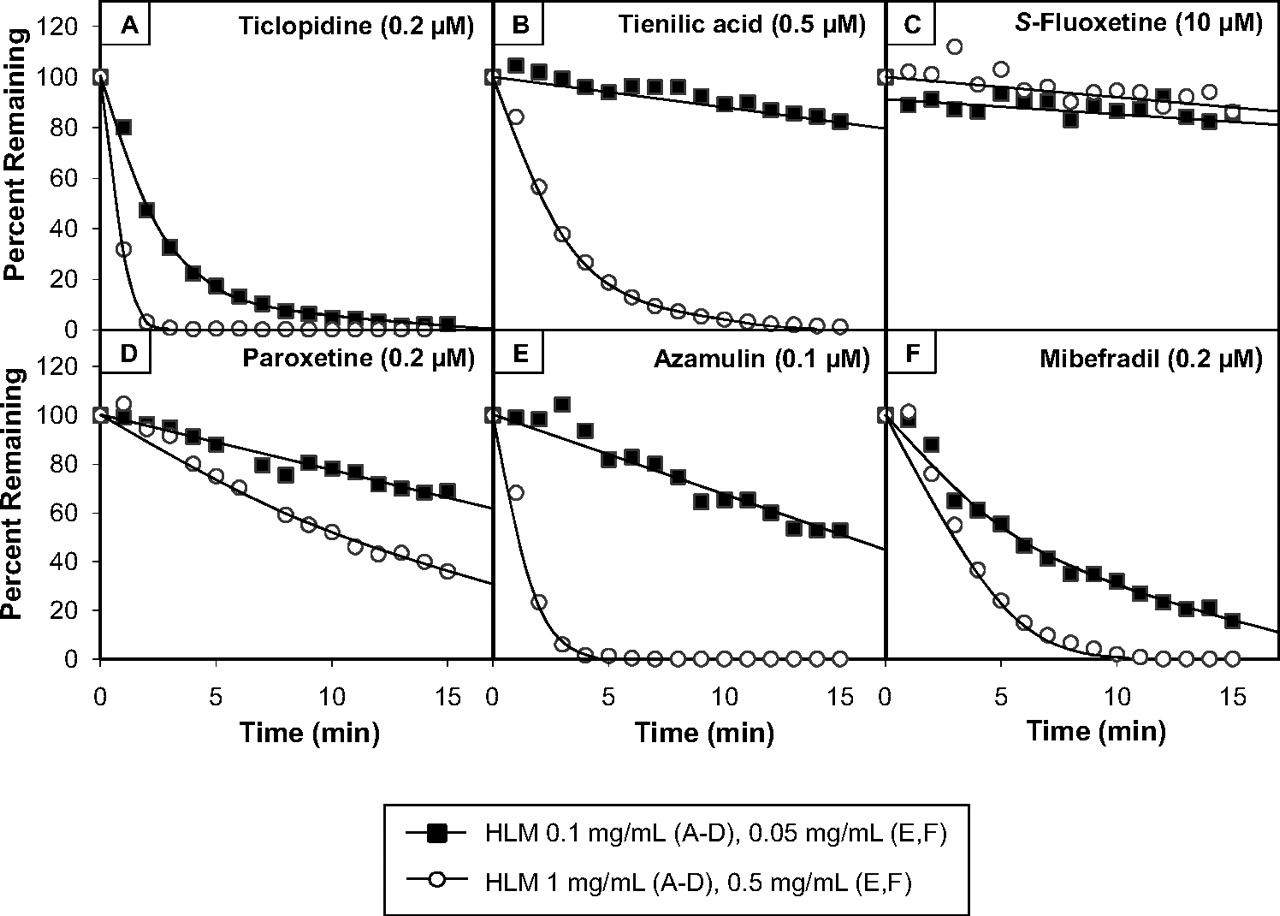
Of the six MDIs that caused less cytochrome P450 inactivation when incubated with a relatively high concentration of HLMs (ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil), only one of them, S-fluoxetine, was metabolically stable (i.e., less than 30% was consumed during a 30-min incubation with NADPH-fortified HLMs at either 0.1 or 1.0 mg/ml). However, S-fluoxetine bound nonspecifically to HLMs. As shown in Table 5, approximately 46 and 78% of fluoxetine bound to HLMs at 0.1 and 1.0 mg/ml, respectively. These results suggest that the influence of protein concentration on the shifted IC50 values for S-fluoxetine, and only fluoxetine among this particular group of MDIs, should be fully correctable by taking nonspecific binding into account. Table 6 shows the shifted IC50 values for ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil determined by the nondilution method (at 0.05 or 0.1 mg/ml) and the dilution method (at 0.5 or 1.0 mg/ml during the preincubation step). When the values were based on the nominal concentration of inhibitor, the shifted IC50 values determined by the nondilution method were all lower than those determined by the dilution method; the ratio of the two values ranged from 0.044 to 0.38. When the shifted IC50 values were based on the free concentration of inhibitor (fumic), the ratio for CYP2C19 inhibition by S-fluoxetine approached unity (the actual value was 0.94). Based on the nominal concentration of S-fluoxetine, the shifted IC50 values determined at 0.1 and 1.0 mg/ml were 5.3 and 14 μM, respectively, but these changed to 2.9 and 3.1 μM based on the free concentration of fluoxetine (Table 6). We also measured the rate of CYP2C19 inactivation (kobs) over a wide range of S-fluoxetine concentrations (3–100 μM) by the dilution method with a 10- and 40-fold dilution (in which case the preincubation concentration of HLMs was 1.0 and 4.0 mg/ml, respectively) to mimic the experimental design commonly used to measure KI and kinact. As shown in Fig. 8, when kobs was based on the nominal concentration of S-fluoxetine, the apparent rate of inactivation of CYP2C19 was greatly diminished at the higher protein concentration. However, when the data were processed based on the free concentration of S-fluoxetine, which was calculated according to both Hallifax and Houston (2006) and Austin et al. (2002), the rate of CYP2C19 inactivation at 4.0 mg/ml was essentially the same as that at 1.0 mg/ml HLMs. Incidentally, the nonspecific binding of S-fluoxetine to HLMs at 1.0 mg/ml was predicted to be 68% by Hallifax and Houston (2006) and 88% by Austin et al. (2002), which bracket our experimentally determined value of 78% (Table 5).
Shifted IC50 values and IC50 ratios corrected for observed microsomal binding
All IC50 values are rounded to two significant figures.
The kinetics of CYP2C19 inactivation (kobs) by S-fluoxetine based on the nominal concentration (left) and free concentration of inhibitor (center and right) each with a 10- and 40-fold dilution step. S-Fluoxetine (3–100 μM) was preincubated at 37°C for 3, 6, 9, 15, and 30 min with NADPH-fortified human liver microsomes (at 1.0 and 4.0 mg/ml) as described under Materials and Methods, after which the samples were diluted 10- or 40-fold (to 0.1 mg/ml) before measuring CYP2C19 activity (Table 1). These conditions represent the experimental design commonly used to measure KI and kinact. The left graph represents the direct plot of the initial rates of inactivation of CYP2C19 (kobs) based on the nominal total concentration of S-fluoxetine. The middle and right graphs show the impact of correcting the concentration of S-fluoxetine for nonspecific binding to microsomes based on the method of Hallifax and Houston (2006) (middle graph) and Austin et al. (2002) (right graph).
Effects of Decreasing Protein Concentration on P450 Inactivation.
