


Abstract
Valdecoxib is a potent and specific inhibitor of cyclooxygenase-2, which is used for the treatment of rheumatoid arthritis, osteoarthritis, and the dysmenorrhea pain. Eight male human subjects each received a single 50-mg oral dose of [14C]valdecoxib. Urine, feces, and blood samples were collected after administration of the radioactive dose. Most of the radioactivity in plasma was associated with valdecoxib and the hydroxylated metabolite of valdecoxib (M1). The estimated terminal half-life for valdecoxib was about 7 h. About 76.1% of the radioactive dose was recovered in urine and 18% of the radioactive dose was recovered in feces. Valdecoxib was extensively metabolized in human, and nine phase I metabolites were identified. The primary oxidative metabolic pathways of valdecoxib involved hydroxylation at either the methyl group to form M1 or N-hydroxylation at the sulfonamide moiety to form M2. Further oxidation of M1 led to the formation of several other phase I metabolites. Oxidative breakdown of the N-hydroxy sulfonamide function group in M2 led to the formation of corresponding sulfinic acid and sulfonic acid metabolites. The O-glucuronide conjugate of M1 andN-glucuronide conjugate of valdecoxib were the major urinary metabolites, which accounted for 23.3 and 19.5% of the total administered dose, respectively. The remaining urinary metabolites were glucuronide conjugates of other phase I metabolites. Only 3% of the administered dose was recovered in urine as unchanged parent, suggesting that renal clearance is insignificant for valdecoxib. Absorption of valdecoxib was excellent since the recovery of unchanged valdecoxib in feces was <1% of the administered dose.
The enzyme, cyclooxygenase, is responsible for the conversion of arachidonic acid to prostaglandin H2, a key step in the generation of proinflammatory eicosanoid mediators (Vane et al., 1994; Vane and Botting, 2000). Now it is well known that cyclooxygenase is present in two forms; one form is constitutive (COX-11) and is widely expressed in nearly all tissues throughout the body, whereas the other form is inducible (COX-2) and is predominantly expressed in inflamed tissues (Needleman and Isakson, 1997; Golden and Abramson, 1999). Therefore, selective inhibition of COX-2 should maintain the anti-inflammatory effects while reducing adverse effects (gastrointestinal bleeding, platelet effect) of traditional nonsteroidal agents due to their nonselective inhibition of COX-1 (Masferrer et al., 1990, 1994; Seibert et al., 1999).
Celecoxib (Fig. 1) is the first selective COX-2 inhibitor developed in the United States and is widely available as a prescription drug in the United States and Europe, as well as in other countries (Goldenberg, 1999; Clemett and Goa, 2000; Goldstein et al., 2000; Leese et al., 2000; Megeff and Strayer, 2000; Steinbach et al., 2000).
Chemical structure of celecoxib and valdecoxib.
Valdecoxib (Fig. 1), a diarylsubstituted isoxazole, is chemically distinct from celecoxib. Compared with celecoxib, valdecoxib possesses even greater potency and high specificity for COX-2 inhibition. As a novel COX-2 inhibitor, valdecoxib demonstrated analgesic, anti-inflammatory, and antipyretic properties in pharmacological studies in animals (Talley et al., 2000). Valdecoxib (BEXTRA) has approved by US Food and Drug Administration for the relief of signs and symptoms of osteoarthritis and adult rheumatoid arthritis and for the treatment of primary dysmenorrhea.
The purpose of this study was to determine the disposition of [14C]valdecoxib after a single oral dose of 50-mg [14C]valdecoxib to human subjects. The selection of a 50-mg dose was to cover the anticipated clinic dose of 40 mg for the acute pain management indication.
Materials and Methods
Radiolabeled Valdecoxib, Dosage Forms and Drug Administration.
[14C]valdecoxib (50 mg with a specific activity of 1.99 μCi/mg with radioactive purity of >97%; Pharmacia, Skokie, IL) was formulated at the clinical pharmacology unit of the Evanston Hospital (Evanston, IL) as a fine suspension in 57.5 ml of a mixture of apple juice/polyethylene glycol 400/Tween 80 (50:7.25:0.25, v/v) and administered orally to 8 male volunteers for the study. The use of the apple juice was mainly to mask the bitter taste of valdecoxib. The volunteers were then instructed to drink two additional cups of 50 ml of apple juice immediately following the dose administration and to consume 180 ml of water at 1, 2, and 3 h postdose. All subjects remained in an upright position for 4 h postdose. This study was reviewed and approved by the local institutional review board, and informed consent was obtained from each volunteer before the study.
Sample Collection.
Blood samples were collected at 30 min, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, 24, 36, 48, 60, and 72 h postdose and centrifuged to obtain plasma and red blood cells. Urine was collected at the following intervals: −12 to 0 h (12 h before dosing), 1 to 2 h, 2 to 3 h, 3 to 4 h, 4 to 8 h, 8 to 12 h, 12 to 24 h, 24 to 48, 48 to 72, 72 to 96, 96 to 120, 120 to 144 and 144 to 168 h postdose. All individual urine samples collected from the study were refrigerated. At the end of each collection interval, the total urine volume collected was measured and recorded.
Individual fecal samples and fecal wipes were collected beginning 12 h predose and at 24 h intervals through 8:00 AM on day 10 postdose. The collection date, time, and weight of the sample were recorded. Samples were frozen immediately. All samples were stored at −20°C before analysis.