The five metabolically unstable MDIs, namely ticlopidine, tienilic acid, paroxetine, azamulin, and mibefradil, were evaluated in a nondilution assay for their ability to inactivate cytochrome P450 at the usual concentration of HLMs (0.1 mg/ml for CYP2B6, 2C9, and 2C19 and 0.05 mg/ml for CYP3A4) and at one-tenth the usual concentration to test the hypothesis that lowering the concentration of HLMs increases the degree of P450 inactivation (as evidenced by a lower shifted IC50 value) and, hence, increases the magnitude of the IC50 shift. As shown in Table 7, decreasing the concentration of HLMs by a factor of 10 lowered the shifted IC50 values for CYP2B6 inactivation by ticlopidine (from 47 to 11 nM), CYP2C9 inactivation by tienilic acid (from 66 to 13 nM), CYP2D6 inactivation by paroxetine (from 51 to 25 nM), and CYP3A4 inactivation by azamulin (from 38 to 13 nM) and mibefradil (from 27 to 23 nM). The IC50 values for direct inhibition did not change appreciably (they remained within a factor of 2) when the concentration of HLMs was decreased 10-fold. Accordingly, lowering the concentration of HLMs by a factor of 10 increased the magnitude of the IC50 shift for ticlopidine (from 3.8- to 20-fold), tienilic acid (from 15- to 40-fold), paroxetine (from 15- to 25-fold), and azamulin (from 5.5- to 25-fold) (Table 7). It did not increase the magnitude of the IC50 shift for mibefradil because lowering the concentration of HLMs from 0.05 to 0.005 mg/ml caused a slight decrease in both the IC50 for direct inhibition (IC50 curve B) and the IC50 for MDI (IC50 curve C).
Effects of decreasing microsomal protein on the shifted IC50 values and magnitude of the IC50 shifts for extensively metabolized MDIs
IC50 shift experiments were performed by preincubating selected inhibitors with HLM at the stated concentrations for 30 min with or without NADPH. After the preincubation step, marker substrate reactions (5 min) were initiated by the addition of marker substrate ([S] = Km) without the incorporation of a dilution step. All values were rounded to two significant figures.
Effect of Substrate Incubation Time on IC50 Values.
It was noted above that the IC50 values for both direct inhibition (curve B) and MDI (curve C) determined by Obach et al. (2007) were slightly but consistently lower than those determined by Perloff et al. (2009) and us. These systematic differences appear to reflect differences in two experimental conditions, namely the concentration of HLMs and the incubation time with the P450 marker substrate. The experimental conditions used by each of the three groups when performing the dilution assay are summarized in Table 8, which shows that the values for both IC50 curves B and C determined by Obach et al. (2007) are generally lower than those determined by Perloff et al. (2009) and us, whereas the magnitude of the IC50 shift is generally smaller. Obach et al. (2007) generally used a lower concentration of microsomal protein and a longer substrate incubation time to measure cytochrome P450 activity than either Perloff et al. (2009) or we did. Because all three groups used a 10-fold dilution [with the exception of a 5-fold dilution for the CYP2C19 assay used by Perloff et al. (2009)], the fact that Obach et al. (2007) used a lower final concentration of HLMs means they also used a lower concentration of HLMs during the preincubation step in the dilution assay. Based on the results obtained with ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil (Table 7; Fig. 6), the lower concentration of HLMs used by Obach et al. (2007) would be expected to produce lower shifted IC50 values (IC50 curve C), as is indeed the case for all MDIs with the single exception of diltiazem (and this exception was only observed when CYP3A4 activity was measured with midazolam, not testosterone) (Table 8). However, a lower concentration of HLMs does not explain why the IC50 values reported by Obach et al. (2007) for direct inhibition (IC50 curve B) are consistently lower than those determined by us and Perloff et al. (2009), but this could be caused by the longer substrate incubation time, which would allow for a greater degree of “unintentional” MDI. (Ideally, no MDI should occur during the determination of IC50 curve B, which is a measure of direct inhibition.) To test this possibility, we determined IC50 values for direct inhibition (IC50 curve B) under our standard conditions (detailed in Table 1) and at one-fourth the protein concentration but at 4 times the substrate incubation time (to keep the overall extent of substrate metabolism the same). As shown in Fig. 9, decreasing the concentration of HLMs and increasing the substrate incubation time caused a decrease in IC50 curve B for all MDIs examined with the exception of diltiazem (and only when CYP3A4 was measured with midazolam). The results in Fig. 9 suggest that the longer incubation time with the P450 marker substrate (during which the inhibitor is present along with HLMs and NADPH) allows each MDI additional time to cause unintentional cytochrome P450 inactivation during the measurement of IC50 curve B.
Comparison of incubation conditions along with IC50 values (and the fold shift) with and without NADPH (curves B and C) for the dilution methods
All IC50 values are based on the final or postdilution concentrations.
Effect of the incubation time with P450 marker substrate in the presence of a metabolism-dependent inhibitor on IC50 curve B, a measure of the direct inhibition of cytochrome P450. IC50 values for direct inhibition (IC50 curve B) were determined for furafylline (0.03–30 μM), ticlopidine (0.1–100 μM), gemfibrozil glucuronide (0.03–30 μM), tienilic acid (0.01–5 μM), S-fluoxetine (0.03–30 μM), paroxetine (0.03–30 μM), azamulin (0.1–100 μM), diltiazem (0.1–100 μM), and verapamil (0.1–100 μM) as described under Materials and Methods. Condition X was based on the concentration of HLMs listed in Table 1 with a 5-min substrate incubation period. Condition Y was achieved by decreasing the concentration of HLMs by a factor of 4 and increasing the substrate incubation time by a factor of 4 so that the incubation time with marker substrate was increased to 20 min but the overall extent of substrate metabolism remained the same. The values represent the ratio of the IC50 values determined under each condition.
Can the Nondilution Method Fail to Detect MDI?
When the dilution method is used and when the data for MDI (IC50 curve C) are processed “incorrectly” based on the final concentration of inhibitor, the magnitude of the IC50 shift for all of the MDIs examined in this study was greater than that determined by the nondilution method (Table 4). Leaving aside the issue of whether the method of data processing is valid or not, this raises the question of whether the IC50 shifts determined by the nondilution method can be too small to identify MDIs. The nondilution method certainly succeeded in detecting an IC50 shift for all the MDIs examined in this study; all of them exceeded our IC50 shift cut-off value of 1.5. Nevertheless, we sought a case in which the dilution method (with incorrect data processing) could detect MDI and in which the nondilution method could not. The case we selected was the MDI of CYP2C19 by ticlopidine, and this choice was based on a recommendation from Scott Obach and Bob Walsky (Pfizer, Inc., Groton, CT), to whom we are most grateful. Ticlopidine was the most metabolically labile of all the MDIs examined in this study; it was completely metabolized after a 30-min incubation even with 0.1 mg/ml HLMs (Table 5; Fig. 7). We have also shown that the nondilution method can detect the MDI of CYP2B6 by ticlopidine (Tables 4 and 6). However, ticlopidine is also an MDI of CYP2C19, but it inactivates CYP2C19 less efficiently than it does CYP2B6 (Venkatakrishnan and Obach, 2007). The kinact/KI for P450 inactivation is 530 min−1 · mM−1 for CYP2B6 but only 23 min−1 · mM−1 for CYP2C19 (Venkatakrishnan and Obach, 2007).
We examined ticlopidine as an inhibitor of CYP2C19 under four conditions: 1) the nondilution method at 0.1 mg/ml (the usual protein concentration), 2) the nondilution method at 0.01 mg/ml (one-tenth the usual protein concentration), 3) the dilution method at 1.0 mg/ml during the preincubation step (and a final concentration of 0.1 mg/ml), and 4) the dilution method at 0.1 mg/ml during the preincubation (and a final concentration of 0.01 mg/ml). For the dilution method, the data for IC50 curve C were processed based on both the final concentration of ticlopidine (as is commonly done) and the initial inhibitor concentration. The results are shown in Fig. 10. With the nondilution method at 0.1 mg/ml, ticlopidine inhibited CYP2C19 and caused a 1.8-fold shift in IC50 curves (from 1.05 to 0.59 μM), which exceeded our cut-off value of 1.5 (Fig. 10A). With the corresponding dilution method (with a predilution and postdilution protein concentration of 1.0 and 0.1 mg/ml, respectively), the shift was 1.7-fold (from 0.57 to 0.34 μM) when the data were processed based on the final inhibitor concentration (Fig. 10C). It is noteworthy that, in theory, the IC50 shift for the dilution method should have been 18-fold, i.e., 10 times the value obtained by the nondilution method because of the incorrect data processing. With the nondilution method at 0.01 mg/ml (one-tenth the usual protein concentration), ticlopidine caused a 6.7-fold shift in IC50 values (from 0.57 to 0.085 μM) (Fig. 10B). With the corresponding dilution method (with a predilution and postdilution protein concentration of 0.1 and 0.01 mg/ml, respectively), the shift was 10.7-fold (from 0.78 to 0.073 μM) when the data were processed based on the final inhibitor concentration (Fig. 10D). Once again, in theory, the IC50 shift for the dilution method should have been 67-fold. Regardless of whether the initial (preincubation) protein concentration was 1.0 or 0.1 mg/ml, when the shifted IC50 values from the dilution experiments were processed based on the initial inhibitor concentration, IC50 curve C was shifted to the right of IC50 curve B (Fig. 10E) or essentially not shifted (Fig. 10F).

An evaluation of ticlopidine as a metabolism-dependent inhibitor of CYP2C19 by the nondilution method (at 0.1 and 0.01 mg/ml) and by the dilution method (at 10 times the initial protein concentration). As described under Materials and Methods, IC50 values for the inhibition of CYP2C19 by ticlopidine were determined under four conditions: 1) the nondilution method at 0.1 mg/ml (A); 2) the nondilution method at 0.01 mg/ml (B); 3) the dilution method at 1.0 mg/ml during the preincubation step and a final concentration of 0.1 mg/ml; and 4) the dilution method at 0.1 mg/ml during the preincubation and a final concentration of 0.01 mg/ml). For the dilution experiments, the IC50 curves for direct inhibition (IC50 curve B; gray circles) were always based on the final (postdilution) concentration of inhibitor. The IC50 curves for metabolism-dependent inhibition (IC50 curve C; black circles) were based either on the final (postdilution) concentration of inhibitor (C and D) or the initial (predilution) concentration of inhibitor (E and F).
Discussion
The potential for drug candidates to function as MDIs of cytochrome P450 is usually assessed in vitro by examining whether a 30-min incubation of the drug candidate with HLMs in the presence of NADPH increases its inhibitory potency (i.e., lowers the value of IC50 curve C) relative to a 30-min incubation in the absence of NADPH (IC50 curve B) or a zero time preincubation (IC50 curve A). A recent PhRMA consensus paper (Grimm et al., 2009) reported that approximately half the researchers surveyed incorporate into this experimental design a dilution step whereby the samples, after being preincubated with a relatively high concentration of HLMs (usually 10-fold higher) for 30 min, are diluted (e.g., 10-fold) before measuring cytochrome P450 activity. The rationale for the dilution step is that it improves the detection of MDI by increasing the magnitude of the IC50 shift ostensibly by lessening the direct inhibitory effect of the drug candidate (Silverman, 1995; Atkinson et al., 2005; Obach et al., 2007; Polasek and Miners, 2007; Fowler and Zhang, 2008; Grime et al., 2009; Grimm et al., 2009; Perloff et al., 2009). In the present study, we examined several known MDIs of cytochrome P450 and determined IC50 values B and C (representing direct inhibition and MDI, respectively) by the nondilution and dilution methods. The results of this study can be summarized as follows.
When the dilution method is used, the IC50 values for direct inhibition (IC50 curves A and B) vary with the dilution factor unless they are based on the final (postdilution) concentration of inhibitor, as shown in Table 3 and Fig. 2 for fluconazole. This finding can be rationalized on the basis that direct inhibition occurs only in the presence of the P450 marker substrate, which occurs after the dilution step (Fig. 1). If the IC50 values are based on the initial concentration of inhibitor, they increase in proportion to the dilution factor (Table 3; Fig. 2).
In contrast, the IC50 values for MDI determined by the dilution method (IC50 curve C) vary with the dilution factor unless they are based on the initial (predilution) concentration of inhibitor, as shown for diltiazem and methimazole (Table 3; Figs. 2 and 3). This finding can be rationalized on the basis that MDI occurs during the preincubation of the inhibitor with NADPH-fortified HLMs, which occurs before the dilution step (Fig. 1). It also can be rationalized on the basis that when the dilution method is used to measure the kinetics of inactivation (KI and kinact), the data are universally processed based on the initial concentration of inhibitor. When the shifted IC50 values are based on the final concentration of inhibitor, as they commonly are, the values decrease in proportion to the dilution factor (Table 3; Figs. 2 and 3).
In its commonly used format (with all data processed on the final concentration of inhibitor), the dilution method does not produce higher values for IC50 curve B (i.e., it does not produce less direct inhibition as is commonly assumed) but produces instead lower values for IC50 curve C, which produces a proportionally larger IC50 shift. The lower shifted IC50 values give the impression that incubating MDIs with high concentrations of HLMs during the preincubation phase of the dilution assay leads to more P450 inactivation, but this is erroneous and misleading. It disguises the reality that, in many cases, increasing the concentration of HLMs results in considerably less P450 inactivation (Table 4; Figs. 4⇑–6) because of nonspecific binding of the inhibitor to microsomes and/or inhibitor depletion, as shown for ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil (Table 5; Fig. 7).
In some, but not all, cases, the shifted IC50 values determined by the dilution method match those determined by the nondilution method provided the former is based on the initial concentration of inhibitor. This is true for all the MDIs shown in Fig. 4 (left) (furafylline, gemfibrozil glucuronide, diltiazem, troleandomycin, and verapamil). However, the shifted IC50 values determined by the dilution method do not match those determined by the nondilution method even if the former are based on the initial inhibitor concentration in those cases when the MDI binds nonspecifically to HLMs and/or is metabolically labile, which is true for all the MDIs shown in Fig. 4 (right) (ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil).
When an MDI is suitably metabolically stable (<30% loss, based on the FDA's criteria) but binds nonspecifically to HLMs, the IC50 values determined by the dilution method can be matched with those determined by the nondilution method by (a) multiplying the value by the dilution factor and (b) basing the IC50 value determined by both the dilution and nondilution methods on the free concentration of inhibitor (fumic). This was demonstrated for the MDI of CYP2C19 by S-fluoxetine (Table 6; Fig. 8).
Increasing the concentration of HLMs can result in a significant decrease in the extent of P450 inactivation due to nonspecific binding and/or inhibitor depletion. A true increase in the extent of P450 inactivation can be achieved by decreasing the concentration of HLMs, as shown for ticlopidine, tienilic acid, S-fluoxetine, paroxetine, and azamulin (Table 7; Fig. 10). A low concentration of HLMs used in the nondilution assay produces a true decrease in shifted IC50 values and a true increase in the magnitude of the IC50 shift, as shown in Table 7 and Fig. 10.
Increasing the incubation time with P450 marker substrate (from 5 to 20 min) can result in a sizeable decrease in the value of IC50 curve B (and curve A) because of unintended MDI of cytochrome P450 during the assessment of direct inhibition, as shown in Fig. 9 [and as previously highlighted by Ghanbari et al. (2006)]. This can reduce the magnitude of the IC50 shift.
Based on the results of this study, we recommend that the identification of MDIs be based on a shift in IC50 values determined by the nondilution method because it offers several advantages over the dilution method: it is technically easier because it involves fewer steps; the method of data processing is unambiguous (there is no predilution and postdilution inhibitor concentration to choose from); and the problems associated with nonspecific binding and inhibitor depletion are reduced compared with the dilution method. We further recommend that 1) the concentration of HLMs in the nondilution assay be relatively low (0.1 mg/ml or less), 2) the preincubation time with the drug candidate (potential inhibitor) be relatively long (i.e., 30 min or longer), 3) the incubation time with P450 marker substrate be relatively short (5 min or less), and 4) the concentration of drug candidate range up to 10 times the total plasma Cmax at steady state (Cmax, ss) and the P450 marker substrate concentration be roughly equal to Km to permit an appropriate estimate of the IC50 value for direct inhibition (IC50 curves A and B).
The results in Table 7 and Fig. 10 establish that the degree of P450 inactivation and, hence, the magnitude of the IC50 shift can be increased by decreasing the concentration of HLMs from 0.1 mg/ml (the concentration widely used in the nondilution method) to 0.01 mg/ml. Accordingly, an argument could be made that the nondilution method should be conducted with HLMs at 0.01 mg/ml (or even less). Unfortunately, decreasing the concentration of HLMs to 0.01 mg/ml may pose an analytical challenge, especially for low turnover substrates such as S-mephenytoin (CYP2C19). This analytical challenge should not be solved by increasing the incubation time with the P450 substrate, which should be kept relatively short (5 min or less) to prevent unintended P450 inactivation during the substrate incubation period (Fig. 9).