Plasma Profiling.
Plasma samples were extracted with acetonitrile/water (90:10). The ratio of plasma to the solvent was 1:4 (v/v) for the first extraction; the precipitate was extracted with 2 ml of solvent three more times. The 16-h plasma samples were pooled from all 8 subjects due to low individual radioactivity concentrations at that time. The extracts were combined and evaporated under a gentle stream of nitrogen at room temperature to near dryness. The residue was then reconstituted in the initial HPLC mobile phase for HPLRC profiling using Method A. The extraction efficiencies of radioactivity from plasma samples (up to 12 h postdose samples) ranged between 104 and 112%, which was similar to the extraction efficiency obtained from the [14C]valdecoxib-spiked standard plasma samples (109 ± 3%). For plasma samples collected at 16 h postdose, the corresponding extraction efficiency was 93.6 ± 2.7%. Concentrations of valdecoxib and M1 in plasma were calculated based on their HPLRC peak area ratios and the total radioactivity of plasma.
Red Blood Cell Profiling.
The RBC samples were extracted using the same extraction procedure as for plasma and profiled using Method A. The extraction efficiencies of radioactivity from RBC samples collected at 1 and 4 h postdose were 90.9 and 98.3%, respectively.
Urine Profiling.
0- to 24- and 24- to 48-h urine samples were loaded onto a preconditioned C18 Bond Elut column (1 cc, 100 mg, Varian Sample Preparation Products, Harbor City, CA). Preconditioned C18 Bond Elut columns (3 cc, 1 g) were used to extract 110 ml of 48- to 96-h urine pooled from 8 subjects. The loaded column was washed with one-half column volume of water and then eluted with one column volume of methanol. The methanol eluates were dried under a gentle stream of nitrogen at room temperature and then reconstituted in the initial HPLC mobile phase for HPLRC profiling using Method A. The mean percentage of radioactivity recovered from the 0- to 24-h urine samples using the C18 Bond Elut column extraction procedure was 96.6%.
Incubation of Urine with β-Glucuronidase and Base.
The β-glucuronidase incubation sample was prepared by adding ∼200 units of β-glucuronidase (made from bovine liver, Sigma-Aldrich, St. Louis, MO) to 1 ml of a mixture of urine and 0.2 M sodium acetate buffer (pH 5.0; 1:1, v/v). The base hydrolysis incubation sample was prepared by mixing urine with 0.2 M glycine buffer (pH 10.0) (1:1, v/v). All incubation samples were covered with punctured parafilm and incubated at 37°C for 16 h. The incubated samples were profiled by HPLRC using Method B.
Fecal Profiling.
Based on sample weight, fecal samples were mixed with 2- to 3-fold excess of acetonitrile/water (80:20) followed by homogenization and centrifugation. This process was repeated three times. The supernatants obtained in each step were combined and evaporated under a gentle stream of nitrogen at room temperature. The dried samples were reconstituted in water and loaded onto preconditioned C18 Bond Elut columns (3 cc, 1 g). The loaded columns were washed with water (1 ml) and then eluted with methanol (3 ml). The methanol eluates were dried under a gentle stream of nitrogen at room temperature and then reconstituted in the initial HPLC mobile phase for HPLRC profiling using Method A. The acetonitrile extraction recovery from fecal homogenates was 38.1% based on the combustion analysis. In contrast, the acetonitrile extraction recovery of spiked fecal homogenate with [14C]valdecoxib and [14C]SC-66905 was complete. Therefore, in estimating of the percent dose recovered as valdecoxib and M1 (SC-66905), the acetonitrile extraction recovery was assumed as complete. Based on these results, it was judged that the remaining fecal radioactivity was not extractable with acetonitrile solution. Further attempts to increase the extraction yield with alternative extraction solvents (methanol or acetone) yielded extracts that were unsuitable for HPLRC profiling, and therefore, acetonitrile extracts were used for the final fecal profiling.
HPLRC.
The HPLRC system consisted of a Hewlett Packard series 1050 autosampler, pump, UV detector (Hewlett Packard Analytical Direct, Wilmington, DE) and Flo–OneA-500 radioactivity (PerkinElmer Life Sciences, Boston, MA). The following HPLC methods were used in the study.
Method A.
The HPLC column was a Novapak C18 column (3.9 × 150 mm, 4 μm; Waters Corp, Milford, MA). Mobile phase A consisted of acetonitrile, methanol, and 25 mM sodium acetate (pH 4.0) at a ratio of 1:2:27 (v/v/v). Mobile phase B consisted of acetonitrile, methanol, and 25 mM sodium acetate (pH 4.0) at a ratio of 1:2:3 (v/v/v). The HPLC gradient started at the initial condition of 100% mobile phase A and progressed to 100% mobile phase B over 25 min. The system was returned to initial conditions and allowed to re-equilibrate for 15 min before the next run. The flow rate of mobile phase was 1.0 ml/min. Eluates from the HPLC column were mixed with FLO-SCINT III (PerkinElmer Life Sciences) at a ratio of 1:3 (v/v) for radioactivity detection.
Method B.