The results of this study raise several questions about the conduct of MDI studies by the dilution method, and they have implications for the development of cut-off criteria for MDI.
The Dilution Method Artificially Lowers the Shifted IC50 Value, but Is This Important?
Yes it is because the shifted IC50 value (the absolute value for IC50 curve C) has considerable utility, and this utility is lost if the absolute shifted IC50 value varies depending on whether it was determined by the nondilution method or the dilution method, and further varies depending on the actual dilution factor. Shifted IC50 values can be used to estimate kinact/KI (a measure of the efficiency of P450 inactivation) based on the following equation (eq. 5) from Maurer et al. (2000):
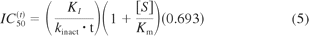 where t is the incubation time with the MDI and [S]/Km is the ratio of the concentration of P450 marker substrate relative to Km (the Michaelis-Menten constant). Maurer et al. (2000) validated this relationship experimentally by examining the kinetics of nitric-oxide synthase inactivation by two MDIs [NG-monomethyl-l-arginine and N(5)-(-iminoethyl)-l-ornithine] based on a nondilution method. The relationship was also validated for the MDI of P450 enzymes by Berry and Zhao (2008), although they derived a modified version of the Maurer equation (eq. 6) to take into account differences in the concentration of HLMs to measure IC50 shifts (by a nondilution assay) and the concentration of HLMs to determine kinact and KI (which incorporated a 10-fold dilution):
where t is the incubation time with the MDI and [S]/Km is the ratio of the concentration of P450 marker substrate relative to Km (the Michaelis-Menten constant). Maurer et al. (2000) validated this relationship experimentally by examining the kinetics of nitric-oxide synthase inactivation by two MDIs [NG-monomethyl-l-arginine and N(5)-(-iminoethyl)-l-ornithine] based on a nondilution method. The relationship was also validated for the MDI of P450 enzymes by Berry and Zhao (2008), although they derived a modified version of the Maurer equation (eq. 6) to take into account differences in the concentration of HLMs to measure IC50 shifts (by a nondilution assay) and the concentration of HLMs to determine kinact and KI (which incorporated a 10-fold dilution):
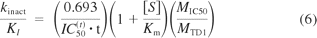 where MIC50 is the concentration of protein in the assay to determine IC50 and MTDI is the concentration of protein in the assay to measure kinact and KI. The ability to estimate kinact and KI from shifted IC50 values [based on a modified Cheng and Prusoff (1973) equation] was also validated by Krippendorff et al. (2009) both theoretically (in silico) and experimentally using a nondilution approach in which the rate of formation of fluorescent metabolites (one for CYP1A2 and one for CYP3A4) was measured in situ at 2-min intervals (the “progress curve” method). Obach et al. (2007) reported a strong empirical correlation between shifted IC50 values and kinact/KI (as would be expected from the equations above). The KI values were determined by the dilution method, and they were based on the initial concentration of inhibitor (as they should be). The shifted IC50 values were also determined by the dilution method, but these were based on the final inhibitor concentration; consequently, the shifted IC50 values are approximately one-tenth the values based on the initial inhibitor concentration (and many of them are likely to be one-tenth of those determined by the nondilution method). Consequently, if investigators determine shifted IC50 values by the nondilution method and use the relationship between shifted IC50 and kinact/KI published by Obach et al. (2007), their estimates of kinact/KI could be off by as much as 1 order of magnitude, assuming a dilution factor of 10 were used.
where MIC50 is the concentration of protein in the assay to determine IC50 and MTDI is the concentration of protein in the assay to measure kinact and KI. The ability to estimate kinact and KI from shifted IC50 values [based on a modified Cheng and Prusoff (1973) equation] was also validated by Krippendorff et al. (2009) both theoretically (in silico) and experimentally using a nondilution approach in which the rate of formation of fluorescent metabolites (one for CYP1A2 and one for CYP3A4) was measured in situ at 2-min intervals (the “progress curve” method). Obach et al. (2007) reported a strong empirical correlation between shifted IC50 values and kinact/KI (as would be expected from the equations above). The KI values were determined by the dilution method, and they were based on the initial concentration of inhibitor (as they should be). The shifted IC50 values were also determined by the dilution method, but these were based on the final inhibitor concentration; consequently, the shifted IC50 values are approximately one-tenth the values based on the initial inhibitor concentration (and many of them are likely to be one-tenth of those determined by the nondilution method). Consequently, if investigators determine shifted IC50 values by the nondilution method and use the relationship between shifted IC50 and kinact/KI published by Obach et al. (2007), their estimates of kinact/KI could be off by as much as 1 order of magnitude, assuming a dilution factor of 10 were used.
The “artificially” low shifted IC50 values produced by the dilution method can also be a source of confusion when the efficiency of P450 inactivation is considered in terms of the number of moles of inhibitor required to inactivate each mole of P450 enzyme (i.e., the partition ratio). As an example, the inactivation of CYP2C9 by tienilic acid is presented here. The partition ratio for the inactivation of CYP2C9 by tienilic acid is ∼12 (Lopez-Garcia et al., 1994), meaning 13 molecules of tienilic acid are consumed for each molecule of CYP2C9 inactivated. Based on P450 quantitation by mass spectrometry (Kawakami et al., 2011), the specific content of CYP2C9 in HLMs is approximately 80 pmol of CYP2C9/mg protein, hence an incubation containing HLMs at 0.1 mg/ml contains roughly 8 pmol of CYP2C9 (i.e., 8 nM enzyme). This represents the situation in our typical nondilution assay (Table 1), and under these conditions, the shifted IC50 value for CYP2C9 inactivation by tienilic acid was 66 nM (Table 4). Assuming for the sake of simplicity the incubation volume was 1 ml, this means 66 pmol of tienilic acid were required to inactivate half of the 8 pmol of CYP2C9 present in the incubation, which is roughly 17 pmol of tienilic acid/pmol CYP2C9. When we used the dilution method, the initial concentration of HLMs was 1.0 mg/ml; hence, the initial concentration of CYP2C9 was roughly 80 pmol/ml. Based on the initial concentration of tienilic acid, the shifted IC50 value for CYP2C9 inactivation was 570 nM (Table 4); hence, 570 pmol of tienilic acid inactivated half the 80 pmol of CYP2C9 in the incubation, which is roughly 14 pmol tienilic acid/pmol CYP2C9, a value that agrees well with that determined by the nondilution method (17 pmol of tienilic acid/pmol CYP2C9). It also agrees with the experimentally determined partition ratio of ∼12 (Lopez-Garcia et al., 1994; Johnson, 2008), which is not surprising because tienilic acid is completely metabolized by HLMs at 1.0 mg/ml. If these same calculations are performed with the shifted IC50 value based on the final concentration of tienilic acid, the shifted IC50 value decreases by a factor of 10 to 57 nM (Table 4). Now it appears that half of the 80 pmol of CYP2C9 (the amount of CYP2C9 present in the preincubation stage, which is when the enzyme inactivation takes place) is inactivated by only 57 pmol of tienilic acid (under conditions in which tienilic acid is completely metabolized); hence, tienilic acid could be mistaken for having a partition ratio close to zero. In other words, if the shifted IC50 value for tienilic acid is based on the final (postdilution) concentration of inhibitor, the dilution assay gives the erroneous impression that tienilic acid is almost a perfect MDI of CYP2C9; one that inactivates the enzyme during a single catalytic cycle. Determining the “true” shifted IC50 value (or the magnitude of the true IC50 shift) also has implications for developing criteria for MDI cut-off values, which is discussed later.
If Increasing the Concentration of HLMs Increases the Metabolism of the MDI, How Can This Result in Less P450 Inactivation?
Increasing the concentration of HLMs increases the metabolism of all MDIs, and yet, in many cases, this results in less cytochrome P450 inactivation (Figs. 4⇑–6). How can increasing the metabolism of the inhibitor result in less MDI? To appreciate the reason for this phenomenon, one must consider the influence of partition ratio. For example, consider a hypothetical study with an MDI of CYP3A4 (or any other enzyme) that has a partition ratio of 9, meaning that 10 molecules of inhibitor are consumed for each molecule of CYP3A4 inactivated. The other nine molecules of metabolite are released from the active site either because they fail to inactive CYP3A4 (they miss their target, so to speak) or because they are noninhibitory metabolites. [Note that a partition ratio of 9 is a realistic number for MDIs of many cytochrome P450 enzymes (Johnson, 2008).] The hypothetical study is conducted with such a low concentration of HLMs that the incubation contains a single molecule of CYP3A4. If 10 molecules of inhibitor were added to the incubation, that single molecule of CYP3A4 would be inactivated (i.e., there would be 100% inactivation), and it may take complete metabolism of the inhibitor to achieve this. If the concentration of HLMs was increased 10-fold (as would be done in the preincubation phase of the dilution method), there would be 10 molecules of CYP3A4. If 10 molecules of inhibitor were added, all of the inhibitor would be metabolized, but only one of the 10 molecules of CYP3A4 would be inactivated (i.e., there would be 10% inactivation). The other nine molecules of CYP3A4 would be spared simply because the concentration of HLMs was increased 10-fold. This illustrates the important principle that when the molar ratio of inhibitor to enzyme falls below the partition ratio, the inhibitor will be completely metabolized before the enzyme is inactivated. In the hypothetical example, all 10 molecules of CYP3A4 could be inactivated by increasing the concentration of inhibitor 10-fold (so that each molecule of CYP3A4 could metabolize 10 molecules of inhibitor), but there is little point performing the dilution assay if it necessitates a corresponding increase in inhibitor concentration.
Implications for Cut-off Value Criteria.
The FDA has specified criteria and cut-off values, such as [I]/Ki < 0.1 [where [I] is total drug concentration (plasma Cmax at steady state) and Ki is the direct (reversible) inhibition constant] for assessing the potential of direct-acting inhibitors to cause clinically relevant cytochrome P450 inhibition, but the agency has not done so for MDI (referred to as time-dependent inhibition in the FDA draft guidance document). The FDA states that “any time-dependent and concentration-dependent loss of initial product formation rate indicates mechanism-based inhibition” and that this finding should be followed up with human in vivo studies (FDA draft Guidance for Industry, 2006). The PhRMA consensus paper on MDI indicates that most respondents use the magnitude of the IC50 shift to assess whether a drug candidate is an MDI of cytochrome P450, but the reported cut-off values range from 1.2 to 10 (Grimm et al., 2009). The lower and upper values likely reflect the cut-off values for respondents using the nondilution method and dilution method, respectively. It is curious that, if the upper cut-off value for the dilution method (10) were corrected for the most commonly used dilution factor (10) so that it might be applied to the nondilution method, the corrected cut-off value would be unity, which would occur when there was no shift between IC50 curves C and B. This is the first hint that cut-off values cannot be scaled between the nondilution method and the dilution method simply by applying the dilution factor.
Using the nondilution method, our criteria for a positive result for MDI is a 1.5-fold shift between IC50 curve C and either IC50 curve A or B. If we applied this same cutoff to the dilution method (one that incorporated a 10-fold dilution step), this cut-off value should theoretically be set to 15 (10 times larger to account for the 10-fold “artificial” increase in IC50 shift). For each of the 14 MDIs shown in Table 4, the IC50 shift with the nondilution method exceeds 1.5. The MDIs that caused the smallest IC50 shifts were verapamil-CYP3A4 (3.5-fold shift) and ticlopidine-CYP2B6 (3.8-fold shift). However, when the dilution method was used, and when the data were processed based on the final concentration of inhibitor (which amplifies the shift), three MDIs failed to meet a cutoff of 15, namely ticlopidine-CYP2B6 (5.1-fold shift), azamulin-CYP3A4 (11-fold shift), and mibefradil (7.5-fold shift), and one only just exceeds it: tienilic acid-CYP2C9 (16-fold shift). In other words, four MDIs that clearly exceed the cut-off value of 1.5 in the nondilution assay would be classified as non-MDIs or marginal MDIs with the dilution assay if the cutoff were set to 15. Therefore, the cutoff for the nondilution method (1.5-fold) cannot be applied to the dilution method simply by adjusting the cut-off value for the magnitude of the dilution. The situation is even worse in the case of the inactivation of CYP2C19 by ticlopidine (Fig. 10). Based on the nondilution method at 0.1 mg/ml, ticlopidine caused a 1.8-fold shift (which narrowly exceeded the 1.5-fold cutoff). Likewise, ticlopidine caused a 1.7-fold shift in the dilution assay (with a 10-fold dilution) when the data were processed based on the final concentration of ticlopidine. This suggests that, even though the dilution method magnifies the IC50 shift for several MDIs by a factor of 10 (for a 10-fold dilution), it would be necessary for the dilution method to have the same cut-off value (1.5) as the nondilution method to classify ticlopidine as an MDI of CYP2C19.
Cut-off values should theoretically vary according to the fold dilution. For example, if the cutoff for the nondilution method were 1.5, the cutoff for 10-, 20-, 30-, and 40-fold dilution should theoretically be 15, 30, 45, and 60, respectively. The foregoing established that this simple relationship cannot be applied without the dilution method failing to identify certain MDIs (those that bind to microsomes and/or are extensively metabolized). The problem is further complicated when one considers not just the fold dilution but the actual concentration of HLMs in the preincubation phase. As shown in Table 8, the initial concentration of HLMs in the dilution method can be as high as 3 mg/ml and as low as 0.25 mg/ml, a 12-fold difference in absolute protein concentration. The problems with nonspecific binding and inhibitor depletion are not the same in these two cases. Therefore, is not surprising that industry cutoffs for MDI vary widely from a 1.2-fold shift to a 10-fold shift in IC50 curves. Developing acceptance criteria and cutoffs for MDI will be extremely difficult with the dilution method because the magnitude of the IC50 shift depends on so many factors, but it seems reasonable to assume that they could be developed with the nondilution method.
Could the Nondilution Assay Fail to Identify an MDI?
When the dilution method is used and the data are processed based on the final concentration of the inhibitor, the magnitude of the IC50 shift is larger than that produced by the nondilution method, as shown in Table 4 and Fig. 3. In many cases, it is 10 times larger (e.g., all the MDIs shown in Fig. 4, left), but in other cases, it is only slightly larger (e.g., all the MDIs shown in Fig. 4, right). This raises the question, could the nondilution method fail to identify an MDI that could otherwise be detected by the dilution method? We have yet to identify an MDI that causes a detectable shift in the dilution assay but not in the nondilution assay (provided we apply a cut-off value of 1.5 to both assays). In an attempt to identify such a compound, we evaluated the inactivation of CYP2C19 by ticlopidine. As shown in Fig. 10, ticlopidine caused a 1.8-fold shift in the nondilution assay (at 0.1 mg/ml) and a 1.7-fold shift in the dilution assay (with an initial protein concentration of 1.0 mg/ml). When the nondilution assay was conducted with a lower concentration of HLMs (0.01 mg/ml), ticlopidine caused a 6.7-fold shift (Fig. 10). Therefore, in general, it is highly unlikely that an MDI that can be identified in the dilution assay could not be identified in the nondilution assay because the true degree of P450 inactivation increases with decreasing protein concentration.
What About Other Assays That Incorporate a Dilution Step?
A dilution step is used in three types of cytochrome P450 inhibition study: 1) an evaluation of IC50 shifts to identify MDIs; 2) a determination of the kinetics of P450 inactivation (the measurement of KI and kinact); 3) an assessment of reversibility of MDI (to determine whether MDI involves irreversible P450 inactivation).