The HPLC column was a Novapak C18 column (3.9 × 150 mm, 4 μm; Waters Corp.). Mobile phase A consisted of acetonitrile and 25 mM ammonium acetate (pH 4.0) at a ratio of 10:90 (v/v). Mobile phase B consisted of acetonitrile and 25 mM ammonium acetate (pH 4.0) at a ratio of 50:50 (v/v). The HPLC gradient started at the initial condition of 100% mobile phase A and progressed to 100% mobile phase B over 20 min. The HPLC was programmed back to 100% mobile phase A over a 2-min period and re-equilibrated to 100% mobile phase A for 15 min before the next run. The flow rate of mobile phase was 1.0 ml/min.
LC-MS/MS.
LC-MS/MS analyses were carried out on a Hewlett Packard 1100 autosampler and HPLC pump coupled to a Quattro II triple quadruple mass spectrometer (Micromass, Altrincham, UK) using HPLC Method B. Approximately 20% of the eluate was diverted to the mass spectrometer via a 1:4 T-splitter, and the remaining eluate was mixed with FLO-SCINT III at a ratio of 0.8:3 (v/v) for on-line radioactivity detection. The HPLRC response was recorded in real time by the mass spectrometer data system, which provided simultaneous detection of radioactivity and MS data. The mass spectral analysis was performed in negative ion electrospray ionization mode. The capillary and cone voltages were set at 3.6 and 30 V. CID studies were performed using collision energy of 26 eV and a collision cell of argon gas with the pressure at 2 × 10−3 mBar.
NMR.
A Bruker AMX-500 NMR instrument (Bruker Instruments, Billerica, MA) equipped with a 5-mm broadband indirect detection probe was used. The solvent used was deuterated methanol. Urine metabolites (V-G, M1-G, M2-G, and M3-G) were first fraction-collected from HPLC and then were dried for NMR analysis.
Liquid Scintillation Counting.
Total radioactivity was measured with liquid scintillation spectrometers (Mark III; Tracor Analytic, Elk Grove, IL). Chemical quenching was corrected using the automatic external standard channel ratio method.
Results
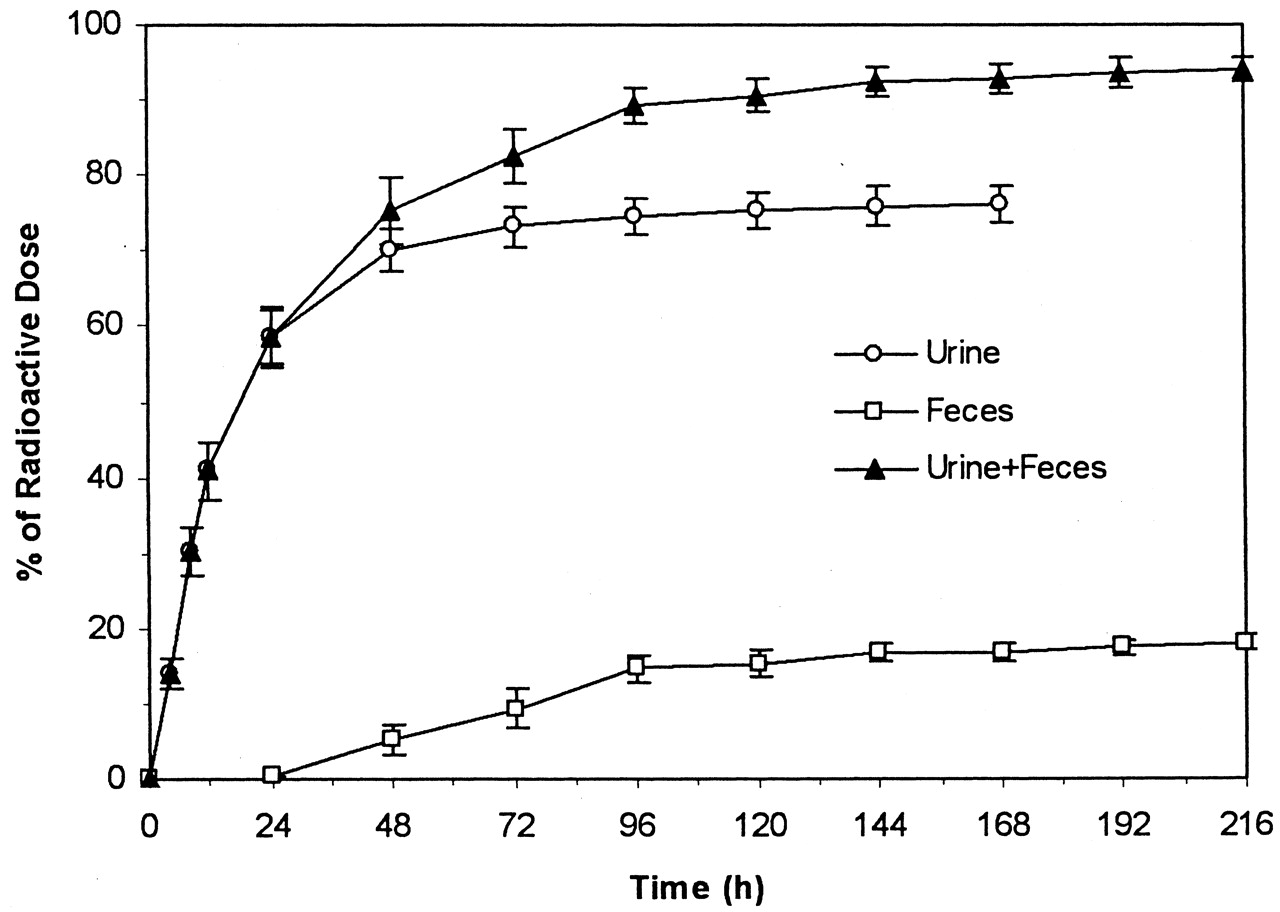
Total Recovery of Radioactivity in Urine and Feces.