These three studies differ in one important aspect: the first one is performed to identify whether a drug candidate is an MDI, whereas the latter two are performed after a drug candidate has been identified as an MDI. One practical outcome of this difference is that cytochrome P450 activity is usually measured with a low substrate concentration ([S] = Km) in the first study (IC50 shifts) but with a high substrate concentration in the other two studies (KI and kinact measurements and an assessment of reversibility). When the dilution method is used to measure KI and kinact, the method of data processing is not contentious; KI is universally based on the initial (predilution) concentration of inhibitor, as it should be. However, all three methods involve an initial incubation of the inhibitor with a relatively high concentration of HLMs (and NADPH), and the problems surrounding nonspecific binding and inhibitor depletion that complicate the assessment of IC50 shifts by the dilution method also come into play when the dilution method is used to measure KI and kinact and assess MDI reversibility. Several of these issues have been investigated previously (Yang et al., 2005; Ghanbari et al., 2006; Van et al., 2006, 2007).
Should the Dilution Method Be Used When Measuring KI and kinact?
The argument for incorporating a dilution step in the measurement of KI and kinact is that it helps to reduce the direct inhibitory effect of MDIs that also cause potent direct inhibition of cytochrome P450. This implies that a dilution step would be particularly useful for potent direct-acting inhibitors that also function as MDIs. Of the MDIs evaluated in this study, those that can also be classified as potent direct-acting inhibitors (those with an IC50 curve B ≤ 1 μM) are ticlopidine (0.18 μM), tienilic acid (1 μM), paroxetine (0.73 μM), azamulin (0.2–0.9 μM), and mibefradil (0.93 μM), as shown in Table 4. All of these compounds are metabolically labile (Fig. 7) and cause considerably less cytochrome P450 inactivation when incubated with a high concentration of HLMs (Fig. 6). Therefore, the decision to incorporate a dilution step in the determination of KI and kinact should not be made simply on the basis that a particular MDI also happens to be a potent direct-acting inhibitor. A dilution step should be incorporated in the measurement of KI and kinact only after it has been established that the shifted IC50 value obtained with the dilution method [at 1.0 mg/ml (or the intended concentration of HLMs in the preincubation step)] is the same as the shifted IC50 value obtained with the nondilution method (at 0.1 mg/ml or less). Alternatively, an ultracentrifugation experiment like that shown in Fig. 6 could be performed to verify that the degree of cytochrome P450 inactivation does not markedly decline when the concentration of HLMs is increased 10- or 25-fold.
The dilution method can also increase the apparent KI value when the high concentration of HLMs causes a significant decrease in the free concentration of inhibitor (due to nonspecific binding). As shown for S-fluoxetine (Table 6; Fig. 8), the impact of microsomal binding can be corrected by expressing shifted IC50 and kobs values (and hence KI values) based on the free concentration of inhibitor (fumic), which can either be measured experimentally or calculated from physicochemical parameters (Austin et al., 2002; Hallifax and Houston, 2006; Gertz et al., 2008).
Can a Dilution Step Be Used to Assess MDI Reversibility?
MDI of cytochrome P450 can involve the formation of metabolites that are 1) more potent reversible (direct-acting) inhibitors than the parent compound or 2) irreversible inhibitors that bind covalently to the protein or heme moiety of cytochrome P450 (or bind coordinately to the ferrous heme iron, in which case the inhibition is said to be quasi-irreversible) (Ogilvie et al., 2008). The inhibitory effect of the former can be lessened by diluting or dialyzing the sample, whereas the inhibitory effect of the latter cannot. The dilution method always involves incubating the drug candidate with a relatively high concentration of HLMs, and this is also commonly done with the dialysis method to offset protein loss due to nonspecific binding of microsomes to the dialysis membrane. When these methods are used, it is often assumed that the degree of P450 inactivation observed at a low concentration of HLMs will occur to the same extent at a 10- to 25-fold higher concentration. The results shown in Fig. 6 for ticlopidine, tienilic acid, S-fluoxetine, paroxetine, azamulin, and mibefradil show that this is a poor assumption. In all these cases, the extent of cytochrome P450 inactivation progressively declined as the concentration of HLMs increased from 0.1 to 3.0 mg/ml. Consequently, all of these irreversible MDIs can easily be misidentified as reversible MDIs when assessed by the dilution method or when the assessment by dialysis involves incubating the inhibitor with a high concentration of HLMs.
Could the Dilution Assay Fail to Identify an MDI?
When the potential for a drug candidate to cause MDI is assessed in vitro based on a leftward shift of IC50 curve C relative to curve B, the nondilution and dilution method can both identify MDIs provided the data from the dilution method are all processed based on the final concentration of inhibitor. However, the dilution method is something of a balancing act. On the one hand, it gains sensitivity by boosting the IC50 shifts by processing the data for IC50 curve C based on the final inhibitor concentration, but on the other hand, it often loses sensitivity because the high concentration of HLMs in the preincubation assay can result in extensive inhibitor depletion and/or a marked decrease in the free concentration of inhibitor. Accordingly, when the dilution method is used, there is a greater need to ensure that incubating the drug candidate with a high concentration of HLMs in the presence of NADPH for 30 min does not cause extensive metabolism and/or a marked decrease in free concentration. It is perhaps fortunate that many of the drug candidates that have been evaluated previously by the dilution method were screened for metabolic stability in NADPH-fortified HLMs, but this is unlikely to apply to all drug candidates that have been evaluated by the dilution method in the past.
We do not know whether any drug candidates assayed by the dilution method have been misclassified as non-MDIs, but this could occur if the cut-off criteria were set to 10, as at least one respondent reported in the PhRMA consensus paper (Grimm et al., 2009). With a cut-off value of 10, ticlopidine would not be identified as an MDI of CYP2B6 and CYP2C19 (IC50 shift = 5.1 [0.20 → 0.039 μM] and 1.7 [0.57 → 0.34 μM], respectively), and mibefradil (IC50 shift 7.5 [0.46 → 0.061 μM]) would not be identified as an MDI of CYP3A4 (Table 4; Fig. 10). The results obtained with mibefradil are illuminating because mibefradil (Posicor) is the only drug withdrawn from the U.S. market largely on the basis of its ability to cause irreversible inactivation of CYP3A4 (Prueksaritanont et al., 1999). When incubated with HLMs at 0.1 mg/ml, mibefradil (0.2 μM) caused ∼70% inactivation of CYP3A4 (Fig. 6). At 1.0 mg/ml, the extent of inactivation decreased to a mere 10%. At 2.0 and 3.0 mg/ml, mibefradil caused no detectable inactivation of CYP3A4. These results argue strongly against the use of high concentrations of HLMs to measure the MDI of cytochrome P450.
Many researchers have established automated procedures to evaluate MDI by the dilution method, and they have developed a large database on MDI. Accordingly, many researchers will be understandably reluctant to switch immediately to a nondilution method. Although we do not advocate the dilution method for identifying MDIs, the answers to the following questions are intended to guide those researchers who will continue to use the dilution method.
Should Differential Data Processing Be Applied to the Dilution Method?
In other words, when the dilution method is used to identify MDI of cytochrome P450, should IC50 curves B and C be based on the final and initial concentration of inhibitor, respectively? Perhaps somewhat surprisingly, the answer to this question is no. When the dilution method is used to identify MDIs of cytochrome P450, all of the data should be processed based on the final concentration of inhibitor. Differential data processing can be applied only after a drug candidate has been identified as an MDI; it cannot be incorporated into a procedure to identify MDIs on the basis of an IC50 shift.
Should All the Data from the Dilution Assay Be Processed Based on the Initial Concentration of Inhibitor to Prevent the Artificial Lowering of the Shifted IC50 Values?
No, this approach is arguably worse than processing all the data based on the final inhibitor concentration because it would only serve to increase the IC50 values for direct inhibition, and an artificial increase in these values would undermine the FDA's criteria for assessing the potential of direct-acting inhibitors to cause clinically significant inhibition of cytochrome P450 (which is based on [I]/Ki <0.1, where Ki can be estimated from the IC50 value for direct inhibition).