Total recovery of the administered radioactive dose was 94.1% (Fig.2). Most of the administered radioactivity was recovered in urine, which accounted for 76.1% of dose, the remainder being recovered in feces, which accounted for18% of dose.

Cumulative mean (±S.E.) recoveries of administered radioactive dose in human urine and feces following a single oral administration of 50 mg of [14C]valdecoxib to each male subject.
Profiling of Plasma.
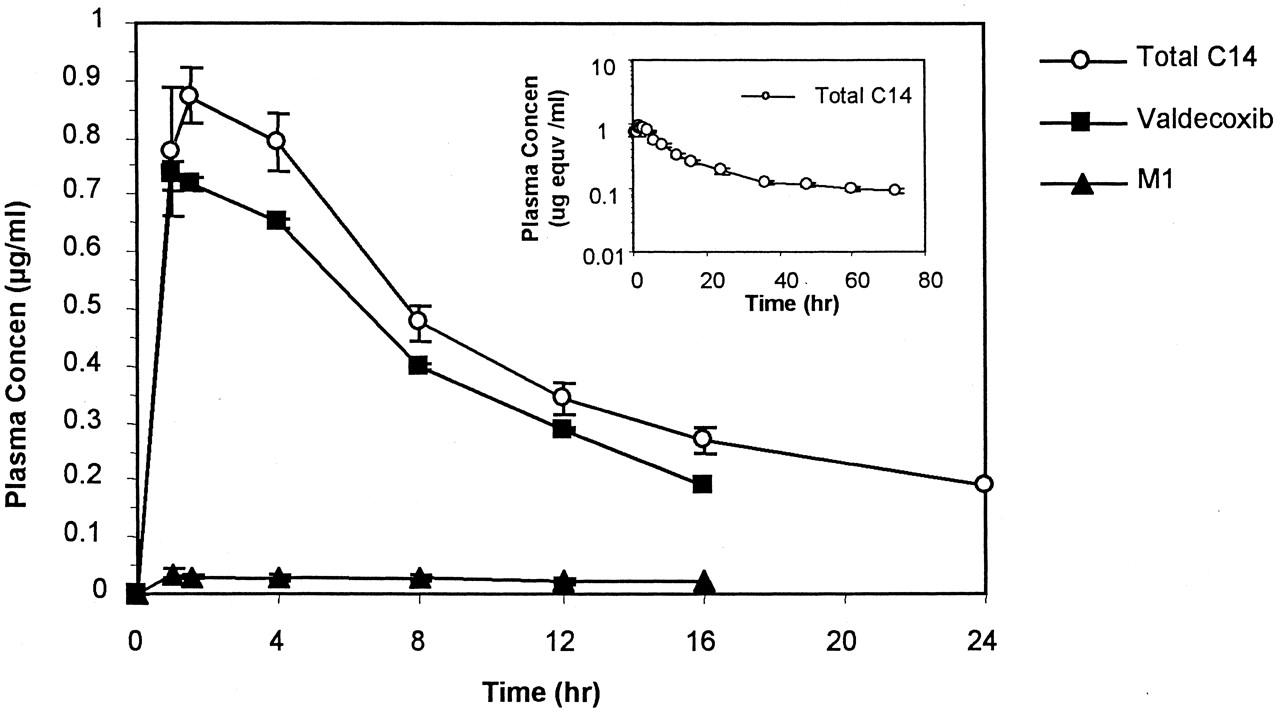
Following oral dosing, radiolabeled material was rapidly absorbed from the intestinal tract into the systemic circulation, with aCmax value of 1.04 μg Eq/ml observed in plasma at 1.7 h postdose (Fig.3). The HPLRC plasma-profiling results are shown in Fig. 4. Most of the radioactivity in plasma was associated with valdecoxib and the hydroxylated metabolite of valdecoxib (later designated asM1), and the remaining radioactivity was probably due to other undetected radiolabeled metabolites. The structure of M1 is shown in Fig. 5. Based on the HPLRC profiling results and the total radioactivity in plasma, the plasma concentration-time profiles of valdecoxib and M1 were constructed, and the results are shown in Fig. 3. The estimated terminal half-life for valdecoxib was about 7 h. Plasma concentrations of M1 were much lower than those of valdecoxib at each sampling time.

Mean (±S.E.) plasma concentration-time profiles of total C14, valdecoxib and M1 following a single oral administration of 50 mg of [14C]valdecoxib to each male subject.
The unit for plasma concentration of total C14 was microgram equivalent of [14C]valdecoxib/ml. Semilog scale is used for the insert.

Typical plasma HPLRC profiles following a single oral administration of 50 mg of [14C]valdecoxib to each male subject.

Proposed metabolic pathways of valdecoxib in human (★ denotes uniformly labeled 14C phenyl ring).
Percent of dose excreted as each metabolite during 0 to 96 h postdose collection period is indicated.
Profiling of Red Blood Cells.
The HPLRC profiling of red blood cells indicated that all radioactivity in red blood cells was associated with valdecoxib at 1 and 4 h postdose. The average ratios of RBC/plasma concentrations of total radioactivity were 4.25 at 1 h and 3.46 at 4 h postdose, indicating radioactivity was more extensively partitioned into RBCs than plasma.
Urine and Fecal Profiling.
Representative HPLC profiles of urine and fecal samples are shown in Fig. 6. The mean percentages of dose excreted as each metabolite are included in Fig. 5. M1 was a minor urinary metabolite but its O-glucuronide conjugate,M1-G, was the major urinary metabolite, accounted for 23.3% of the administered dose. The N-glucuronide conjugate of the parent compound, valdecoxib (V-G), accounted for 19.5% of the administered dose. The other urinary glucuronide conjugate metabolites were M2-G, M3-G, M5-G, andM9-G. Their chemical structures are shown in Fig. 5. Small amounts of M4, M6, and M9 were found in the urine. M7, a sulfinic acid metabolite formed by the oxidative breakdown of the N-hydroxy sulfonamide group in M2, was detected in urine in a trace amount. M8, a sulfonic acid metabolite formed by oxidation of M7, was also detected in urine in a trace amount. All of the remaining identified metabolites were present in urine in trace amounts only.

Representative urine and fecal HPLRC profiles following a single oral administration of 50 mg of [14C]valdecoxib to each male subject.
HPLRC profiling of fecal extracts (Fig. 6) showed the presence of many small peaks. Among these small peaks, valdecoxib, M1, andM3 were identified by LC-MS/MS. The remaining peaks were not identified. Overall, less than 1% of the administered radioactive dose was recovered as valdecoxib, M1, and M3 in fecal samples.
Incubation of Urine with β-Glucuronidase and Base.
Aliquots of urine samples were incubated with β-glucuronidase or with basic buffer to determine the presence of glucuronide- or sulfate-conjugated metabolites (since the purchased bovine liver β-glucuronidase also contained some arylsulfatase activity). β-Glucuronidase incubation of urine samples showed the conversion ofM2-G, M5-G, M1-G, and M3-Gto M2, M5, M1, and M3, respectively (Fig. 7), which suggests that M2-G, M5-G, M1-G, andM3-G were O-glucuronide conjugates. Base incubation of urine showed the conversion of V-G to valdecoxib. The fact that V-G could not be hydrolyzed by β-glucuronidase but could be hydrolyzed by base suggests thatV-G is an N-glucuronide conjugate, which was confirmed by LC-MS/MS.

HPLRC profiles of urine (0–24 h) without treatment, with β-glucuronidase treatment and with base treatment.
Metabolite Structure Elucidation by MS and NMR.
Valdecoxib was detected in plasma, urine, and feces. The HPLC retention time of the valdecoxib peak was the same as that of the authentic valdecoxib standard. A deprotonated molecular ion with anm/z value of 313 was present in the negative ion mass spectrum of the samples isolated from plasma, urine, and feces which was consistent with that of the valdecoxib standard. The CID spectrum of m/z 313 generated a series of product ions, which was the same as those from the valdecoxib standard. These results confirmed that the assignment of valdecoxib peak in the HPLRC profile of plasma, urine, and feces was correct.