In summary, we have compared the dilution and nondilution methods for identifying MDIs of cytochrome P450 (based on IC50 shifts) and shown that the ability of the dilution method to produce relatively large IC50 shifts is a consequence of basing the shifted IC50 values (IC50 curve C) on the final concentration of inhibitor. This artificial lowering of shifted IC50 values (and the corresponding augmentation of IC50 shifts) disguises the fact that, for many MDIs, the dilution method actually decreases the extent of P450 inactivation due to inhibitor depletion and/or a decrease in the free concentration of inhibitor. When drug candidates are evaluated in vitro for their ability to cause MDI based on a leftward shift in IC50 curve C relative to IC50 curve A or B, we recommend that such experiments be conducted with a nondilution approach (with HLMs at 0.1 mg/ml or less) with a cut-off value of 1.5 (for IC50 C/ IC50 A or B). The nondilution method offers several advantages over the dilution method: it is, technically, easier; the method of data processing is unambiguous (there is no predilution and postdilution inhibitor concentration to choose from), and the problems associated with nonspecific binding and inhibitor depletion are reduced. Furthermore, the shifted IC50 values determined by the nondilution method are true values (i.e., they are not affected by the dilution factor), and they can be used to estimate KI/kinact (the efficiency of enzyme inactivation). Perhaps more importantly, the magnitude of the IC50 shift determined by the nondilution method is not augmented by a dilution factor or markedly diminished by inhibitor