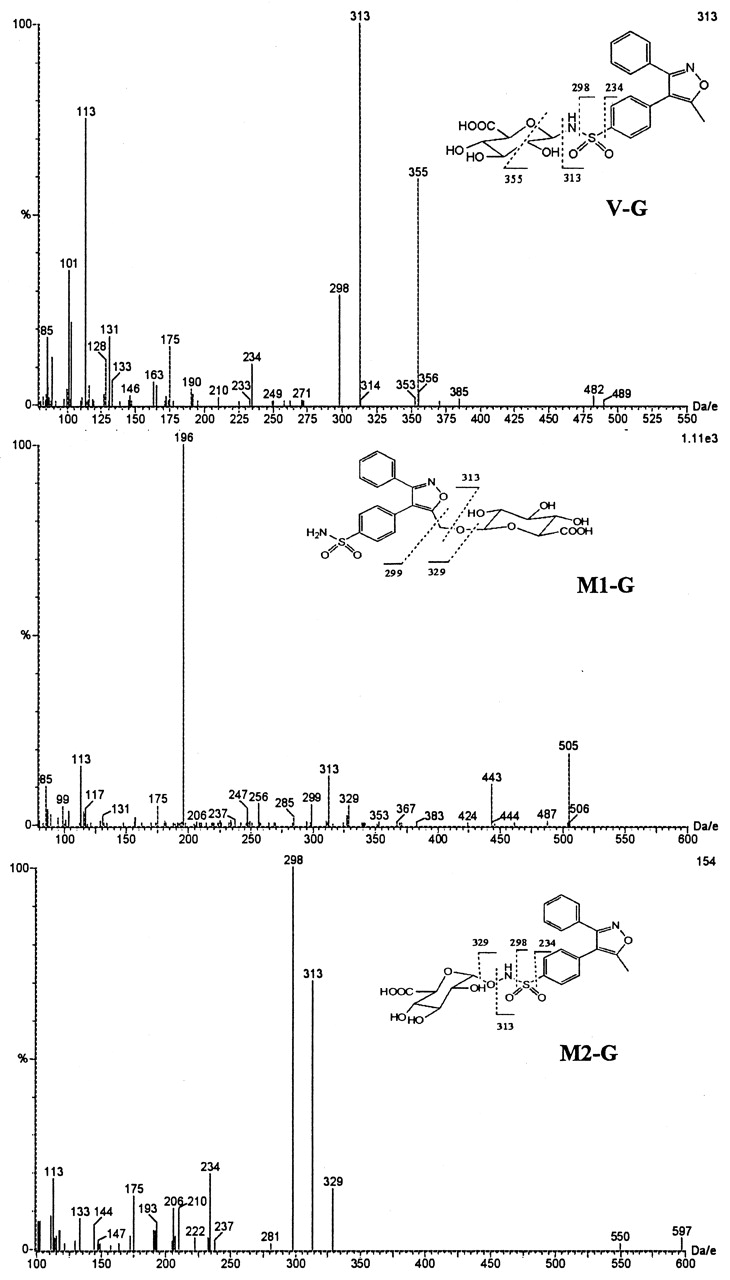
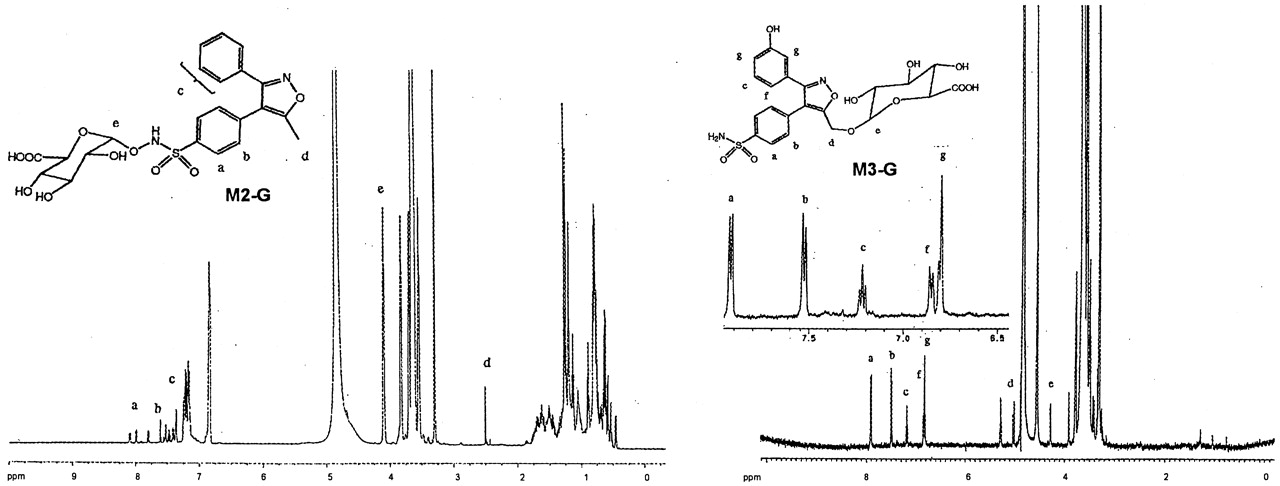
Valdecoxib-N-glucuronide (V-G) was only found in urine samples. The negative ion mass spectrum of V-G showed a deprotonated molecular ion at m/z 489, 176 Da higher than that of valdecoxib, indicating that V-G was a glucuronide conjugate of valdecoxib. The CID spectrum of m/z489 generated a series of product ions at m/z355, 313, 298, 234, 175, and 113 (Fig.8). The product ions atm/z 313, 298, and 234 were formed by sequential losses of 176 (dehydroglucuronic acid), 15 (NH), and 64 (SO2) daltons from m/z 489. These results suggested that the glucuronide conjugation of valdecoxib had occurred at the sulfonamide group. The product ion atm/z 355 was formed by a loss of 134 Da (CHOH-CHOH-CH-COOH-O from the glucuronic acid moiety) from the deprotonated molecular ion. The product ions atm/z 175 and 113 were generated from the subsequent losses of 18 (H2O) and 62 (HOCH2-CH2OH) daltons from glucuronic acid. The structure of V-G was further elucidated by proton NMR analysis (Fig. 9). The proton chemical shift of the methyl group, 2.51 ppm, was very similar to the shift observed for the methyl group of valdecoxib (2.48 ppm), indicating no change at the methyl moiety. The chemical shifts of the protons on the phenyl ring with the sulfonamide moiety were slightly different from the corresponding shifts observed for valdecoxib. The chemical shift of the protons ortho to the sulfonamide was shifted downfield compared with that observed for valdecoxib. The signal of the protons meta to the sulfonamide group was shifted to higher field (0.06 ppm) due to the attachment of glucuronic acid at the nitrogen. Consistent with the proposed structures of V-G and M2-G, the chemical shift of the C-1 glucuronide proton of V-G (4.52 ppm; doublet, J = 8.8 Hz) is up field of the chemical shift of the C-1 proton in the N-hydroxy glucuronide, M2-G (4.80 ppm, doublet, J = 8.6 Hz). The large three-bond couplings indicate that the C-1 protons in both V-G and M2-G have axial-axial relationships with the C-2 protons, and thus both structures have been assigned with the glucuronide groups attached in the equatorial positions. Based on the MS and NMR data, V-G was identified as anN-glucuronide conjugate of valdecoxib.

Representative CID MS spectra (negative ion mode) of isolated valdecoxib metabolites from urine.

Proton NMR spectra ofN-glucuronide conjugated valdecoxib ( V-G ),valdecoxib, M1-G , and M1.
M1 was found in plasma, urine and feces. The deprotonated molecular ion at m/z 329 and the CID spectrum ofM1 matched those of the synthetic standard. The CID spectrum of m/z 329 showed a major product ion atm/z 196, which was formed by the loss of C8H7NO via a five-member ring rearrangement mechanism (Fig. 8). Based on these data,M1 was identified as the 5-methyl hydroxylated metabolite of valdecoxib.
M1-G was found in urine and could be converted toM1 upon incubation with β-glucuronidase, suggesting it is a glucuronide conjugate. M1-G exhibited a deprotonated molecular ion at m/z 505, 176 Da higher than M1, confirming that M1-G was a glucuronide conjugate of M1. The CID spectrum of m/z 505 generated a series of product ions at m/z 443, 329, 313, 299, 175, 113 and a base peak at m/z of 196 Da, which was similar to that of M1 (Fig. 8). The sequential losses of 176 (dehydroglucuronic acid), 16 (O), and 14 (CH2) daltons from the m/z 505 in the CID spectrum ofM1-G suggested that glucuronide conjugation ofM1-G occurred at the 5-hydroxymethyl group of M1. Detailed NMR analysis (Fig. 9) confirmed the structural assignment ofM1-G.
The CID spectrum of M2 (m/z 329) showed a product ion at m/z 298 and an intense product ion at m/z 234 (Fig. 9), which matched with the mass spectrum of a synthesized standard. Based on its CID spectrum as well as a detailed NMR analysis (Fig.10) of its glucuronide conjugate (M2-G), M2 was identified as anN-hydroxy sulfonamide metabolite of valdecoxib.
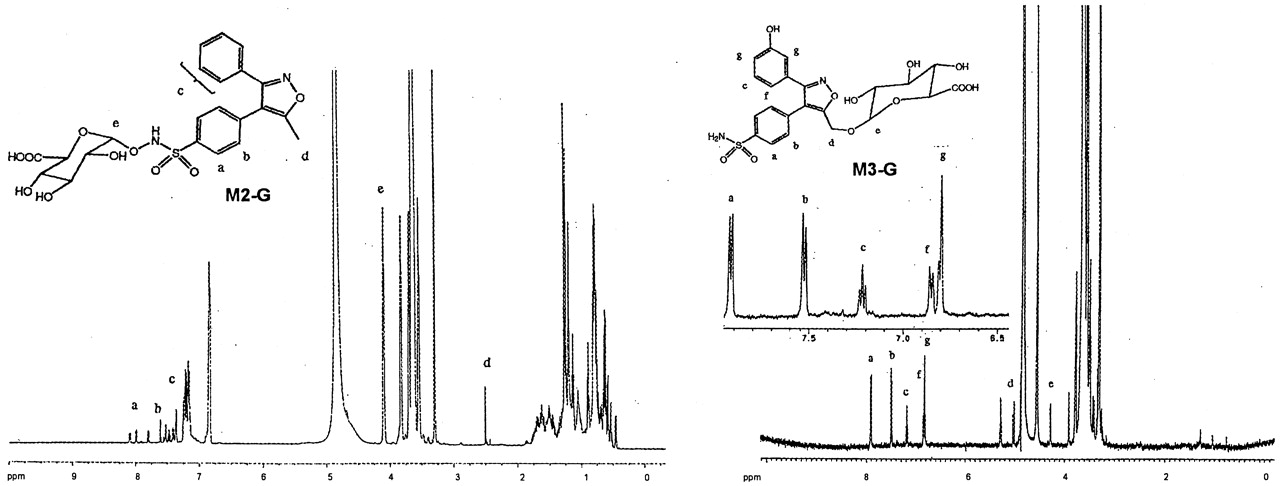
Proton NMR spectra of M2-G andM3-G.
The N-hydroxy sulfonamide function group in M2could undergo oxidative breakdown to form the corresponding sulfinic and sulfonic metabolites. The chemical mechanism of this reaction was reported using N-hydroxybenzenesulfonamide as a model compound (Grzesiok et al., 1994; Ruben et al., 1995). Indeed, a small amount of M7, a sulfinic acid metabolite, and M8, a sulfonic acid metabolite, were detected in human urine.
M2-G was also found in urine and could be converted toM2 upon incubation with β-glucuronidase, suggesting it is a glucuronide conjugate. M2-G showed a deprotonated molecular ion at m/z 505, which was 176 Da higher than that of M1, but it had a different HPLRC retention time than M1-G, suggesting that the site of glucuronide conjugation was different between M2-G and M1-G. The CID spectrum of m/z 505 generated a series of product ions at m/z 329, 313, 298, 234, 175, and 113 (Fig. 8) which were also different from the product ions ofM1-G. The subsequent losses of 176 (dehydroglucuronic acid), 16 (O), 15 (NH), and 64 (SO2) daltons from them/z 505 ion in its CID spectrum suggested thatM2-G was a glucuronide conjugate of hydroxylated valdecoxib with glucuronide conjugation occurring at the N-hydroxy sulfonamide group of valdecoxib. The structure of M2-G was further confirmed by NMR analysis (Fig. 10). The proton chemical shift of the methyl group, 2.51 ppm, was very similar to the shift observed for the methyl group of valdecoxib (2.48 ppm), indicating that no changes occurred in the methyl group. The proton chemical shifts of the 3-phenyl ring were also very similar to those observed for valdecoxib. The proton chemical shifts on the sulfonamide phenyl ring were significantly different from the corresponding shifts observed for valdecoxib. The chemical shift of the protons ortho to the sulfonamide was downfield from the shift observed for valdecoxib. The chemical shift of the protons meta to the sulfonamide group was shifted to lower field (0.03 ppm) due to the glucuronide residue linked through oxygen. The assignment of the structure ofM2-G was consistent with the observation that the chemical shift of the C-1 proton of the glucuronic acid group was further down field (4.8 ppm) when the glucuronide residue was attached through the oxygen atom of the N-hydroxy sulfonamide moiety. Based on the MS and NMR data, the structure of M2-G was identified as a glucuronide conjugate of the N-hydroxy metabolite of valdecoxib.