depletion and/or microsomal binding, therefore they can be used to develop in vitro acceptance criteria and cut-off values to guide the selection of those drug candidates that should be evaluated in vivo for their ability to cause MDI of cytochrome P450.
Authorship Contributions
Participated in research design: Parkinson, Kazmi, Buckley, Paris, and Ogilvie.
Conducted experiments: Kazmi and Yerino.
Contributed new reagents or analytic tools: Holsapple, Toren, and Otradovec.
Performed data analysis: Parkinson, Yerino, Kazmi, Buckley, Paris, and Ogilvie.
Wrote or contributed to the writing of the manuscript: Parkinson, Kazmi, Buckley, Paris, and Ogilvie.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.111.038596.
-
ABBREVIATIONS:
- P450
- cytochrome P450
- FDA
- U.S. Food and Drug Administration
- HLMs
- human liver microsomes
- LC/MS/MS
- liquid chromatography/tandem mass spectrometry
- KI
- inhibitor concentration that causes half the maximal rate of enzyme inactivation
- kinact
- maximal rate of enzyme inactivation
- Km
- substrate concentration supporting half the maximal rate of an enzyme-catalyzed reaction
- fumic
- unbound fraction at various concentrations of HLMs
- MDI
- metabolism-dependent inhibition
- PhRMA
- Pharmaceutical Research and Manufacturers of America.
- Received February 7, 2011.
- Accepted April 27, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
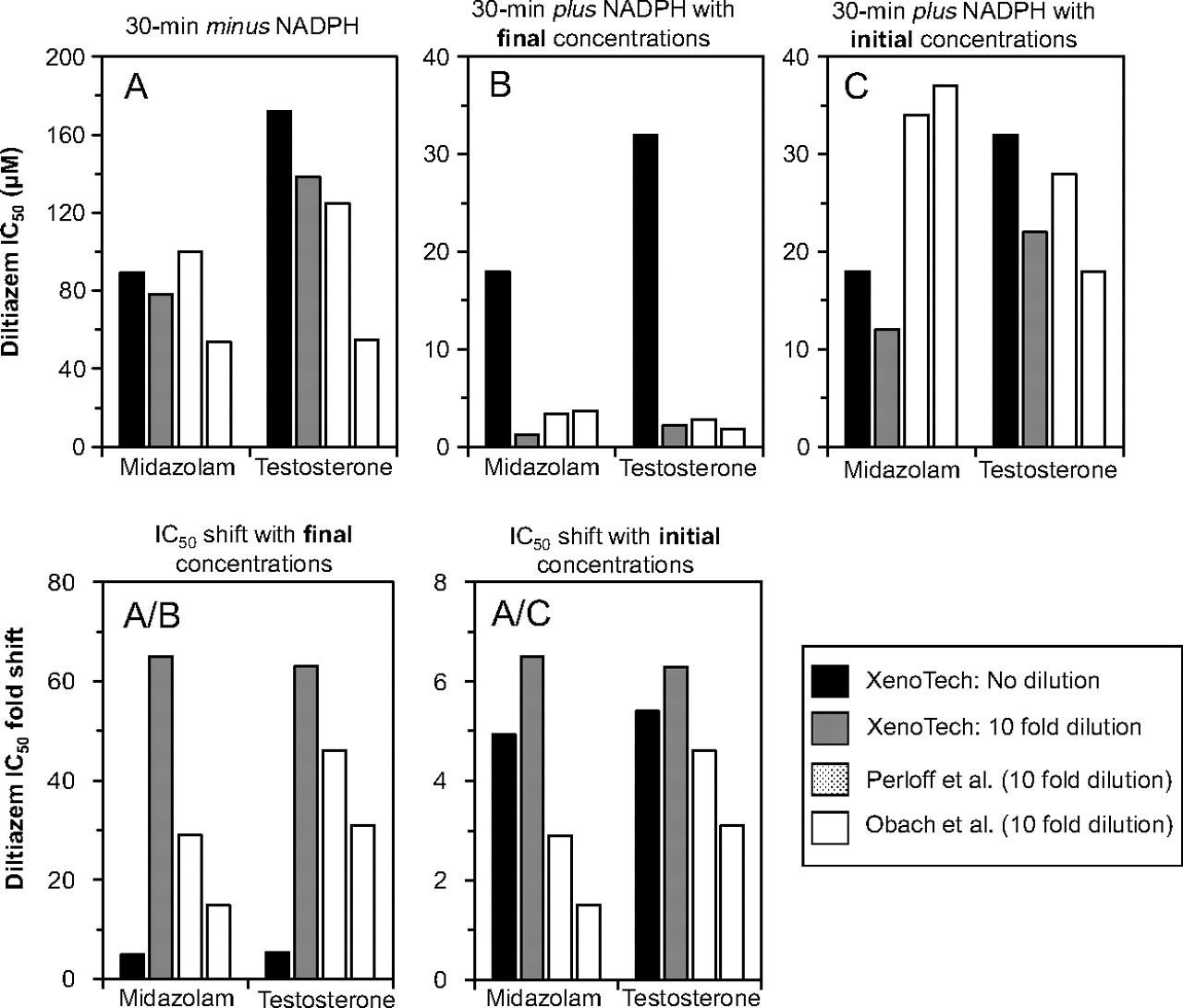{kind=link}
{kind=link}
{kind=link}
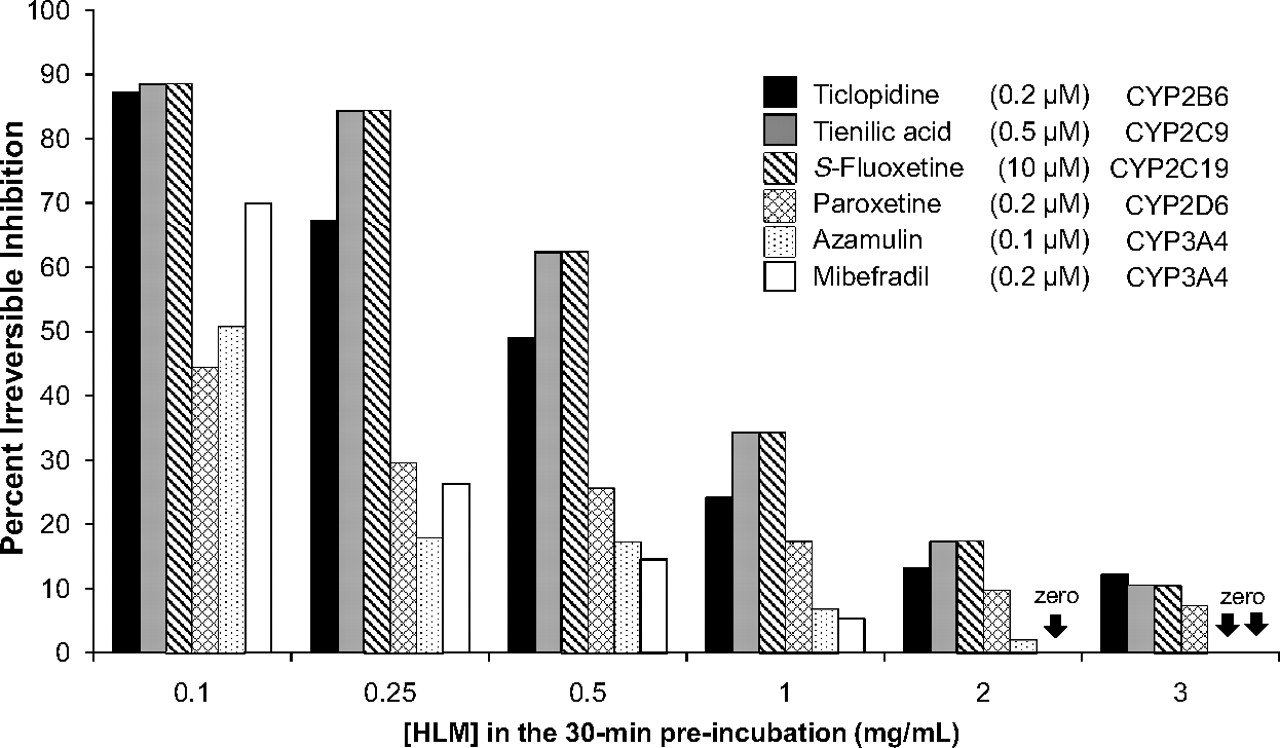{kind=link}
{kind=link}
{kind=link}
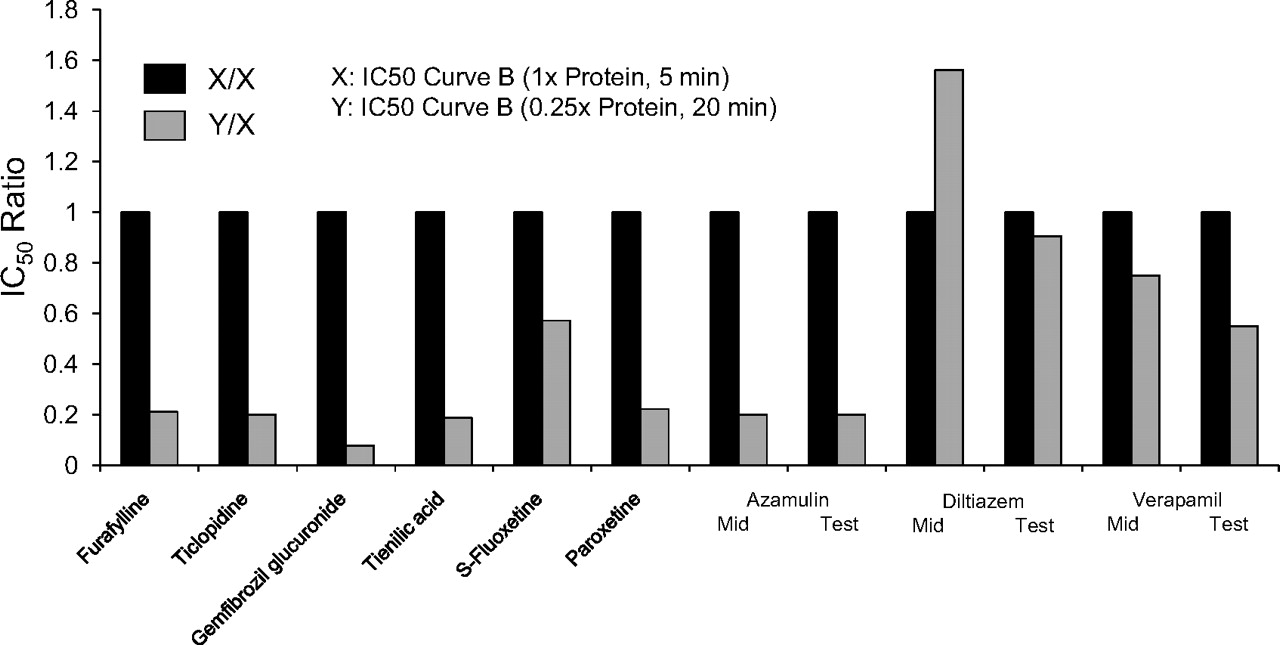{kind=link}
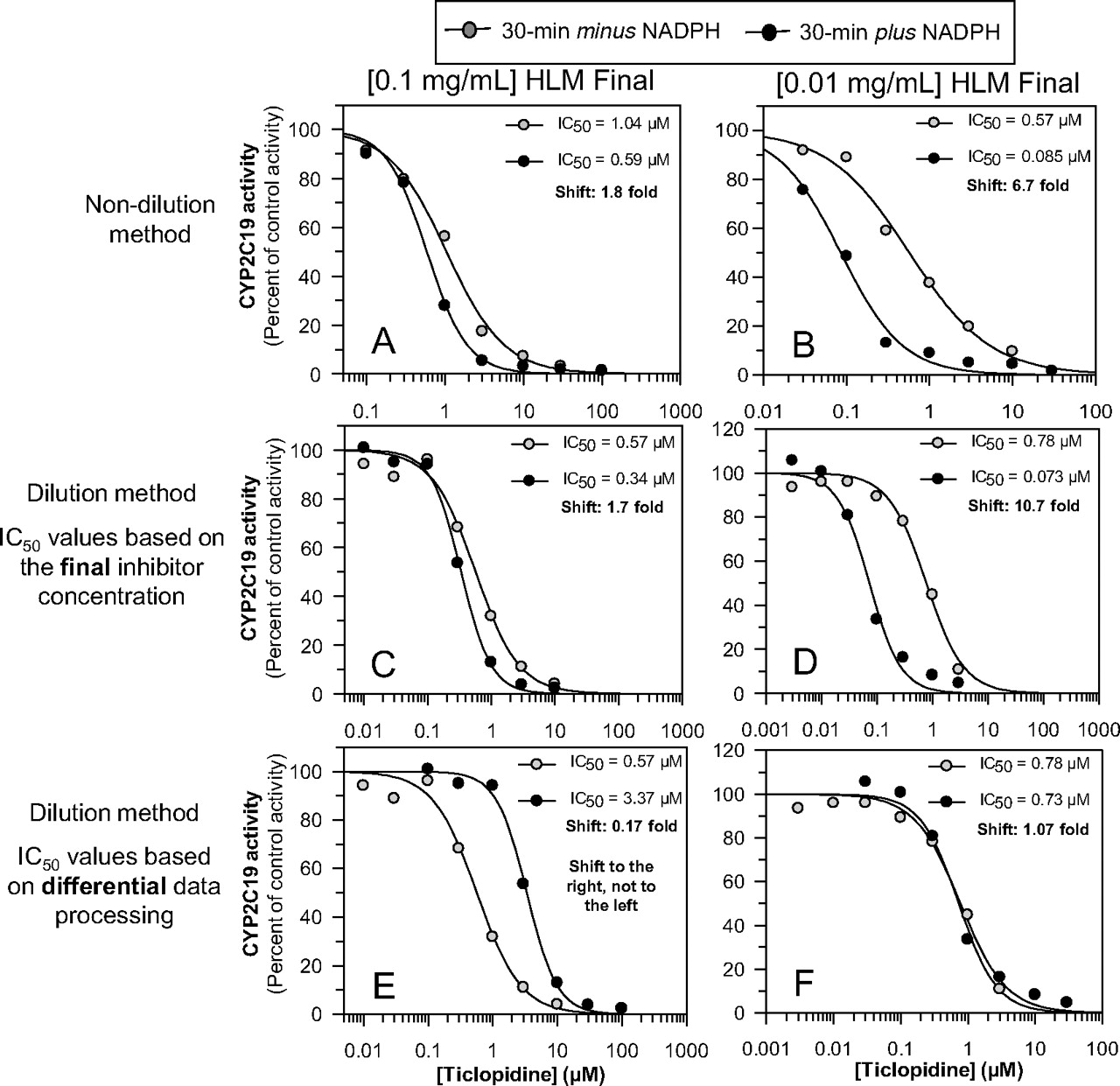{kind=link}