Similar detailed chemical structure analyses were performed forM3, M3-G, M4, M5,M5-G, M6, M7, M8,M9, and M9-G. Selected CID spectra data obtained from these metabolites are shown in Table1. The NMR spectrum of M3-G is shown in Fig. 10. The CID MS spectra and NMR spectrum were consistent with the structure assignment for these metabolites shown in Fig. 5.
CID mass spectra data of valdecoxib metabolites in human
Valdecoxib Metabolic Pathway.
The metabolic pathways of valdecoxib in human are summarized in Fig. 5. The phase I metabolism of valdecoxib in human involved hydroxylation at the 5-methyl group to form M1, hydroxylation at themeta position of the 3-phenyl group to form M9 orN-hydroxylation at the sulfonamide function group to formM2. The further oxidation of M1 led to the formation of M4, a carboxylic acid metabolite andM6, an isoxazole ring opened metabolite. M1 orM9 could be further hydroxylated to form the same di-hydroxylated metabolite, M3, in which hydroxylation occurred at both the meta position of the 3-phenyl group and the 5-methyl group of valdecoxib. Hydroxylation either at the 5-methyl group of M2 or at the sulfonamide function group ofM1 led to the formation of the same metabolite,M5. The oxidative breakdown of the N-hydroxy sulfonamide group in M2 led to the formation of the sulfinic acid metabolite, M7, which was further oxidized toM8, a sulfonic acid metabolite.
Phase II metabolism is the major elimination pathway for valdecoxib, since glucuronide conjugates accounted for ∼80% of the radioactivity recovered in urine. O-glucuronide conjugation occurred at the hydroxyl group, which led to the formation of M1-G,M2-G, M3-G, M5-G, and M9-G.N-glucuronide conjugation at the sulfonamide function group led to the formation of the N-glucuronide of valdecoxib (V-G).
Discussion
Valdecoxib and its hydroxylated metabolite, M1, were the main radioactive components circulating in blood and in plasma. However, as indicated in Fig. 4, plasma concentrations of M1were much lower than the corresponding plasma concentrations of valdecoxib. Given the lower potency of M1 in a rat adjuvant arthritis model (Talley et al., 2000), it is expected that contributions of M1 to the overall efficacy following oral administration of valdecoxib would be relatively minor.
Valdecoxib was extensively metabolized. The phase I metabolic pathway for valdecoxib involved several oxidation reactions, which were mainly mediated by the CYP3A4 isoform based on in vitro experiments (E. Burton, private communication). Nine phase I metabolites were found. Three (M1, M2, and M9) were formed by monohydroxylation of valdecoxib. Two other phase I metabolites were formed by di-hydroxylation of valdecoxib (M3 andM5). Two other metabolites, M4 and M6, were formed by further oxidation of M1. The remaining two metabolites,M7 and M8, were formed from oxidative breakdown of the N-hydroxy sulfonamide group in M2.
Conjugation pathways played an important role in the elimination of valdecoxib in human. Only a small percentage of the dose was recovered in urine as unchanged valdecoxib or as nonconjugated phase I metabolites. The glucuronide conjugates of valdecoxib and its hydroxylated metabolites were the major urinary metabolites of valdecoxib. The O-glucuronide of M1 andN-glucuronide of valdecoxib accounted for 23.3 and 19.5% of the administered dose, respectively. The N-glucuronide pathway is often species dependent (Lee, 1998), which is also true for valdecoxib (N-glucuronide of valdecoxib was found in the monkey but absent in the rat and the dog).
Less than 1% of the administered radioactive dose was recovered as unchanged valdecoxib in feces, suggesting that the absorption of valdecoxib from the suspension formulation was excellent. This was later confirmed in a separate study, in which the absolute bioavailability of valdecoxib (BEXTRA) in human subjects is 83% (see BEXTRA package insert, available at www.bextra.com).
Acknowledgment
Special thanks to Ming Chang for LC-MS/MS work and Elizabeth Hajdu for NMR work.
Footnotes
- Abbreviations used are::
- valdecoxib
- 4-(5-methyl-3-phenyl-isoxazol-4-yl)benzenesulfonamide (code name, SC-65872)
- COX
- cyclooxygenase
- HPLC
- high-performance liquid chromatography
- HPLRC
- high-performance liquid radiochromatography
- LC-MS/MS
- liquid chromatography-tandem mass spectometry
- CID
- collision-induced dissociation
- NMR
- nuclear magnetic resounance
- M1-G
- M2-G, M3-G… , glucuronide conjugate of M1, M2, M3…
- V-G
- N-glucuronide conjugate of valdecoxib
- RBC
- red blood cell
- SC-66905
- 4-[5-(hydroxymethyl)-3-phenylisoxazol-4-yl]benzenesulfonamide
- Received March 20, 2002.
- Accepted June 12, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}